STA
T Thh
DEP ABILITY h
heeTTaammiillNN I
Innppaarrttiiaall
M
M
A
A
(
(
P
P
ARTMEN KMC KOVA
INDICAT
D
Diisssseerrttaatt N
NaadduuDDrr..MM l
lffuullffiillllmmeenn
A
A
S
S
T
T
E
E
R
R
P
P
h
h
a
a
r
r
m
m
a
a
c
c
A APP
NT OF PH CH COLL AI ESTAT COIMBA
TING STU
t
tiioonnSSuubbmmii M
M..GG..RR..MMeedd n
nttffoorrtthheeaa
I
I
N
N
P
P
H
H
A
A
c
c
e
e
u
u
t
t
i
i
c
c
a
a
l
l
A
A
P
PRRIILL--220011
HARMACE LEGE OF P TE, KALA ATORE -UDIES BY i
itttteeddttoo d
diiccaallUUnniivv a
awwaarrddoofftthh
A
A
R
R
M
M
A
A
C
C
A
A
n
n
a
a
l
l
y
y
s
s
i
i
s
s
)
)
2 2 EUTICAL PHARMA APATTI R 641048. Y RP-HPL v
veerrssiittyy,,CChhee thheeDDeeggrreeee
C
C
Y
Y
)
)
L ANALY ACY OAD, LC e ennnnaaii.. e eooffSTA
T Thh
DEP ABILITY h
heeTTaammiillNN I
Innppaarrttiiaall
M
M
A
A
(
(
P
P
ARTMEN KMC KOVA
INDICAT
D
Diisssseerrttaatt N
NaadduuDDrr..MM l
lffuullffiillllmmeenn
A
A
S
S
T
T
E
E
R
R
P
P
h
h
a
a
r
r
m
m
a
a
c
c
A APP
NT OF PH CH COLL AI ESTAT COIMBA
TING STU
t
tiioonnSSuubbmmii M
M..GG..RR..MMeedd n
nttffoorrtthheeaa
I
I
N
N
P
P
H
H
A
A
c
c
e
e
u
u
t
t
i
i
c
c
a
a
l
l
A
A
P
PRRIILL--220011
HARMACE LEGE OF P TE, KALA ATORE -UDIES BY i
itttteeddttoo d
diiccaallUUnniivv a
awwaarrddoofftthh
A
A
R
R
M
M
A
A
C
C
A
A
n
n
a
a
l
l
y
y
s
s
i
i
s
s
)
)
2 2 EUTICAL PHARMA APATTI R 641048. Y RP-HPL v
veerrssiittyy,,CChhee thheeDDeeggrreeee
C
C
Y
Y
)
)
L ANALY ACY OAD, LC e ennnnaaii.. e eooffK.M.C.H. College of Pharmacy,
Kovai Estate, Kalapatti Road,
Coimbatore -641048. (T.N)
CERTIFICATE
This is to certify that, the work embodied in the thesis entitled “DEVELOPMENT AND VALIDATION FOR THE DETERMINATION OF RELATED SUBSTANCE IN IRINOTECAN HCl FORMULATION AND ITS STABILITY INDICATING STUDIES BY RP- HPLC” is a
bonafide research work carried out by Mr. Rambabu Cherukuri (Reg. No:
26107226), Student in Master of Pharmacy, Department of Pharmaceutical Analysis, K.M.C.H. College of Pharmacy, Coimbatore, Tamilnadu, under the guidance of Prof. Dr. A. Rajasekaran, Dept of Pharmaceutical Analysis, K.M.C.H. College of Pharmacy during the academic year 2011-2012.
Date:
Place: Coimbatore.
Signature,
Prof. Dr. A. Rajasekaran, M. Pharm, Ph.D.,
Department of Pharmaceutical Analysis,
K.M.C.H. College of Pharmacy,
Coimbatore -641048.
CERTIFICATE
This is to certify that, the work embodied in the thesis entitled “DEVELOPMENT AND VALIDATION FOR THE DETERMINATION OF RELATED SUBSTANCE IN IRINOTECAN HCl FORMULATION AND ITS STABILITY INDICATING STUDIES BY RP- HPLC” is a
bonafide research work carried out by Mr. Rambabu Cherukuri (Reg. No:
26107226), Student in Master of Pharmacy, Department of Pharmaceutical Analysis, K.M.C.H. College of Pharmacy, Coimbatore, Tamilnadu, under my supervision and guidance during the academic year 2011-2012.
I am fully satisfied with his performance and work with great pleasure. I forward this Dissertation work for evaluation.
Date:
Place: Coimbatore.
Signature,
Prof. Dr. A. Rajasekaran, M. Pharm, Ph.D.,
Guide
I am here by stating that, to the best of my knowledge and belief, the project report entitled “DEVELOPMENT AND VALIDATION FOR THE DETERMINATION OF RELATED SUBSTANCE IN IRINOTECAN HCl FORMULATION AND ITS STABILITY INDICATING STUDIES BY RP- HPLC” being submitted for the partial fulfillment of Master of Pharmacy in Pharmaceutical Analysis for the academic year 2011-2012 of KMCH. College of Pharmacy affiliated to The Tamilnadu Dr. M.G.R. Medical
University carried out under the guidance of Prof. Dr. A. Rajasekaran, M.
Pharm, Ph.D., at the Department of Pharmaceutical Analysis, KMCH College of Pharmacy, Coimbatore.
I abide that all the data presented in this report will be treated with
utmost confidentiality.
Signature,
Date: Rambabu Cherukuri
This is to certify that, the work embodied in the thesis entitled “DEVELOPMENT AND VALIDATION FOR THE DETERMINATION OF RELATED SUBSTANCE IN IRINOTECAN HCl FORMULATION AND ITS STABILITY INDICATING STUDIES BY RP- HPLC” submitted
by Mr. Rambabu Cherukuri (Reg. No: 26107226), to The Tamilnadu Dr.
M.G.R. Medical University, Chennai, in partial fulfillment for the Degree of Master of Pharmacy, in Pharmaceutical Analysis, is a bonafide research work carried out by the candidate at K.M.C.H. College of Pharmacy, Coimbatore,
Tamilnadu, the same was evaluated by us during academic year 2011-2012.
Examination Center: KMCH College of Pharmacy, Coimbatore.
Date:
Internal Examiner External Examiner
My dissertation entitled “DEVELOPMENT AND VALIDATION FOR THE DETERMINATION OF RELATED SUBSTANCE IN IRINOTECAN HCl FORMULATION AND ITS STABILITY INDICATING STUDIES BY RP- HPLC” would not have been a feasible one without the grace of god almighty who gave me morale till the completion of my project.
To begin with I would like to thank Dr. Nalla G. Palanisamy, Chairman, and Dr. Thavamani D. Palanisamy, Managing Trustee, K.M.C.H. College of Pharmacy, Coimbatore for all the facilities, which have been provided to us at the institution, enabling me to do work of this magnitude.
I always remain indebted to my Academic Guide Dr. A. Rajasekaran, M. Pharm, Ph.D., Principal, K.M.C.H College of Pharmacy for his constant insight, guidance, countless serenity, encouragement and painstaking efforts in my project work. I am indebted to his kindness and never failing co – operation and provided facilities.
My esteemed and beloved staff Mr. J. Dharuman, M. Pharm., Professor, Department of Pharmaceutical Analysis, KMCH College of Pharmacy, for their sensible help and suggestions.
Sivakumar, M. Pharm., Assist. Professor, Dept. Pharm. Chemistry, Mrs. S. Hurmath Unnissa, M. Pharm., Assist. Professor, Dept. Pharm. Chemistry, Mr. C. Sundaramoorthi, M. Pharm., Professor, Dept. Pharm. Biotechnology and Mr. R. Arivukkarasu, M. Pharm., Assist. Professor, Dept. Pharmacognasy, for their sensible help and suggestions.
My special thanks to all teaching and non-teaching staff members of KMCH College Pharmacy, Coimbatore, Library and computer lab faculties who directly or indirectly gave a helping hand during the course of study.
I express my sincere thanks to all my classmates, senior, juniors, and friends for their timely help and co-operation.
Last but not the least; I thank all those persons who directly or indirectly helped me in this project work to finish with great success.
