Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Rearrangements in the 5⬘
Nontranslated Region and Phylogenetic
Analyses of Cucumber Mosaic Virus RNA 3 Indicate
Radial Evolution of Three Subgroups
MARILYN J. ROOSSINCK,1* LEE ZHANG,2ANDKARL-HEINZ HELLWALD2†
Plant Biology Division, The Samuel Roberts Noble Foundation, Ardmore, Oklahoma 73402,1and
Department of Plant Pathology, Cornell University, Ithaca, New York 148532
Received 22 February 1999/Accepted 16 April 1999
Cucumber mosaic virus (CMV) has been divided into two subgroups based on serological data, peptide
mapping of the coat protein, nucleic acid hybridization, and nucleotide sequence similarity. Analyses of a number of recently isolated strains suggest a further division of the subgroup I strains. Alignment of the 5ⴕ
nontranslated regions of RNA 3 for 26 strains of CMV suggests the division of CMV into subgroups IA, IB, and II and suggests that rearrangements, deletions, and insertions in this region may have been the precursors of the subsequent radiation of each subgroup. Phylogeny analyses of CMV using the coat protein open reading frame of 53 strains strongly support the further division of subgroup I into IA and IB. In addition, strains within each subgroup radiate from a single point of origin, indicating that they have evolved from a single common ancestor for each subgroup.
Cucumber mosaic virus(CMV), genusCucumovirus, family Bromoviridae, is a positive-sense RNA plant virus with a tri-partite genome (for a review see reference 12). RNAs 1 and 2 encode the nonstructural proteins involved in viral replication. RNA 3 encodes a movement protein and the coat protein (CP), which is translated from a subgenomic mRNA, RNA 4. A fifth open reading frame (ORF), the 2b ORF, is also en-coded on RNA 2. It has been implicated in virus movement and symptom severity (3–5). CMV has an extremely broad host range (approximately 1,000 species), and numerous strains of CMV have been described. Serological data, peptide mapping of the CP, and nucleic acid hybridization divided CMV strains into two subgroups, designated I and II. Sequence analysis of a representative strain from each subgroup verified the desig-nations (12). Over the past several years a large number of additional strains have been described and partially sequenced, and recent analysis of the CP genes of several subgroup I strains (2) suggests that they can be further divided into two groups.
While divergence and speciation of RNA viruses probably occurs rapidly and frequently, little is known about the mech-anisms of these events (14). Clearly, rapid mutation rates can play a role (6, 7), but other events like reassortment in viruses with divided genomes and RNA-RNA recombination are probably also important driving forces in RNA virus evolution (9, 15). Previously it was shown that phylogenetic estimates for the Cucumovirusgenus were different when the ORFs from different RNAs were used for analysis, suggesting that reas-sortment of the three genomic segments may have played an important role in the speciation of this genus (16). A naturally occurring reassortant between CMV and Peanut stunt virus (PSV), genus Cucumovirus, supported this theory (16). Al-though speculation about the role of RNA recombination in
the speciation of RNA viruses has been common, the capacity for rapid change by mutation in RNA virus genomes has made the footprints of previous recombination events difficult to uncover. In one study comparing two members of the genus Bromovirus, family Bromoviridae, however, evidence for past recombination events in the 5⬘ nontranslated regions (NTRs) of RNAs 3 was noted (1). Among the Bromoviridae, the 5⬘
NTRs of RNAs 1 and 2 are conserved, both within and be-tween viruses of the same species. The 5⬘NTRs of RNAs 3 are less similar to the 5⬘NTRs of RNAs 1 and 2 of the same virus but are conserved between viruses.
In this study, the 5⬘NTRs of RNA 3 for 26 strains of CMV were aligned. The NTRs fall into three groups, one corre-sponding to subgroup II strains and two correcorre-sponding to sub-group I strains, suggesting further division of the subsub-group I strains into IA and IB.
To confirm the subgrouping, a phylogenetic analysis was done on CMV strains. Most of the sequence data available for CMV are for the CP gene. A limited number of strains have had the entire RNA 3 analyzed, and still fewer have had the complete sequence of RNAs 1 and 2 determined. All previous taxonomic considerations for CMV have been based on the CP. Here we used the sequences of the CP genes of 53 strains of CMV and of the CP gene from the ER strain of PSV (11), anotherCucumovirusspecies, to estimate the phylogenetic re-lationships of CMV. The analysis clearly supports the same subdivision of subgroup I strains into IA and IB groupings as seen in the 5⬘NTR analysis.
