Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Expression of an E1A/E7 Chimeric Protein Sensitizes Tumor Cells to
Killing by Activated Macrophages but Not NK Cells
Tanya A. Miura,
1Han Li,
1Kristin Morris,
1Sharon Ryan,
1Kristine Hembre,
1James L. Cook,
2and John M. Routes
1,3*
Departments of Medicine and Immunology, National Jewish Medical and Research Center, Denver, Colorado 802061;
Departments of Medicine and Microbiology-Immunology and the Cancer Center, University of Illinois at Chicago
College of Medicine, Chicago, Illinois 606122; and Departments of Medicine and Immunology and the
Cancer Center, University of Colorado Health Sciences Center, Denver, Colorado 802623
Received 13 November 2003/Accepted 31 December 2003
Adenovirus (Ad) E1A and human papillomavirus (HPV) E7 express homologous conserved regions (CRs) that mediate their shared biological functions. Despite their similarities, the expression of E1A sensitizes tumor cells to killing by NK cells and macrophages but the expression of E7 does not, a factor that may contribute to the dissimilar oncogenicities of Ad and HPV. This study was undertaken to define molecular differences between E1A and E7 that are responsible for the ability of E1A and the inability of E7 to sensitize cells to killing by NK cells and macrophages. Genetic mapping studies using human fibrosarcoma cells (H4) that stably expressed mutant forms of E1A showed that only those forms of E1A that interacted with the transcriptional coadaptor protein p300 sensitized cells to killing by NK cells and macrophages. E7 lacks the N-terminal p300-binding region present in E1A. Therefore, a chimeric E1A/E7 gene was constructed that included the N terminus and the CR1 (p300-binding) domain of E1A fused to CR2 and the C-terminal sequences of E7. The E1A/E7 protein interacted with p300 and pRb and immortalized primary mouse embryo fibroblasts (MEF). The expression of E1A/E7 sensitized H4 and MEF cells to killing by activated macrophages but not to killing by NK cells. Therefore, N-terminal differences between E1A and E7 that map to the E1A-p300 binding region accounted for differences in their abilities to sensitize cells to killing by macrophages. However, regions in addition to the E1A-p300 binding region are required to sensitize cells to killing by NK cells.
Adenoviruses (Ad) and human papillomaviruses (HPV) are common human pathogens that express functionally analogous oncoproteins. Two viral oncoproteins are consistently ex-pressed upon cellular transformation in HPV-induced malig-nancies or Ad-transformed cells (HPV E7 and E6; Ad E1A and E1B). The E1A and E7 oncoproteins express homologous conserved regions (CRs), CR1 and CR2. These CRs interact with and inhibit cellular growth regulatory proteins (pRb, p107, p130, and cyclin A) (20, 27, 37–40, 52). CR1 and CR2 of E1A and E7 are interchangeable for cellular immortalization (6). Through different molecular mechanisms, the Ad E1B-55K and HPV E6 oncoproteins inhibit the function of p53, thereby complementing E1A and E7 in inducing cellular trans-formation.
Despite the functional similarities in their transforming on-coproteins, Ad do not appear to be oncogenic in humans. In contrast, HPV are responsible for ⬎95% of cervical carcino-mas (5, 25). The inability of Ad to be oncogenic in humans is notable because Ad are fully competent to transform human cells (24) and Ad-transformed human cells form tumors in immunodeficient mice (13). These observations suggest that factors in addition to cellular transformation by Ad or HPV determine the oncogenicity of these viruses.
The difference in the abilities of E1A and E7 to elicit an antitumor immune response is one factor that likely
contrib-utes to the dissimilar oncogenicities of Ad and HPV. In im-munocompetent mice, tumor cells that express Ad5 E1A were observed to be over 1,000-fold less tumorigenic than tumor cells that express HPV type 16 (HPV16) E7 or HPV16 E7 and E6 (47). In contrast, these same E1A- and E7-expressing cell lines are equivalently tumorigenic in CD3ε-transgenic mice, which do not have NK cells or T cells. These data establish that the ability of E1A to induce an NK cell- and T-cell-mediated immune response, not the in vivo growth characteristics of E1A-expressing cells, directly influences primary tumor devel-opment.