Yours sincerely,
Rambabu cherukuri
UV Ultra violet
IP Indian Pharmacopoeia
USP United States Pharmacopoeia
M.W Molecular weight
e.g. Example
% Percentage
PDA Photo Diode Array
ACN Acetonitrile
MET Methanol
mg Milligram
g gram
µg Microgram
mL Milliliter
w/w Weight by weight
µg/mL Microgram per milliliter
°C Degree Celsius
t Time
Abs. Absorbance
Conc. Concentration
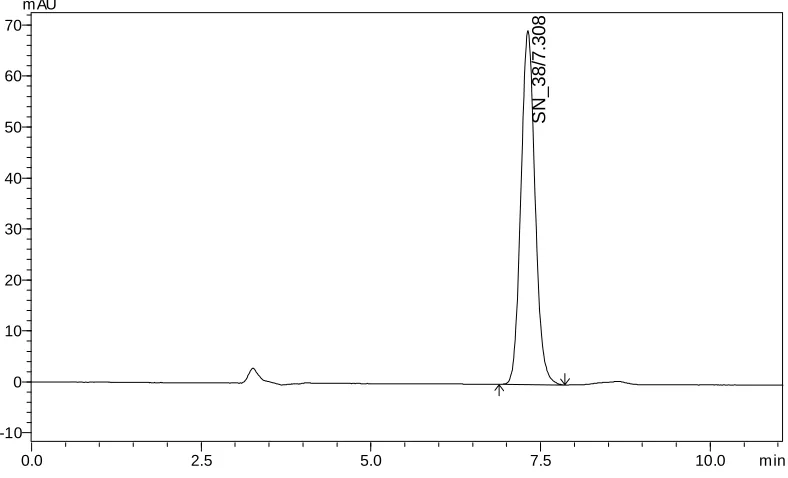
SN-38 7-ethyl 10-hydroxy camptothecin
min−1 Per one minute
CHAPTER
CONTENT
PAGE
1
Introduction
01
2
Literature
review
28
3
Drug
profile
34
4
Aim
and
objective
39
5
Plan
of
work
41
6
Experimental
44
7
Results
and
discussion
74
8
Summary
and
conclusion
87
9
Bibliography
89
10
Glossary
94
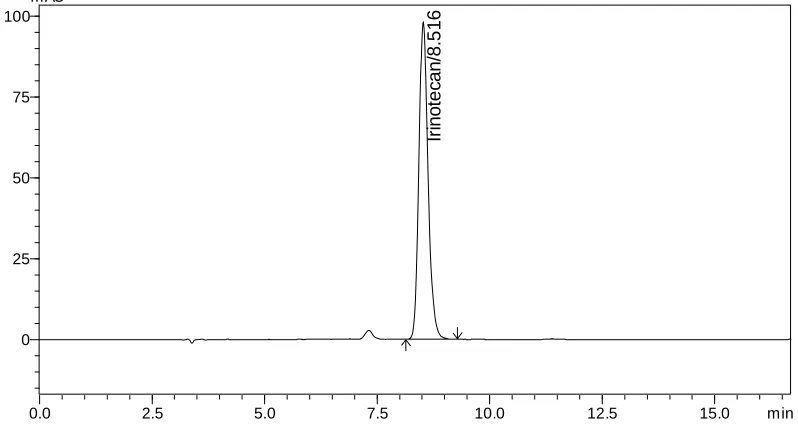
validated for the determination of Irinotecan HCl and its related substance (SN-38) in pharmaceutical dosage forms as per ICH guidelines. The separation achieved on a reversed phase Phenomenex Luna C18 Column, 5µ, 250 × 4.60 mm as a stationary phase and 0.5% trichloro
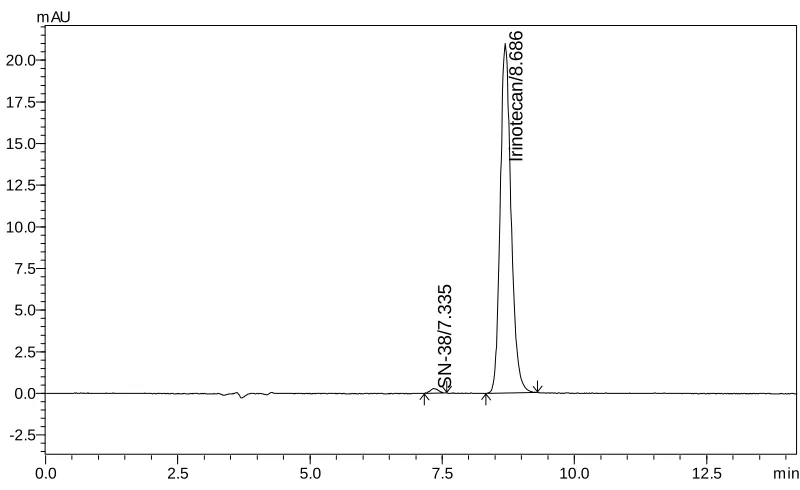
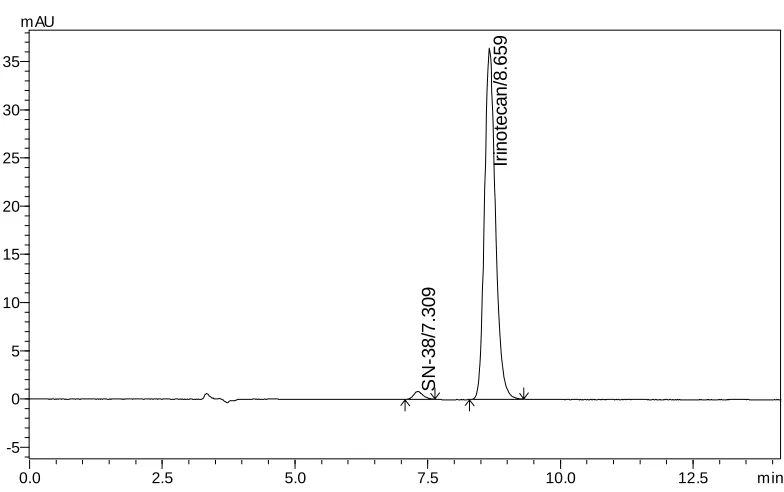
acetic acid: Acetonitrile: Methanol (60: 20: 20 v/v/v) as mobile phase at a flow rate of 1.0 mL/min. The UV detection was performed at 372 nm. The retention time (Rt) for Irinotecan HCl
and SN-38 was found to be 8.65 and 7.30 min respectively.The detector response was linear in the concentration range of 30-150 µg/mL. The respective linear regression equation being Y= 5233.x + 13299 with R2 0.999. The percentage of Irinotecan HCl in pharmaceutical dosage form was found to be 100.5% and the percentage of related substance (SN-38) in formulation was found to be 0.19%. The limit of detection of Irinotecan was 0.014 µg/mL and the limit of quantification was 0.045 µg/mL. The results of the study showed that, the proposed RP-HPLC method was simple, rapid, precise, accurate and stability indicated, which can be used for the routine determination of Irinotecan HCl and its related substance (SN-38) in pharmaceutical dosage form.
CHAPTER-1
Chapter 1 INTRODUCTION (1-24) IMPURITY STUDIES (1-15)
mpurities in a drug substance (a new chemical entity of therapeutic interest) or drug product (a drug substance formulated into a suitable product for administration to patient) may cause serious side effects and hence we need to isolate and characterize impurities.
The following definition of impurity is currently under consideration by the regulatory bodies, which is likely to be included in the future guidance:
Impurity: any entity of the drug substance (bulk material) or drug product (final container product) that is not the chemical entity defined as the drug substance, an excipient, or other additives to the drug product.
Related substances: These substances are structurally related to the drug substance and may be identified or unidentified degradation products or impurities arising from a manufacturing process or during storage of a material.
Impurity profile has become essential as per various regulatory requirements. Various regulatory authorities like ICH, USFDA, Canadian Drug and Health Agency are emphasizing on the purity requirements and the identification of impurities in Active Pharmaceutical Ingredient’s (API’s). Qualification of the impurities is the process of acquiring and evaluating data that establishes biological safety of an individual impurity; thus, revealing the need and scope of impurity profiling of drugs in pharmaceutical research The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) has also published guidelines for validation of methods for analyzing impurities in new drug substances, products, residual solvents and microbiological impurities.[2-5]
As per ICH guidelines Impurities are listed alphabetically as given below.