MATERIALS AND METHODS
Source of sequence data.Sequence data for RNA 3 and the CP were obtained from GenBank (accession numbers are shown in Table 1). The cloning of LS-CMV has been described elsewhere (17). The cDNA clone of RNA 3 was sequenced by standard dideoxy sequencing techniques using Sequenase (U.S. Biochemical). K-CMV RNA 3 was cloned by using a cDNA cloning kit (Amer-sham) and a primer specific to the 3⬘end of subgroup I CMV strains. The 5⬘642 nucleotides of the K-CMV RNA 3 cDNA clone (pK302) were derived by reverse transcription-PCR using avian myeloblastosis virus reverse transcriptase, 30 ther-mocycles withTaqpolymerase, primerNheI (5⬘CACGCTAGCTGTGGTACCG G3⬘), and 5⬘-terminal primer (BamHI site; T7 RNA polymerase promoter-GT AATCTTACCACTG). Plasmid pK302 was sequenced by using a Taq DyeDeoxy Terminator cycle sequencing kit (Perkin-Elmer/Applied Biosystems, Foster City, * Corresponding author. Mailing address: The S. R. Noble
Founda-tion, Plant Biology Division, P.O. Box 2180, Ardmore, OK 73402-2180. Phone: (580) 223-5810. Fax: (580) 221-7380. E-mail: mroossinck @noble.org.
† Present address: Institute of Phytomedicine, University of Hohen-heim, 70593 Stuttgart, Germany.
6752
on November 9, 2019 by guest
http://jvi.asm.org/
Calif.). The products of the reaction were separated electrophoretically, and the data were processed by an ABI model 373A automated DNA sequencer (Perkin-Elmer/Applied Biosystems). DNA sequences were assembled and edited with the PC/Gene program ASSEMGEL (IntelliGenetics, Mountain View, Calif.).
5ⴕNTR and phylogenetic analyses.Nucleotide sequences of the 5⬘NTR or of the CP gene were initially aligned with the Pileup program of the Wisconsin Package, version 9.0 (Genetics Computer Group, Madison, Wis.). Alignments were edited with the SEQUENCE editing and analysis program, version 3.0.4, from Gary Olsen. Phylogenetic analyses were performed with PAUP 4.0b1 (Smithsonian Institution). Characters were either unordered or assigned user-defined weights, and gaps were treated as a fifth character state. All analyses were tested by at least 100 bootstrap replicates for confidence levels, and branches with less than 70% bootstrap support were collapsed. The larger data set was analyzed by the heuristic algorithm in the parsimony setting. Smaller data sets were also analyzed by the branch-and-bound algorithm. In addition, all data sets were analyzed in the maximum likelihood setting, using a fast branch swap-ping search. Character state changes were calculated by using MacClade 3.0
(developed by D. Maddison and W. Maddison; obtained from Sinauer Associ-ates, Inc.).
Nucleotide sequence accession numbers.The sequences of LS-CMV RNA 3 and K-CMV RNA 3 were deposited in GenBank with accession no. AF127976 and AF127977, respectively.
RESULTS
Analysis of the 5ⴕNTR of RNA 3.A set of 26 strains of CMV that have sequence data available for the entire RNA 3 were used for the 5⬘ NTR analysis. When these were aligned with PileUp, the alignment was very poor, even when the gap pen-alties were adjusted. The use of color-highlighted fonts for the A, C, G, and U nucleotides assisted in constructing a better alignment and revealed several interesting features of these sequences (Fig. 1A). The subgroup II 5⬘NTR sequences are nearly identical, with only strains Trk7 and M2 showing minor differences compared with the other four strains. However, the subgroup I strains are more divergent and can be clearly di-vided into two further subgroups (Fig. 1). A number of motifs can be identified in the 5⬘NTRs. Boxes A and F, at the 5⬘and 3⬘ termini, respectively, of the NTR, are highly conserved among all strains. Box D is a tandem repeat in all of the subgroup I sequences but is not found in subgroup II (Fig. 1A). A more striking observation concerns the differences in the 5⬘ NTRs of these RNAs. Boxes B, C, and E contain regions where there is considerable subgroup-specific variation. Boxes B and E contain motifs in subgroup I strains that appear in another position in subgroup II strains. This rearrangement is most obvious when we compare the second subgroup I set, designated IB, and the subgroup II strains (1. and 2. in Fig. 1A and B). If two rearrangement events are invoked in the sub-group IB consensus sequence, the resulting sequence is very similar to that for the subgroup II 5⬘ NTRs (Fig. 1B, RIB) requiring only several deletions of short regions of nucleotides to generate the extant sequence. An alignment of the consen-sus sequence for subgroups IA and IB shows two deletions, one in the IA sequence and the other in the IB sequence (Fig. 1C), suggesting that these subgroups could have arisen from a pro-genitor CMV by separate deletion events.
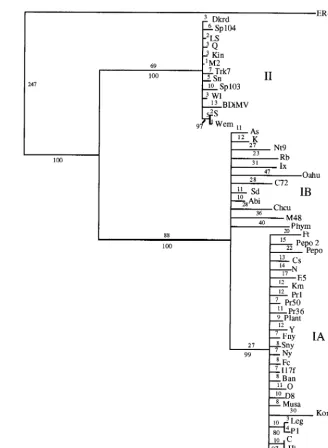
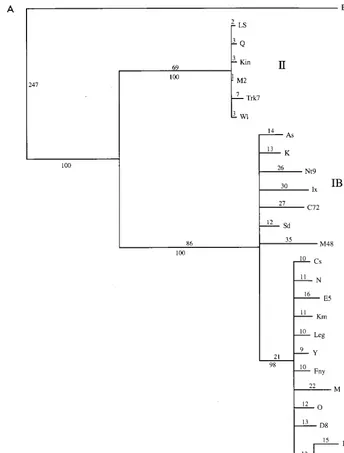
Phylogenetic analyses. The initial data set containing 53 CMV taxa was analyzed by both a heuristic bootstrap search using the maximum parsimony setting and a fast branch swap-ping search using maximum likelihood. The CP gene of the ER strain of PSV was used as an outgroup. The topologies of both trees were essentially identical, with three major clades coin-ciding with strains from CMV subgroups IA, IB, and II (Fig. 2 and data not shown). The large size of the data set precluded a more detailed analysis, but a subset of the entire group, using the 26 strains from the 5⬘ NTR study, was analyzed in more detail, using a branch-and-bound search (Fig. 3A). The topol-ogies of the two trees were essentially identical, with very little internal branching within the groups. To analyze the within-group relationships, each within-group was subjected to a more rig-orous analysis with 1,000 bootstrap replications of a branch-and-bound search. For these analyses, two members of the most closely related group were used as an outgroup: LS- and Q-CMV for subgroup I; K- and Nt9-CMV for subgroup IA; Fny- and O-CMV for subgroup IB; and Fny- and K-CMV for subgroup II. The results from maximum parsimony and max-imum likelihood analyses gave nearly identical results. In all cases, the groups have identical patterns of relationship to the complete analysis, and the CMV strains appear to have a “star phylogeny” pattern of evolution (Fig. 2 and 3).