• By-product
• Degradation product
• Interaction product
• Intermediate
• Penultimate intermediate
• Related product
• Transformation product
By-products: The unplanned compounds generated in the reaction to produce API are generally called by-products. For example during the synthesis of paracetamol we get diacetylated paracetamol which is the unexpected product called by-product.
Degradation products: The compounds produced as a result of decomposition of the material of interest or API is often called degradation products. Due to degradation, impurities may also be formed. Degradation of beta lactum ring in penicillin and cephalosporin are the examples of degradation products. (4-formylphenyl) propionic acid, (4-methylphenyl) propionic acid , 2-(4-ethylphenyl) propionic acid , 4- isobutylacetophenone , 2-(4-n-propylphenyl) propionic acid and 2-(4-n-butylphenyl) propionic acid are the reported degradation products of ibuprofen.
Interaction products: This term is slightly more inclusive and more difficult to evaluate than the two previously described, i.e., by-products and degradation products, in that it takes into account interactions that could possibly occur between various involved chemicals – purposely or inadvertently.
Intermediates: The planned compounds produced during synthesis of the desired substance are called intermediates, especially if they have been isolated and characterized.
Penultimate intermediate: As the name implies, this is the last compound in the synthetic chain just preceding the production of the ultimate desired compound.
Related products: As suggested previously, the term “related products” tends to imply that the impurity is similar to the drug substance, and it thus tends to downplay the negativity frequently attached to the term “Impurity”.
1.1.Classification of impurities
According to ICH guidelines, impurities can be broadly classified in to three categories for the drug substance produced by chemical synthesis:
• Organic impurities (starting materials, process-related products, inter- mediators, and degradation products).
• Inorganic impurities (salts, catalysts, ligands, and heavy metals or other residual metals).
• Residual solvents (organic and inorganic liquids used during production and/or recrystallization).
1.2. Need to Isolate and Characterize Impurities
be readily apparent; however, on further thought it will become clear that if we are to ensure that the accurate amount of the drug substance is being administered to the patient, we must assess its purity independent of the extraneous materials. Therefore, any extraneous material present in the drug substance or active ingredient must be considered an impurity even if it is totally inert or has superior pharmacologic properties, so that an appropriate evaluation of its content in the drug product can be made. The control of low-level impurities is of great importance when a drug is taken in large quantities. Impurities can also affect the purity of API or can be harmful to patients.
1.2. Sources of Impurities
Impurities can originate from various sources, the most obvious source of impurities is from synthesis of API, where intermediates and by-products may be carried into the API as impurities or become a source of other impurities resulting from them. Following are the sources of the impurities
1.3.1. Crystallization- related impurities
Polymorphism is the term used to denote crystal systems where a substance can exist in different crystal packing arrangements, all of which have the same elemental composition. It is also possible to have crystal systems where the substance exists in different crystal packing arrangements, each of which has a different elemental composition; this phenomenon is known as solvatomorphism. The nature of the crystal structure of a given material can influence the following properties:
• Conductivity
• Crystal hardness
• Crystal shape and color
• Density
• Diffusivity
• Dissolution rate
• Enthalpy of transitions
• Heat capacity
• Heat of solution
• Hygroscopicity
• Latent heat of fusion
• Melting or sublimation properties
• Phase diagrams
• Rates of reactions
• Refractive index
• Solubility
• Surface tension
• Viscosity
• Volume
1.3.2. Stereochemistry-related impurities
Substances having similar chemical structure but having different spatial orientation are considered as stereo-chemical impurities in the API. Stereoisomerism is possible in molecules that have any of the following characteristics:
• One or more center of chirality
• Helicity (e.g., helical nature of tertiary structure of proteins, polysaccharides and nucleotides)
• Planar chirality(e.g., polycyclophanes)
• Axial chirality (e.g,.spiranes with cyclic skeleton )
• Torsional chirality (e.g., torsion about double or single bonds like cis and trans isomers and rotomers)
• Topological asymmetry (e.g., catenanes).
1.3.3 Residual solvents
Water is commonly present in drug products. As a result, water is by far the most commonly found volatile impurity in drug products, and most of the time it is not even considered an impurity. A number of solvents that are used for the synthesis of the API or formulation of the drug product can be present in the drug product. The content of these solvents, which are commonly called organic volatile impurities (OVI), is generally determined by the OVI methods specified in the compendia.
1.3.4. Synthetic intermediates and by-products
In addition to the residual solvents, polymorphic, solvatomorphic, and chiral impurities mentioned previously, impurities in a pharmaceutical compound or a new chemical entity (NCE) can originate during the synthetic process from raw materials, intermediates, and/or by-products. The solvents used in synthesis are also likely to involve a number of impurities that may extend from trace levels to critical quantities that can react with various chemicals used in the synthesis, to give rise to other impurities.
1.3.5. Formulation-related impurities
As mentioned before, many impurities in a drug product can originate from excipient used to formulate a drug substance. In addition, a drug substance is subjected to a variety of conditions in the process of formulation that can cause its degradation or have other undesirable reactions. For example if heat is used for drying purpose or for some other reason, it can hasten degradation.
1.3.6. Impurities arising during storage
1.3.7. Method related impurity
Terminal sterilization by autoclave for the diclofenac injectables known to produse impurity of 1-(2, 6-dichlorophenyl) indolin-2-one. Intramolecular cyclic reaction of diclofenac sodium cause the formation of indolinone impurity during autoclave method.
1.3.8. Mutual interaction amongst ingredients
In multi component dosage forms there may be a chance to get mutual interaction, particularly this occurs in multivitamin dosage forms. the presence of nicotinamide in a formulation containing four vitamins (nicotinamide, pyridoxine, riboflavin, and thiamine) can cause the degradation of thiamine to a sub-standard level within a one year shelf life of vitamin B-complex injections.
1.3.9 Functional group related degradations
API containing ester groups can readily be hydrolysed causing the formation of degraded products. For Eg aspirin, benzocaine, benzylpenicillin, oxazepam and lincomycin,cefotaxime, ethyl paraben, and cefpodoxime undergo hydrolysis readily causing the degradation of API.
API containing hydroxyl group directly bonded to an aromatic ring (viz phenol derivatives such
as catecholamines and morphine), conjugated dienes (viz vitamin A and unsaturated free fatty
acids), heterocyclic aromatic rings, nitroso and nitrite derivatives, and aldehydes (especially flavorings) are all susceptible to oxidative degradation.
Some of the API undergoes degradation on exposure to light. API like Ergometrine [27], nifedipine [28], nitroprusside, riboflavin and phenothiazines are veryliable to photo-oxidation.
1.4. ANALYTICAL METHOD DEVELOPMENT
Analytical method development for the quantification of low-level impurities present in pharmaceuticals can be thought of as a three-step process:
2. Screening of chromatographic conditions and phases, typically using the linear-solvent-strength model of gradient elution
3. Optimization of the method to fine-tune parameters related to ruggedness and robustness. This can be accomplished using a factorial optimization approach.
1.5 CHARACTERISATION OF IMPURITIES
1.5.1. SPECTROSCOPIC AND SEPARATION METHODS
The following spectroscopic measurement techniques have been used for characterizing and isolating impurities; most of these are very useful as detectors for chromatographic methods:
• Ultraviolet(UV)
• Infrared(IR)
• Raman spectroscopy
• Mass spectrometry(MS)
• Nuclear magnetic resonance(NMR)
• Capillary electrophoresis (CE)
• Thin-layer chromatography(TLC)
• Gas chromatography (GC)
• High- pressure liquid chromatography (HPLC)
• Supercritical fluid chromatography (SFC)
• Chiral separations
Ultraviolet spectrophotometry (UV) at a single wavelength furnishes minimum selectivity of analysis; however, with the current accessibility of diode array detectors, it is conceivable to obtain sufficient simultaneous information at various wavelengths to assure greater reliability.
Infrared spectrophotometry (IR) affords specific information on some functional groups that offer selectivity and aloe quantification.
Thin-layer chromatography coupled with densitometric detection is a highly sensitive method for quick assessment of the purity of various compounds including reference standards.
Mass spectrometry (MS) provides excellent structural information, and, based on the resolution of the instrument, it may be an effective tool for differentiating molecules with small differences in molecular weight. However, it has finite uses as a quantitative procedure.
Nuclear magnetic resonance spectroscopy (NMR) provides reasonably detailed structural information on a molecule and is an extremely useful method for characterization of impurities; however, its use as a quantitative method is limited.
Capillary electrophoresis is an effective technique in situations where very low quantities of samples are available and high resolution is essential.
Gas chromatography is an extremely useful technique for quantification. It can afford the desired resolution, selectivity and ease of quantification.
High-pressure liquid chromatography is often referred to as high-performance liquid chromatography today. Both terms can be abbreviated as HPLC, and the terms are used interchangeably by chromatographers. The applications of this very effective technique have been significantly expanded for the pharmaceutical chemist by the use of a variety of detectors such as fluorescence, electrometric, MS and so forth.
Supercritical fluid chromatography (SFC) offers some of the advantages of GC in terms of detection and of HPLC in terms of separations, in that volatility of the sample is not of paramount importance.
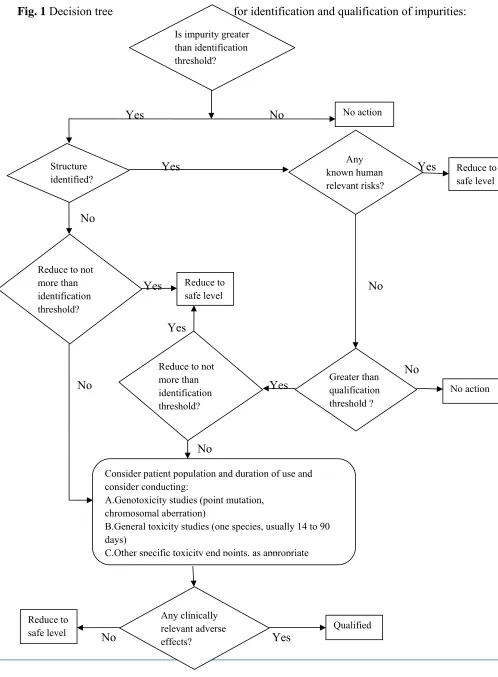
1.6. REGULATORY GUIDANCE ON IMPURITIES
Yes No
Yes es Yes
No
Yes No
Yes
No No
No No Yes
No
No Yes Consider patient population and duration of use and
consider conducting:
A.Genotoxicity studies (point mutation,
chromosomal aberration)
B.General toxicity studies (one species, usually 14 to 90
days)
C.Other specific toxicity end points, as appropriate
Any clinically relevant adverse effects? Qualified Reduce to safe level
Is impurity greater than identification threshold? No action Structure identified? Any known human relevant risks? Reduce to safe level
Reduce to not more than identification threshold? Reduce to safe level Greater than qualification threshold ? No action Reduce to not
[image:23.612.69.567.68.745.2]2. METHOD DEVELOPMENT (16-19)
Numerous methods are required to characterize drug substances and drug products. Specifications may include description; identification; assay (of composite sample); tests for organic synthetic process impurities, inorganic impurities, degradation products, residual solvents, and container extractables; tests of various physicochemical properties, chiral purity, water content, content uniformity, and antioxidant and antimicrobial preservative content; microbial tests; dissolution/disintegration tests; hardness/friability tests; and tests for particle size and polymorphic form. Some of these tests may be precluded, or additional tests may be added as dictated by the chemistry of the pharmaceutical or the dosage form. Due to the variability in specific tests required to fully characterize a pharmaceutical, it is difficult to provide a comprehensive discussion to address all aspects of pharmaceutical development. However, the requisite tests can be broadly subdivided into three main categories:
• Tests concerned with solid-state characterization • Compendial tests
• Quantitative tests to characterize drug substance and drug product composition
2.1 HPLC METHOD DEVELOPMENT STEPS:
The steps will be discussed in the same order as they would be investigated during the method development process. The rational will be illustrated by focusing on developing a stability-indicating HPLC-UV method for related substances (impurities). The principles, however, will be applicable to most other HPLC methods.
The following is a suggested method development timeline for a typical HPLC-UV related substance method. The percentage of time spent on each stage is proposed to ensure the scientist will allocate sufficient time to different steps. In this approach, the three critical components for a HPLC method (sample preparation, HPLC analysis and standardization) will first be investigated individually. Each of these steps will be discussed in more detail in the following paragraphs.
Step 1: Define method objectives and understand the chemistry of analytes
Determination of the goals for method development (e.g., what is the intended use of the method?), and understand the chemistry of the analytes and the drug product.
Step 2: HPLC Initial conditions
Development of preliminary HPLC conditions to achieve minimally acceptable separations. These HPLC conditions will be used for all subsequent method development experiments.
Step 3: Procedure for Sample preparation
Develop the suitable sample preparation scheme for the drug product
Step 4: Standardization
Step 5: Final method optimization or robustness
Identification of the “weaknesses” of the method and optimization of the method through experimental design and understand the method performance with different conditions, different instrument set ups and different samples.
Step 6: Validation of method
The complete method validation according to ICH guidelines
2.1.1. DEFINE METHOD OBJECTIVES
There is no absolute end to the method development process. The question is what is the “acceptable method performance”? The acceptable method performance is determined by the objectives set in this step. This is one of the most important considerations often overlooked by scientists. In this section, the different end points (i.e., expectations) will be discussed in descending order of significance.
2.1.1.1. Analytes:
For a related substance method, determining the “significant and relevant” related substances is very critical. With limited experience with the drug product, a good way to determine the significant related substances is to look at the degradation products observed during stress testing. Significant degradation products observed during stress testing should be investigated in the method development.
Based on the current ICH guidelines on specifications, the related substances method for active pharmaceutical ingredients (API) should focus on both the API degradation products and synthetic impurities, while the same method for drug products should focus only on the degradation products. In general practice, unless there are any special toxicology concerns, related substances below the limit of quantitation (LOQ) should not be reported and therefore should not be investigated.
2.1.1.2. Significant related substances: Linearity, accuracy and response factors should be established for the significant related substances during the method validation. To limit the workload during method development, usually 3 or less significant related substances should be selected in a method.
2.1.1.3. Other related substances: These are potential degradation products that are not significant in amount. The developed HPLC conditions only need to provide good resolution for these related substances to show that they do not exist in significant levels.
2.1.2. Resolution (Rs)
A stability indicating method must resolve all significant degradation products from each other. Typically the minimum requirement for baseline resolution is 1.5. This limit is valid only for 2 Gaussian-shape peaks of equal size. In actual method development, Rs = 2.0 should be used as a minimum to account for day to day variability, non-ideal peak shapes and differences in peak sizes.
2.1.3. Limit of Quantitation (LOQ)
The desired method LOQ is related to the ICH reporting limits. If the corresponding ICH reporting limit is 0.1%, the method LOQ should be 0.05% or less to ensure the results are accurate up to one decimal place. However, it is of little value to develop a method with an LOQ much below this level in standard practice because when the method is too sensitive, method precision and accuracy are compromised.
2.1.4. Precision, Accuracy
2.1.5. Analysis time
A run time of about 5-10 minutes per injection is sufficient in most routine related substance analyses. Unless the method is intended to support a high-volume assay, shortening the run time further is not recommended as it may compromise the method performance in other aspects (e.g., specificity, precision and accuracy.)
2.1.6. Adaptability for Automation
For methods that are likely to be used in a high sample volume application, it is very important for the method to be “automatable”. The manual sample preparation procedure should be easy to perform. This will ensure the sample preparation can be automated in common sample preparation workstations.
2.1.7. UNDERSTAND THE CHEMISTRY
Similar to any other research project, a comprehensive literature search of the chemical and physical properties of the analytes (and other structurally related compounds) is essential to ensure the success of the project.
2.1.8. Chemical Properties
Most sample preparations involve the use of organic-aqueous and acid-base extraction techniques. Therefore it is very helpful to understand the solubility and pKa of the analytes. Solubility in different organic or aqueous solvents determines the best composition of the sample solvent. pKa determines the pH in which the analyte will exist as a neutral or ionic species. This information will facilitate an efficient sample extraction scheme and determine the optimum pH in mobile phase to achieve good separations.
2.1.9. Potential Degradation Products
used in method development, and to determine the sample solvent that gives the best sample solution stability.
In addition, the structures of the analytes will indicate the potential active sites for degradation. Knowledge from basic organic chemistry will help to predict the reactivity of the functional groups. For example, some excipients are known to contain trace level of peroxide impurities. If the analyte is susceptible to oxidation, these peroxide impurities could possibly produce significant degradation products.