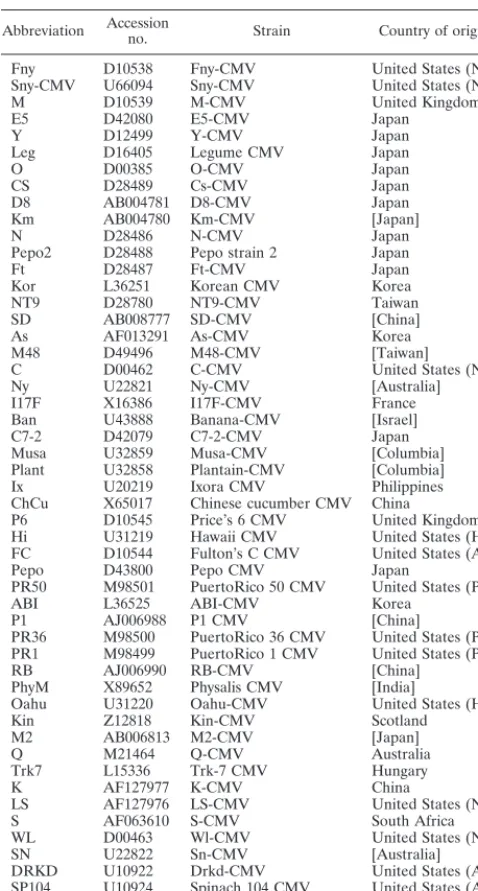
[image:2.612.55.294.83.528.2]The default settings for standard phylogeny estimations are for unordered character states; that is, each type of nucleotide change is given an equal probability of occurring. However, in TABLE 1. Strain abbreviations and GenBank accession numbers
Abbreviation Accessionno. Strain Country of origina
Fny D10538 Fny-CMV United States (NY)
Sny-CMV U66094 Sny-CMV United States (NY)
M D10539 M-CMV United Kingdom
E5 D42080 E5-CMV Japan
Y D12499 Y-CMV Japan
Leg D16405 Legume CMV Japan
O D00385 O-CMV Japan
CS D28489 Cs-CMV Japan
D8 AB004781 D8-CMV Japan
Km AB004780 Km-CMV [Japan]
N D28486 N-CMV Japan
Pepo2 D28488 Pepo strain 2 Japan
Ft D28487 Ft-CMV Japan
Kor L36251 Korean CMV Korea
NT9 D28780 NT9-CMV Taiwan
SD AB008777 SD-CMV [China]
As AF013291 As-CMV Korea
M48 D49496 M48-CMV [Taiwan]
C D00462 C-CMV United States (NY)
Ny U22821 Ny-CMV [Australia]
I17F X16386 I17F-CMV France
Ban U43888 Banana-CMV [Israel]
C7-2 D42079 C7-2-CMV Japan
Musa U32859 Musa-CMV [Columbia]
Plant U32858 Plantain-CMV [Columbia]
Ix U20219 Ixora CMV Philippines
ChCu X65017 Chinese cucumber CMV China
P6 D10545 Price’s 6 CMV United Kingdom
Hi U31219 Hawaii CMV United States (HI)
FC D10544 Fulton’s C CMV United States (AR)
Pepo D43800 Pepo CMV Japan
PR50 M98501 PuertoRico 50 CMV United States (PR)
ABI L36525 ABI-CMV Korea
P1 AJ006988 P1 CMV [China]
PR36 M98500 PuertoRico 36 CMV United States (PR)
PR1 M98499 PuertoRico 1 CMV United States (PR)
RB AJ006990 RB-CMV [China]
PhyM X89652 Physalis CMV [India]
Oahu U31220 Oahu-CMV United States (HI)
Kin Z12818 Kin-CMV Scotland
M2 AB006813 M2-CMV [Japan]
Q M21464 Q-CMV Australia
Trk7 L15336 Trk-7 CMV Hungary
K AF127977 K-CMV China
LS AF127976 LS-CMV United States (NY)
S AF063610 S-CMV South Africa
WL D00463 Wl-CMV United States (NY)
SN U22822 Sn-CMV [Australia]
DRKD U10922 Drkd-CMV United States (AR)
SP104 U10924 Spinach 104 CMV United States (AR)
SP103 U10923 Spinach 103 CMV United States (AR)
Wem L40953 Wem CMV Unknown
aIndividual states or territories in the United States are shown in parentheses
in standard postal code abbreviation; countries shown in brackets are locations of the researchers reporting the sequence, where no description of the strain origin is given.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 1. Nucleotide sequence rearrangements in the 5 ⬘ NTR of CMV. (A) Alignment of the 5 ⬘ NTRs of 26 strains. Bases are shown with colored highlights. Boxes A and F are conserved in all strains. Boxes B, C, and E are variable. Boxes D1 and D2 are direct repeats conserved in all subgroup I strains. Motifs that have apparently been rearranged are indicated as 1. and 2. (B) Alignments and potential rearrangements in the consensus sequences of the 5 ⬘ NTRs of subgroup IB and II. The top portion shows the consensus sequence for subgroup IB. 1. and 2. indicate the two motifs that are rearranged to form the sequence in the lower portion (RIB, rearranged IB) by deletion and insertion at the respective diamonds. The rearranged sequence is shown in the bottom portion, aligned with the co nsensus subgroup II sequence. (C) Nucleotide sequence alignment with potential deletion events indicated, of the subgroup IA and IB consensus sequences.