2.1.10. Sample Matrix
Physical (e.g., solubility) and chemical (e.g., UV activity, stability, pH effect) properties of the sample matrix will help to design an appropriate sample preparation scheme. For example, Hydroxypropyl Methylcellulose (HPMC) is known to absorb water to form a very viscous solution, therefore it is essential to use mostly organic solvents in sample preparation.
2.2. INITIAL METHOD CONDITIONS
The objective at this stage is to quickly develop HPLC conditions for subsequent method development experiments. A common mistake is that scientists spend too much time at this stage trying to get a perfect separation.
2.2.1. Preliminary HPLC Conditions
Computer assisted method development can be very helpful in developing the preliminary HPLC conditions quickly. Since the objective at this stage is to quickly develop HPLC conditions for subsequent method development experiments, scientists should focus on the separation of the significant related substances instead of trying to achieve good resolution for all related substances. These significant related substances should be baseline resolved from each other with Rs > 2.0. After the preliminary method development, the HPLC conditions can be further fine-tuned at a later stage (see section 8, method optimization/ robustness) to achieve the required specificity for the other related substances.
2.2.3. Aged HPLC Column
An aged HPLC column should be used to develop the initial HPLC conditions. Usually it is more difficult to achieve the required resolution with an aged column (e.g., column with about 200 injections). This will reflect the worst case scenario likely to be encountered in actual method uses, and help the long-term method robustness.
In general, develop all methods with HPLC columns from the same vendor. The preferred brand of HPLC column should be selected primarily based on the long term stability and lot to lot reproducibility.
2.4. SAMPLE PREPARATION 2.4.1 Selection of Sample Solvent
2.4.1.1. Accuracy: To investigate the accuracy in sample preparation (i.e., extraction efficiency), prepare a spiked solution by adding known amounts of related substances into a sample matrix. Compare responses of the spike solutions and the neat standard solutions to assess the recovery from the sample preparation. In this stage, since only one particular step is being investigated (i.e., sample preparation), close to theoretical recovery should be observed at this point (e.g., 90-110%).
2.4.1.2. Precision: Use the stressed sample to represent the worst case scenario and perform replicate sample preparations from the same sample composite. Investigate the consistency of the related substance profile (i.e., any missing peaks?) and the repeatability results from these preparations.
Another objective is to determine the sample concentration that gives an acceptable LOQ (Signal to Noise ratio, S/N) in low level spike concentrations. The sample concentration should be low enough to maintain linearity and precision, but high enough to achieve the desired LOQ. For example, if the ICH reporting limit for this drug product is 0.1%, the LOQ of the method should be less than 0.05% (i.e., desired LOQ, in %). By using spike sample solutions of very diluted concentrations for the significant related substances, estimate the concentrations that give a S/N of about 10 for the significant related substances. This estimated concentration is the
approximate LOQ concentration (i.e., estimated LOQ concentration, in μg/mL).
The following equation can be used to estimate the target sample concentration for the method:
Target sample concentration = estimated LOQ concentration (μg/mL) x 1/desired LOQ (%) x 100%
2.5. STANDARDIZATION 2.5.1. Area % method
concentration is usually limited and this will reduce the method sensitivity (i.e., increase LOQ). In general, use this approach as long as the desired LOQ can be achieved.
2.5.2. External Standard method
Use the external standard method if the response of the active pharmaceutical ingredient is not linear throughout the whole range, or the desired LOQ cannot be achieved by the area % method. The concentration of standard solution should be high enough to ensure the standard solution can be prepared accurately and precisely on a routine basis, it should be low enough to approximate the concentration of related substance in the sample solution. In general, the standard concentration should correspond to about 5 % of related substances.
2.5.3. Wavelength Selection and Relative Response Factor
Generate the linearity plot of API and related substances at different wavelengths. At this point, Photodiode Array Detector can be used to investigate the linearity of the active pharmaceutical ingredient and related substances in the proposed concentration range. By comparing the linearity slopes of the active pharmaceutical ingredient and the related substances, one can estimate the relative response factors of the related substances at different wavelengths.
Disregard of whether Area % or External Standard approach is used, if the relative response factors of some significant related substances are far from unity, a response factor correction must be applied. The optimum wavelength of detection is the wavelength that gives the highest
sensitivity (λmax) for the significant related substances and minimizes the difference in response
factors between those of the active pharmaceutical ingredient and the related substances. After the optimum wavelength is determined, use a highly stressed sample (e.g., 5% degradation) to verify that the selected wavelength will give the highest % related substance results.
2.5.4. Overall accuracy
extraction efficiency, choice of wavelength and the bias in standardization influence the calculated related substance result, this is the best way to investigate the accuracy of the method. Overall accuracy reflects the true accuracy of the method.
2.6. METHOD OPTIMIZATION/ ROBUSTNESS
After the individual components of the method are optimized, perform the final optimization of the method to improve the accuracy, precision and LOQ. Use an experimental design approach to determine the experimental factors that have significant impact on the method. This is very important in determining what factors need to be investigated in the robustness testing during the method validation. To streamLine the method optimization process, use Plackett Burmann Design (or similar approach) to simultaneously determine the main effects of many experimental factors.
Some of the typical experimental factors that need to be investigated are:
HPLC conditions: % organic, pH, flow rate, temperature, wavelength, column age. Sample preparation: % organic, pH, shaking/sonication, sample size, sample age.
Calculation/standardization: integration, wavelength, standard concentration, response factor correction.
Typical responses that need to be investigated are:
Results: precision (%RSD), % related substance of significant related substances, total related substances.
Chromatography: resolution, tailing factor, separation of all related substances.
2.7. METHOD VALIDATION 2.7.1. Robustness
varied within a narrow range to reflect normal day to day variation. During the method validation, the purpose is to demonstrate that the method performance will not be significantly impacted by slight variations of the method conditions.
2.7.2. Linearity, Accuracy, Response Factor
Linearity, accuracy and response factors should be established for the significant related substances during the method validation. In order to limit the workload of method development, usually less than 3 significant related substances should be selected in a method. Therefore, the other related substances should not be included in these experiments.
2.7.3. System suitability criteria
It is advisable to run system suitability tests in these robustness experiments. During the robustness testing of the method validation, critical method parameters such as mobile phase composition and column temperature are varied to mimic the day-to-day variability. Therefore, the system suitability results from these robustness experiments should reflect the expected range. Consequently, the limits for system suitability tests can be estimated from these experiments.
3. DEGRADATION STUDIES (20-24)
A degradation product is defined as a chemical change in the drug molecule brought about over time and/or by action of, e.g., light, temperature, pH, or water, or by reaction with an excipient and/or the immediate container/closure system (also called decomposition product).
ICH Guideline on Stability Testing
establish the inherent stability characteristics of the molecule, such as the degradation pathways, and lead to identification of the degradation products and hence support the suitability of the proposed analytical procedures. The detailed nature of the studies will depend on the individual drug substance and type of drug product.
The 1987 Food and Drug Administration (FDA) Stability Guideline defines stability-indicating methodology as:
Quantitative analytical methods that are based on the characteristic structural, chemical or biological properties of each active ingredient of a drug product and that will distinguish each active ingredient from its degradation products so that the active ingredient content can be accurately measured.
3.1.Drug Substance Degradation Studies:
Purposeful degradation studies of the drug substance include appropriate solution and solid-state stress conditions (e.g., acid/base hydrolysis, heat, humidity, oxidation, and light exposure, in accordance with ICH guidelines). Guidelines from the United States Pharmacopoeia (USP), ICH, and FDA provide a brief outline of drug substance conditions. The ICH guidelines specifically state
Stress testing is likely to be carried out on a single batch of material and to include the effect of temperatures in 10 ◦C increments above the accelerated temperature test condition (e.g., 50 ◦C, 60 ◦C, etc.); humidity where appropriate (e.g. 75% RH or greater); oxidation and photolysis on the drug substance plus its susceptibility to hydrolysis across a wide range of pH values when in solution or suspension.
3.1.1. Acid/Base Stress Testing:
likely to introduce acid/base hydrolysis are amides (lactams), esters (lactones), carbamates, imides, imines, alcohols (epimerization for chiral centers), and aryl amines.
To initiate acid/base studies, a preliminary solubility screen of the drug substance is performed. Solubility of at least 1 mg/mL in 1 N acidic and basic condition is recommended for the acid/base stress testing; however, concentrations less than 1 mg/mL can be used if solubility is an issue. In some cases, a co-solvent may be necessary to achieve the target concentration. Special attention should be given to the drug substance structure when choosing an appropriate co-solvent.
Table 2. Guidance on Acid/Base Experimental Setup:
Samples: Drug substance + acid (1 N HCl) Drug substance + base (1 N NaOH) Drug substance “as is”
Acid control (1 N HCl) Base control (1 N NaOH)
Kinetic points: 0 – 1week
3.1.2. Thermal and Thermal/Humidity Stress Testing:
The goal of thermal and thermal/humidity studies is to force the degradation of drug substances over time to determine the primary thermal and/or humidity degradation products.
Table 3. Guidance for Thermal Stress Length of Study:
Temperature Length of storage
Table 4. Guidance for Thermal/Humidity Experimental Setup:
Samples: 70˚C/30% RH (ambient humidity) 70˚C/30% RH
Time points: 0 – 6 week
3.1.3. Oxidation:
Oxidative studies are executed to force the degradation of drug substances to determine the primary oxidative degradation products. Oxidative degradation is a serious stability problem and can cause a major halt in pharmaceutical development. The 1987 Stability Guidelines state that a high oxygen atmosphere should be evaluated in stability studies on solutions or suspensions of the bulk drug substance.
Table 5. Guidance for Oxidative Degradation Experimental Setup:
Samples: Oxygen with initiator Oxygen without initiator Argon with initiator Thermal control
Initiator and antioxidant without drug substance
Time points: 0 – 10 days
3.1.4. Photostability:
electron in the relevant chromophore is raised to an excited state. Light stress conditions can also induce photo oxidation by free radical mechanisms.
Table 6. Guidance for Ultraviolet Degradation Experimental Set Up:
3.1.4.1.Ultraviolet Exposure:
ICH guidelines specify an exposure of 200 watt h/m2 for ultraviolet light confirmatory testing (1× ICH). Recommended stress conditions and time points to be tested are 5× and 10× ICH for solid drug substances
All samples should be taken at the appropriate kinetic time points and protected from any further light exposure. Light measurements should be taken in watts/m2 to be consistent with the ICH guidelines.
3.1.4.2.Fluorescence Exposure:
ICH guidelines specify an exposure of 1.2×106 lux hours for fluorescence (1× ICH). Recommended stress conditions and time points to be tested are 5× and 10× ICH for solid drug substances.
Filters may be useful to determine any wavelength range causing instability. For example, a 400 nm filter in fluorescence experiments to look at the 400–700 nm component and eliminate the UV portion is especially critical in solution experiments. If photodegradation is observed, one can determine whether it is the result of visible or ultraviolet light using appropriate spectral filters.
Samples: Ultraviolet
Thermal foil-wrapped control
Table 7. Guidance for Fluorescence Degradation Experimental Set Up:
Samples: Fluorescence
Thermal foil-wrapped control
Time points: 5× and 10 × ICH* * Note: ICH fluorescent conditions = 1.2 × 106 lux hrs
3.2.Degradation Prediction:
Degradation prediction is extremely helpful for understanding a degradation mechanism. One program that has been particularly useful is CAMEO (computer assisted mechanistic evaluation of organic reactions). CAMEO is a computer program that predicts the products of organic reactions given starting materials, reagents, and conditions. The analyses cover the key degradation conditions, basic/nucleophilic, acidic/electrophilic, radical, oxidative/ reductive and photochemical, as well as mechanistic interpretations of these reactions.
CHAPTER-2
Chapter 2
LITERATURE REVIEW
Shende et al.28 developed an isocratic reversed-phase high-performance liquid chromatographic (RP-HPLC) method for analysis of irinotecan HCl and validated. Separation was achieved on a C18 column with potassium dihydrogen phosphate buffer (pH adjusted to 3.5 with orthophosphoric acid)–acetonitrile–methanol 55:25:20 (v/v) as mobile phase at a flow rate of 1.0
mL min−1. UV detection was performed at 254 nm. The method is simple, sensitive, rapid, and selective, and linear over the range 30–70 μg mL-1 for assay of irinotecan HCl. The precision of the assay method was below 1.0% RSD. Mean recovery was in the range 98–102%. Recovery of the active pharmaceutical ingredient from dosage forms ranged from 99.0 to 101.0. The method is useful for quality control in bulk manufacture and of the pharmaceutical formulation.
Satyanarayana et al.29 have reported a reverse phase HPLC method, developed for the estimation of Irinotecan HCl in tablet dosage form. An X-Terra RP C18, 250 X 4.6 mm i.d, 5 m particle size, with mobile phase consisting of Methanol and 0.01 M Ammonium Acetate containing 0.1% formic acid and methanol in the ratio of 50:50 v/v was used. The flow rate was 1 mL/min and the effluents were monitored at 250 nm. The retention time was 4.69 min. The detector response was linear in the concentration of 120-360 mcg/mL. The respective linear regression equation being Y=166582.24x+86439.5. The limit of detection and limit of quantification was 0.06 and 0.18 mcg/mL respectively. The percentage assay of Irinotecan HCl was 99.09 %. The method was validated by determining its accuracy, precision and system suitability. The results of the study showed that the proposed RP-HPLC method is simple, rapid, precise and accurate, which is useful for the routine determination of Irinotecan HCl in bulk drug and in its pharmaceutical dosage form.
was performed at wavelength 225 nm using Photo Diode Array (PDA) detector at ambient temperature. The method was validated and stability studies were conducted under different conditions. The retention time for irinotecan was around 5.82 minutes. The calibration curves were linear (R2≥0.9998) over a concentration range from 20.0 to 80.0 µg/mL. Limit of detection
(LOD) and Limit of quantitation (LOQ) were 8 ng/mL and 24 ng/mL respectively. The developed method was successfully applied to estimate the amount of irinotecan in injection formulations.
Owens et al.31 developed a method for the determination of CPT-11 and the three metabolites SN-38, SN-38G, and APC in biological matrices. In most of these published methods, chromatography was carried out with fluorescence detection. Liquid chromatography-mass spectrometry methods have also been described, but despite the latter methods providing good results, the instrumentation involved is expensive and not always readily available for routine
drug monitoring.
Takahashi et al.32 found that both CPT-11 and SN-38 were detectable in saliva and that the patterns of their concentration-time curves in plasma and saliva were very similar. To date, however, no bioanalytical assay has been validated for the determination of irinotecan and its metabolites in saliva. This report describes rapid, specific, reliable, and sensitive analytical methods to simultaneously quantify irinotecan and four metabolites (SN-38, SN-38G, APC, and NPC) in human plasma and saliva. These methods, involving the use of an internal standard, were validated according to validation procedures, parameters, and acceptance criteria based on US Pharmacopoeia XXIII guidelines and Food and Drug Administration guidance.
Ali mohammadi et al.33 developed a new simple high performance liquid chromatography (HPLC) method and validated for the simultaneous determination of irinotecan (CPT-11) and two related compounds viz., 7-ethyl-10-hydroxycamptothecin (SN-38) and camptothecin (CPT)
quantitation for all the compounds was 0.1-10 μg/mL. The limit of quantitation for all the compounds ranged between 0.01-0.05 μg/mL. The method has the requisite accuracy, selectivity, sensitivity and precision to assay of CPT-11 and related compounds in pharmaceutical dosage forms and bulk API.
Mohammed Ishaq et al.34 have reported a rapid and sensitive RP-HPLC method with UV detection at 222 nm for routine analysis of Irinotecan in bulk and pharmaceutical formulations. Chromatography was performed with mobile phase containing a mixture of acetonitrile and phosphate buffer in the ratio of 60:40, v/v with flow rate 1.0 mL/min. The calibration graph of Irinotecan was found to be linear over the range of 40 to 120 μg/mL with correlation coefficient of 0.9999. Sensitivity, accuracy, range, precision, robustness, ruggedness, stability, specificity, limit of detection, limit of quantification and system suitability parameters were validated for the developed method. The developed method was successfully applied to estimate the amount of
irinotecan in injection formulations.