on November 9, 2019 by guest
http://jvi.asm.org/
comparisons of viral sequences, certain types of changes are far more common than other types, and these biases can be dif-ferent for difdif-ferent viruses (reviewed in reference 13). Transi-tions between A and G or between C and U are much more common than transversions between A and U. Transversions between C and A or G are extremely rare. Hence the character state changes are probably ordered, such that while a U-to-C change may be a single step that could be given a relative value of 1, an A-to-C change would have a higher cost, and the change may occur more readily by two steps: A to U (value greater than 1) and U to C (value of 1), for example. The PAUP program allows the user to supply a step matrix of character state changes to account for ordered change.
All phylogeny analyses are, at best, estimations. It is not possible to be certain that the relationships indicated are
[image:4.612.132.461.70.519.2]com-pletely accurate. To determine the most likely ordering of character state changes for CMV CP evolution, we estimated the frequency of various changes by constructing four separate trees with one member of each subgroup (ER-PSV as the outgroup in all cases, and different members of CMV sub-groups IA, IB, and II as insub-groups). By using simple trees with only one member of each subgroup the confidence in each tree is very high, and the resulting information about the character state changes is most likely accurate. The frequency of each type of change was calculated from these simple trees by using MacClade 3.0. The character state changes were very similar for each tree, and Table 2 shows the average frequency of each type of change occurring at all homologous sites. These num-bers were used to generate a step matrix of character state changes, such that a greater cost is assigned to changes that are
FIG. 2. Phylogenetic estimation of 53 strains of CMV based on the CP ORF, derived from 100 bootstrap replicates, using the heuristic search method of PAUP 4.0b1. All branches with less than 70% bootstrap support were collapsed. Numbers above the lines indicate branch lengths determined from unordered character state changes (i.e., number of changes); numbers below the lines indicate bootstrap values from the corresponding cladogram.
on November 9, 2019 by guest
http://jvi.asm.org/
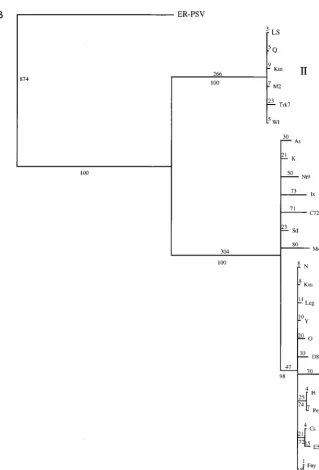
very rare or may require more than one step. The phylogeny estimation was repeated for both the individual groups and the subset of sequences, using the new step matrix. The relation-ships of the strains were not changed, and each subgroup still exhibited a star phylogeny. However, the branches lengths were increased in a nonproportional manner, suggesting that the very rare changes may be more common in some lineages than in others (compare Fig. 3A and B).
DISCUSSION
Mechanisms of RNA-RNA recombination in plant viruses have been studied in the bromoviruses and inTurnip crinkle
virus (reviewed in reference 15). These mechanisms can in-volve either heterologous or homologous recombination. Het-erologous recombination events require a short region of complementarity between two templates, which can then form a short heteroduplex. The polymerase switches from one tem-plate to the other rather than unwinding the heteroduplex region. Homologous recombination uses a copy-choice mech-anism, whereby the replication complex falls off one template, the nascent chain anneals with another template, and the rep-lication proceeds on the new template. The apparent rear-rangements in the 5⬘NTRs of RNA 3 must have occurred via RNA-RNA recombination events. There is no clear evidence, however, for the regions either of complementarity or of
sim-FIG. 3. Detailed analysis of a subset of 26 strains of CMV. (A) Phylogram showing branch lengths when characters are unordered. Numbers represent actual number of nucleotide changes. Bootstrap values from the corresponding cladogram are shown below the branch points. (B) Phylogram generated by using a step matrix of character state changes as described in the text. Branch lengths are proportional to the cost of the nucleotide changes. Numbers represent total relative cost. Bootstrap values from the corresponding cladogram are shown below the branch points.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.127.471.76.529.2]ilarity that are required for these mechanisms, in the extant sequences of the CMV RNAs 3 5⬘NTRs, and hence the mech-anism of recombination that gave rise to the 5⬘ NTRs of the various CMV subgroup RNAs 3 is not apparent. The progen-itor virus could have contained sequences that have since been
lost in all of the extant strains, or the recombination events could have involved novel mechanisms of recombination.