Gogineni Ratna Prasad et al.35developed a simple, specific, accurate and precise reverse phase high performance liquid chromatographic method and validated for the estimation of Irinotecan in tablet dosage form. An Inertsil ODS C-18, 5 μm column having 250 x 4.6 mm internal diameter in isocratic mode with mobile phase containing Methanol: Triethylamine: 1% Orthophosphoric acid in the ratio of 60:5:35 (v/v/v) was used. The flow rate was 1.0mL/min and effluents were monitored at 254 nm. The retention time for Irinotecan was 5.109 min. The method was validated for linearity, accuracy, precision, specificity, limit of detection, limit of quantification and robustness. Limit of detection and limit of quantification were found to be 0.09 ppm and 0.297 ppm respectively. The proposed method was successfully applied for the quantitative determination of Irinotecan in tablet formulation.
column in gradient mode using 0.1 % v/v orthophosphoric acid in water and acetonitrile as the mobile phase. The analytical column was thermo stated at 50°C and flow rate was set at 0.5 mL per min. The eluted peaks were detected by photodiode array (PDA) detector at a wavelength of 254 nm. The retention time of irinotecan was found 2.127 min. Linearity responses in analyte standard peak areas were observed over the concentration ranges of 100 - 500 μg/mL. The limit
of detection and limit of quantification were found 0.0560 and 0.1698 μg/mL, respectively. Percentage recoveries were obtained in the range of 97.5 % and 98.88 %. The proposed method is precise, accurate, selective and reproducible.
Sushama Talegaonkara et al.37developed a simple, sensitive, precise and accurate stability-indicating high performance thin-layer chromatographic method for analysis of irinotecan both as bulk drug and marketed injectables and validated as per ICH guidelines. Chromatographic separation was achieved on LiChrospher aluminum plates precoated with silica gel 60F254 as stationary phase. The solvent system consisted of acetone –ethyl acetate –acetic acid 8.5:1.5:0.1 (v/v/v) and this system were found to give compact spots for irinotecan at Rf value of 0.31 ± 0.02. Densitometric analysis was performed in the absorbance at 366 nm. The linear regression analysis for the calibration plots showed good linear relationship with R2 = 0.9973 ± 0.0013 in the concentration range of 50–500 ng/spot. The % recovery (94.63–101.40%) and precision (≤ 4.30) were found to be satisfactory. Irinotecan was subjected to acid and alkali hydrolysis, oxidation, thermal and ultraviolet radiation treatments. All the peaks of degradation products were well resolved from the standard drug with significantly different retention factor (Rf) values. Developed method effectively separated out the drug from its degradation products and hence can be used as stability-indicating method as well as in routine analysis of irinotecan.
linear in the concentration range of 10.0–2000.0 ng/mL for CPT-11 and 0.5–200.0 ng/mL for 7-ethyl-10-hydroxycamptothecin (SN-38), respectively. The lower limit of quantification (LLOQ) was 10 ng/mL for CPT-11 and 0.5 ng/mL for SN-38. The intra- and inter-day relative standard deviation across three validation runs over the entire concentration range was less than 10.6%. The accuracy determined at three concentrations was within ± 11.4% in terms of relative error. Due to the unavailability of standard for 7-ethyl-10-O-glucuronyl-camptothecin (SN-38G) and
the importance of knowing the concentration of this metabolite, we developed a method for analysis SN-38G by taking advantage of the quantitative conversion of SN-38G to SN-38 using glucuronidase. This enzymatic method of identification and quantitation of gluconated compound can be widely used when the standard for phase II glucuronide metabolites are not available.
CHAPTER-3
Chapter 3 DRUG PROFILE (25-27) IRINOTECAN HYDROCHLORIDE
Irinotecan HCl also called as Irinotecanum available in market in various following Brand names
• Camptosar • CP0 • Irinotecan • CPT-11 • Irinocam • Torsin • campto
Structure:
.HCl .3H2o
Molecular formula:
Molecular weight:
Trihydrate: 677.18 Anhydrous: 623.14
Chemical name:
(S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3, 14-dioxo-1H- Pyranol-[3`, 4`:6,7]-indolizino [1,2-b]quinolin-9-yl-[a,4`-bipiperidine]-1`-carboxylate, monohydrochloride, trihydrate.
Content:
Irinotecan Hydrochloride contains NLT 98.0% and NMT 102.0% of C33H38N4O6·HCl, calculated on the anhydrous basis.
Characteristics:
Appearance: Pale yellow crystalline powder
Solubility: Slightly soluble in water, methanol and ethanol, freely soluble in glacial acetic acid. Identification:
IR
Melting/Freezing Point: 222-223oC Boiling Point: 100°C
PH: 3.5
Mechanism of action:
double-stranded breaks in DNA. As a result, DNA damage is not efficiently repaired and apoptosis (programmed cell death) occurs.
Toxicity:
Gastrointestinal complications such as nausea, vomiting, abdominal cramping, diarrhea, and infection.
Storage:
Stored in Glass containers, Polyethylene or polypropylene containers at 15-30oC, and protected from light.
Category:
• Antineoplastic Agents
• Radiation-Sensitizing Agents • Parasympathomimetics • Enzyme Inhibitors • Prodrugs
• Phytogenic agent
Metab
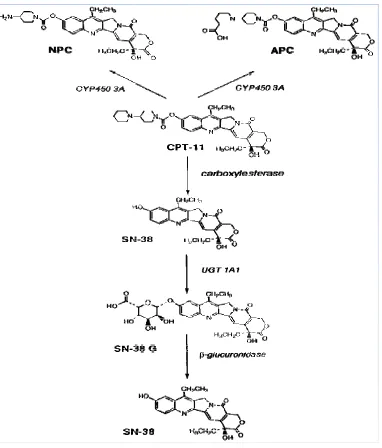
Fig. 2
Metab and it isofor also u latter bolic pathw bolic pathwa ts subsequen rms (UGT1A undergo CY
can be hydro
way of Irinot
ays of CPT-1 nt conversio A7), with de YP3A4-media
olyzed by CE
tecan HCl:
11 showing on to a glu glucuronida ated oxidati E to release
CE-mediate ucuronide de ation by intes ve metaboli
SN-38.
ed formation erivative (SN
stinal β-gluc ism to form
n of the activ N-38G) by curonidase (β m APC and
ve metabolite UGT1A1 a β-Glu). CPT
NPC, of wh
e SN-38 and 1A7 T-11 can
[image:50.612.94.476.130.578.2]CHAPTER-4
Chapter 4
AIM AND OBJECTIVE
SN-38 (7-ethyl-10-Hydroxycamptothecin) is one of the metabolite/related substance present in irinotecan HCl (40-41).The assessment of the limit of SN38 concentrations in humans is very important, since SN38 is 100 to 1,000 fold more cytotoxic than the parent compound. SN-38 is reported to be associated with the severe diarrhea with one of the major side effects causing direct enteric injury (Araki et al., 1993). There is no report for the quantification of SN-38 in irinotecan HCl andon forced degradation studies of irinotecan HCl by HPLC method.
Hence it was proposed to develop and validate a RP-HPLC method for detection and quantification of SN-38 in irinotecan HCl and to perform the forced degradation studies of irinotecan HCl in order to identify and quantify the degradents by HPLC.
CHAPTER-5
Chapter 5 PLAN OF WORK • PHASE -I
1. Optimization of chromatographic conditions,
Selection of wavelength
Selection of initial separation conditions
Selection of mobile phase (pH, peak modifier, solvent strength, ratio and flow rate)
Nature of the stationary phase
Selection of separation method and agent
• PHASE-II
2. Validation of the method
The developed method to be validated using the various validation parameters such as,
Accuracy
Precision
Linearity and of detection (LOD) / Limit of quantitation (LOQ)
Selectivity / Specify
Robustness / ruggedness
System suitability.
• PHASE-III
3. Quantification of Related substance:
• PHASE-IV
4. Performance of forced degradation studies such as Acid degradation
Alkaline degradation Oxidative degradation Photolytic degradation Thermal degradation
CHAPTER-6
Chapter 6 EXPERIMENTAL
6.1 Analytical Method Development and determination of Irinotecan HCl and its related substance 7-ethyl 10-hydroxy camptothecin (SN-38) in marketed formulation
6.1.1 MATERIALS AND INSTRUMENTS USED a) Drug sample & Study products
Irinotecan HCl was obtained from Dr. Reddy’s Laboratories, Hyderabad and 7-ethyl 10-hydroxy camptothecin (SN-38) was obtained from Henan Dongtai Pharm Co., Ltd, China.
Test product:
Irinocam-40 mg (Dr. Reddy’s Laboratories) injection was purchased from the local market.
b) Chemicals and solvents used for estimation: • HPLC grade Water • HPLC grade Acetonitrile • HPLC grade Methanol
• Analytical grade Dihydrogen sulphoxide • Analytical grade Trichloro acetic acid • Analytical grade Distilled water
c) Instruments used:
• Elico pH meter LI 127. • Shimadzu LC-20 AT HPLC.
• Shimadzu 1700 UV Spectrophotometer. • Sonica Ultrasonic cleaner.
D) preparation of standard stock solutions: I. Irinotecan HCl (10 µg):
Ten milligrams (mg) of standard Irinotecan HCl was dissolved in the 10 mL of methanol, then serial dilutions were made with water to get a final concentration of 10 µg/mL.
II. SN-38 (7-ethyl 10-hydroxy camptothecin) 10 µg:
Ten milligrams (mg) of standard SN-38 was dissolved in the 10 mL of Dihydrogen sulphoxide, then serial dilutions were made with methanol to get a final concentration of 10 µg/mL.
6.1.2 OPTIMIZATION OF CHROMATOGRAPHIC CONDITIONS FOR METHOD DEVELOPMENT OF IRINOTECAN HCl AND SN-38 (7-ETHYL 10-HYDROXY CAMPTOTHECIN)
Method development:
The aim of the present study was to develop and validate the RP-HPLC method for the estimation of Irinotecan HCl and SN-38 in the injection dosage form.
Selection of wavelength
An UV-Visible spectrum of 10 µg/mL Irinotecan HCl in methanol and SN-38 in Dihydrogen sulphoxide and methanol was recorded by scanning in the range of 200 nm to 800 nm. From the UV-Visible spectrum wavelength of 372 nm was selected. At this wavelength Irinotecan HCl showed good absorbance.
Selection of chromatographic method
Initial separation conditions
Standard solution: 10 μg/mL of Irinotecan HCl in HPLC grade Methanol.
Equipment
System : Shimadzu gradient HPLC
Pump : LC – 20AT prominence solvent delivery system
Detector : SPD-M20A Prominence Diode array detector
Injector : Rheodyne 7725i with 20 μL loop
Chromatographic condition 1
Stationary phase : Phenomenex Luna C18 Column
Mobile phase : Solvent A: 10 mM ammonium acetate in 100mL HPLC
grade water PH adjusted to 4 with orthophosphoric acid
Solvent B: Acetonitrile and methanol (50:50 v/v)
Solvent ratio : 60:40
Detection : 372 nm
Flow rate : 1.0 mL/min
Sample size : 20 μL
Needle wash : water HPLC grade
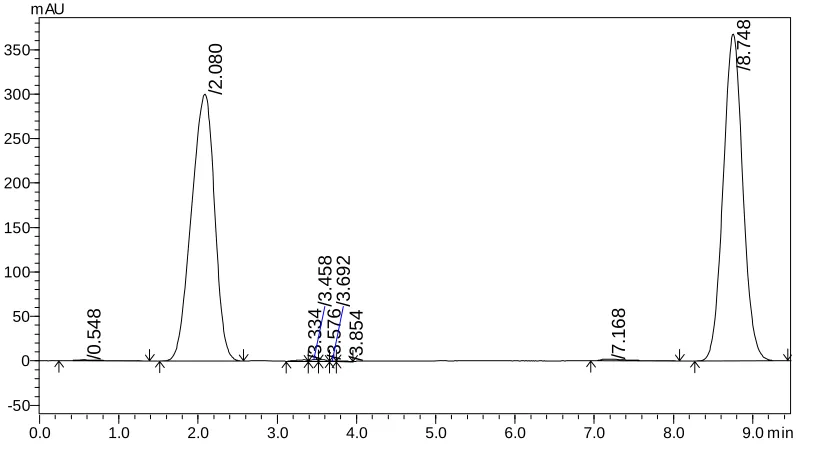
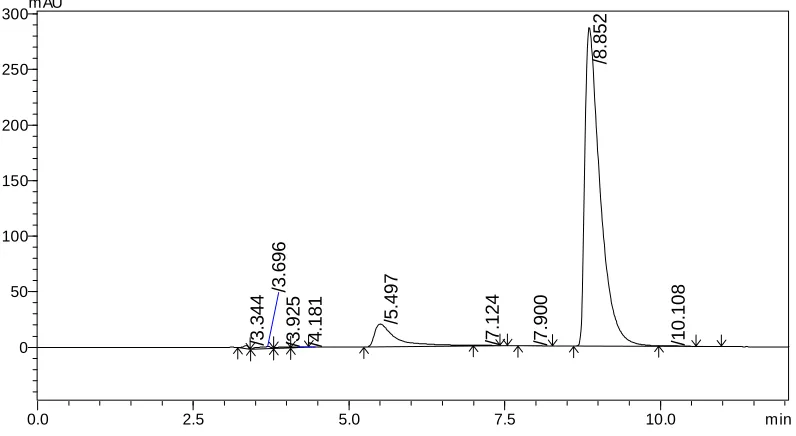
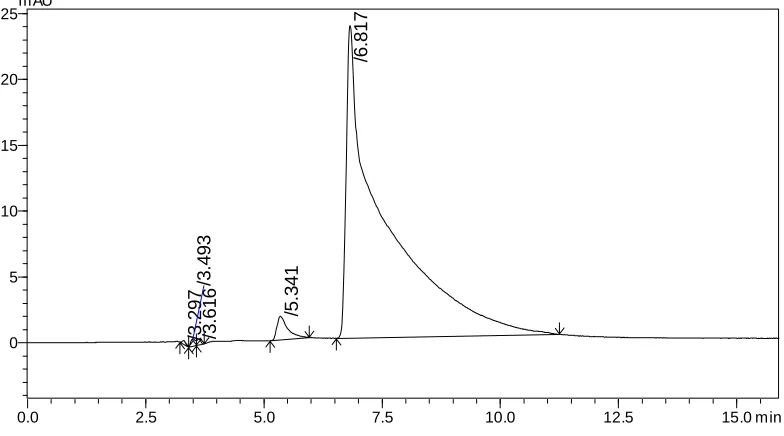
The retention time of Irinotecan HCl andSN-38 were found to be 6.67 and 8.75 min but peak tailing was observed for Irinotecan HCl as shown in Fig. 3 &4. So we adjusted the PH of the buffer solution.
Chromatographic condition 2
Stationary phase : Phenomenex Luna C18 Column
Mobile phase : Solvent A: 10 mM ammonium acetate in 100 mL HPLC
grade water PH adjusted to 5.0 with orthophosphoric acid
Solvent B: Acetonitrile and methanol (50:50 v/v)
Solvent ratio : 60:40
Detection : 372 nm
Flow rate : 1.0 mL/min
Sample size : 20 μL
Needle wash : water HPLC grade
Column temperature : Ambient
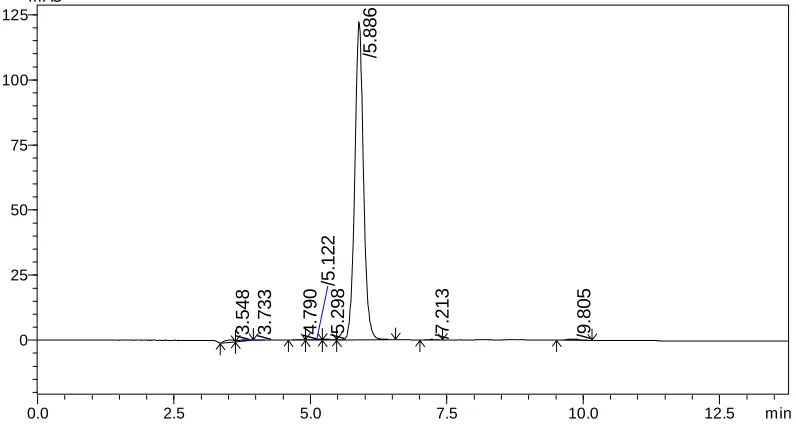
Chromatographic condition 3
Stationary phase : Phenomenex Luna C18 Column
Mobile phase : Solvent A: 10 mM ammonium acetate in 100 mL HPLC
grade water PH adjusted to 4.5 with orthophos