One of the subgroup I CMV strains used in this analysis, C72-CMV, appears to be an intermediate between the IA and IB strains by the 5⬘NTR analysis, with the 5⬘portion (box C) more similar to IB and the 3⬘portion (box E) more similar to IA. However, the phylogenetic analysis clearly places the C72 CP into the subgroup IB viruses. C72 could contain a more ancestral form of the 5⬘NTR, or it could be the product of a more recent recombination event between subgroups IA and IB.
[image:6.612.147.466.71.540.2]Although sequence data are available for RNAs 1 and 2 of only a limited number of these strains, such rearrangements were not apparent in the other 5⬘ NTRs, nor was their any evidence of rearrangements in the 3⬘NTRs or the intergenic region of RNAs 3 (data not shown). It is possible that the TABLE 2. Summary of frequency of specific nucleotide changes in
CMV CP gene evolution
Nucleotide Frequency changes
A C G U
A 0 4 1
C 1 0 6
G 6 0 0
[image:6.612.53.293.656.728.2]U 1 8 1
FIG. 3—Continued.
on November 9, 2019 by guest
http://jvi.asm.org/
rearrangements of the RNA 3 5⬘NTRs were the initial events that gave rise to the various subgroups, which later accumu-lated differences in their various ORFs. Estimations of relat-edness based on the CP genes of these strains have previously relied only on sequence similarity (distance methods), and hence it was not possible to determine if the subgroup II strains are closer to the IA or the IB strains. The phylogeny estimations shown here indicate that the subgroup II strains are the most closely related to the ancestral state, with the IB subgroup falling out as a sister clade to them. This suggests that the 5⬘NTR shown in Fig. 1B (RIB) may be the closest to the ancestral 5⬘NTR and that deletions gave rise to subgroup II, whereas rearrangements gave rise to subgroup IB. Sub-group IA falls as a sister clade to the IB strains, indicating that it arose from IB and is the result of the most recent subspe-ciation event.
The evolutionary patterns indicated by the phylogeny esti-mations of CMV using CP sequences are consistent with a few events in which CMV passed through a narrow bottleneck and was disseminated from that point to many parts of the world. The first radiation, of subgroup II strains, is worldwide. From those strains, a second radiation gave rise to the subgroup IB strains. This distribution may have been more limited, as all subgroup IB strains described so far originated in Asia. From the subgroup IB strains there occurred a third radiation event that gave rise to the subgroup IA strains, and again there was a worldwide distribution. These types of radiation patterns, or star phylogenies, have been noted in recently emerged viruses such asSimianandHuman immunodeficiency viruses, and rapid radiation may be a common outcome of emergence (10). This would suggest that CMV may also have emerged relatively recently. However, without a time line for virus evolution, it is very difficult to speculate about when these events occurred.
An alternate hypothesis that could also be consistent with the data is that the three subgroups of CMV represent the collection of variants around three fitness peaks. This would indicate that, rather than being a recently emerged virus, CMV has simply reached three stable equilibria instead of one. Were this the case, however, one might expect the three subgroups to fall out as sister clades, rather than successive subclades. In addition, our recent studies on mutation frequencies in CMV (to be published elsewhere) suggest that it is a rapidly evolving virus.
The phylograms show another interesting feature of CMV evolution. The branch lengths of the subgroup II strains are quite short in comparison to those for the subgroup I strains, and the subgroup IB strains show the most divergence from each other. This suggests that the subgroup I strains are evolv-ing more rapidly, although it is not possible to put any timeline on this evolution, and hence rates are not directly comparable. These differences may be reflective of the broader host range and higher incidence of subgroup I strains compared with subgroup II strains.
The enormous amount of variation seen in RNA virus ge-nomes has allowed them to be very successful at infecting new hosts and evading the host’s defense responses. Clearly
varia-tion is an advantage, and viruses are believed to exist at the threshold of catastrophe (8). The evolution of RNA viruses is undoubtedly a series of complex events that involves all possi-ble mechanisms to introduce variation, including mutation, reassortment, and recombination. Any or all of these events may play a pivotal role in virus speciation.
ACKNOWLEDGMENTS
We thank Peter Nagy for helpful discussions on RNA recombina-tion, and we thank Stan Flasinski, Joachim deMiranda, Richard Nel-son, William Schneider, and Peter Palukaitis for careful reading of the manuscript.
This work was supported by the S. R. Noble Foundation.
REFERENCES
1.Allison, R. F., M. Janda, and P. Ahlquist.1989. Sequence of cowpea chlo-rotic mottle virus RNAs 2 and 3 and evidence of a recombination event during bromovirus evolution. Virology172:321–330.
2.Chaumpluk, P., Y. Sasaki, N. Nakajima, H. Nagano, I. Nakamura, K. Suziki, K. Mise, N. Inouye, T. Okuno, and I. Furusawa.1996. Six new subgroup I members of Japanese cucumber mosaic virus as determined by nucleotide sequence analysis of RNA3’s cDNAs. Ann. Phytopathol. Soc. Jpn.62:40–44. 3.Ding, S.-W., B. J. Anderson, H. R. Haase, and R. H. Symons.1994. New overlapping gene encoded by the cucumber mosaic virus genome. Virology
198:593–601.
4.Ding, S.-W., W. X. Li, and R. H. Symons.1995. A novel naturally occurring hybrid gene encoded by a plant RNA virus facilitates long distance virus movement. EMBO J.14:5762–5772.
5.Ding, S.-W., B.-J. Shi, W.-X. Li, and R. H. Symons.1996. An interspecies hybrid RNA virus is significantly more virulent than either parental virus. Proc. Natl. Acad. Sci. USA93:7470–7474.
6.Domingo, E., and J. J. Holland.1988. High error rates, population equilib-rium, and evolution of RNA replication systems, p. 3–36.InE. Domingo, J. J. Holland, and P. Ahlquist (ed.), Variability of RNA genomes, 1st ed., vol. III. CRC Press, Boca Raton, Fla.
7.Domingo, E., and J. J. Holland.1994. Mutation rates and rapid evolution of RNA viruses, p. 161–184.InS. S. Morse (ed.), The evolutionary biology of viruses. Raven Press, Ltd., New York, N.Y.
8.Holland, J. J., E. Domingo, J. C. delaTorre, and D. A. Steinhauer.1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol.64:3960–3962.
9.Lai, M. M. C.1992. Genetic recombination in RNA viruses. Curr. Top. Microbiol. Immunol.176:21–32.
10. Myers, G., K. MacInnes, and L. Myers.1993. Phylogenetic moments in the AIDS epidemic, p. 120–137.InS. S. Morse (ed.), Emerging viruses. Oxford University Press, New York, N.Y.
11. Naidu, R. A., G. B. Collins, and S. A. Ghabrial.1991. Nucleotide sequence analysis of a cDNA clone encoding the coat protein gene of peanut stunt virus. Plant Mol. Biol.17:175–177.
12. Palukaitis, P., M. J. Roossinck, R. G. Dietzgen, and R. I. B. Francki.1992. Cucumber mosaic virus. Adv. Virus Res.41:281–348.
13. Ramı´rez, B.-C., P. Barbier, K. Se´ron, A.-L. Haenni, and F. Bernardi.1995. Molecular mechanisms of point mutations in RNA viruses, p. 105–118.In
A. J. Gibbs, C. H. Calisher, and F. Garcı´a-Arenal (ed.), Molecular basis of virus evolution. Cambridge University Press, Cambridge, England. 14. Roossinck, M. J.1997. Mechanisms of plant virus evolution. Annu. Rev.
Phytopathol.35:191–209.
15. Simon, A. E., and J. J. Bujarski.1994. RNA-RNA recombination and evo-lution in virus-infected plants. Annu. Rev. Phytopathol.32:337–362. 16. White, P. S., F. J. Morales, and M. J. Roossinck.1995. Interspecific
reas-sortment in the evolution of a cucumovirus. Virology207:334–337. 17. Zhang, L., K. Hanada, and P. Palukaitis.1994. Mapping local and systemic
symptom determinants of cucumber mosaic cucumovirus in tobacco. J. Gen. Virol.75:3185–3191.