In addition to eliciting a vigorous antitumor immune re-sponse in vivo, the expression of E1A also sensitizes cells to killing by NK cells, macrophages, and the immune effector mechanisms utilized by these cells, including tumor necrosis factor alpha (TNF-␣), TRAIL, Fas, nitric oxide, and perforin, but the expression of E7 does not (11, 14, 19, 32, 36, 46). The sensitivity of E1A- and E7-expressing murine and human tu-mor cells to killing by NK cells and macrophages in vitro directly correlates with their tumorigenicity in vivo (9, 13, 36, 41, 47–50, 53). Thus, several lines of evidence suggest that the ability of the innate immune response to reject E1A-expressing cells but not E7-expressing cells influences the tumorigenicity of these cells.
We hypothesized that despite many shared biological func-tions transduced by the homologous conserved regions, differ-ences exist that account for the ability of E1A and the inability of E7 to sensitize cells to killing by NK cells and macrophages. In order to approach this issue, it was necessary to define the regions of E1A required to sensitize cells to killing by
macro-* Corresponding author. Mailing address: Department of Medicine, National Jewish Medical and Research Center, 1400 Jackson St., Den-ver, CO 80206. Phone: (303) 398-1291. Fax: (303) 398-1806. E-mail: routesj@njc.org.
4646
on November 8, 2019 by guest
http://jvi.asm.org/
phages and NK cells. E1A genetic mapping studies to define the regions of E1A necessary to sensitize cells to lysis by mac-rophages have not been performed. Previous studies using the human fibrosarcoma cell line H4 demonstrated that the ex-pression of E1A-RG2, which does not interact with p300, does not sensitize cells to lysis by NK cells (15). E1A-RG2, which contains a single point mutation in the N-terminal noncon-served region of E1A (Fig. 1), also has a reduced ability to bind pRb (54). The E1A-RG2 mutation may also affect the binding of other cellular proteins that interact with E1A in the N-terminal nonconserved region, such as PCAF and p400.
Therefore, we performed NK and macrophage cytolysis as-says by using H4 cells that expressed mutant forms of E1A that have deletions within CR1 or CR2. The deletions abolished the interactions of E1A with p300, pRb, or both p300 and pRb. These mapping studies demonstrated that the expression of an intact CR1 region of E1A, which encompasses the p300-bind-ing domain, is required to sensitize cells to killp300-bind-ing by NK cells and macrophages. (For the purposes of this paper, we do not distinguish between p300 and the highly homologous transcrip-tional coactivator CBP.) Although the E7 oncoprotein inter-acts with p300, it lacks an N-terminal p300-binding site homol-ogous to that of E1A (3). Therefore, we constructed a chimeric
E1A/E7gene that included the N terminus and the CR1
(p300-binding) domain of E1A fused to the CR2 and C-terminal sequences of E7. Expression of the E1A/E7 chimeric protein sensitized both human and murine cells to killing by activated
macrophages but not to killing by NK cells. These data suggest that N-terminal differences between E1A and E7 were respon-sible for differences in the abilities of these oncoproteins to sensitize cells to killing by macrophages but not to killing by NK cells.
MATERIALS AND METHODS
Plasmids.Plasmids containing mutant Ad5 E1A gene sequences (pLE2) were obtained from S. Bayley (30) (McMaster University, Hamilton, Ontario, Can-ada). Mutant Ad5 E1A genes were cloned from pLE2 plasmids into pLXSN (35). The E1A-⌬p300 (dl1104) mutant has a deletion in amino acids 48 to 60 in CR1, which eliminates binding to p300 (Fig. 1A). The E1A-⌬pRb (dl1108) mutant does not bind to pRb due to a deletion in amino acids 124 to 127 in CR2. The E1A-⌬p300-⌬pRb mutant (dl04/08) contains both the⌬p300 and the⌬pRb mutations (28). p5XhoI-C, which expresses the E1 region of Ad5, was obtained from A. van der Eb (Leiden University, Leiden, The Netherlands) (2). All plasmids except pLXSN-E1A-⌬pRb express both the 12S and the 13S forms of E1A. pLXSN-E1A-⌬pRb expresses only the 12S mRNA. Previous studies have shown that the expression of either the 12S or the 13S form of E1A sensitizes cells to killing by NK cells and macrophages (9–11, 14).
TheE1A/E7chimeric gene was constructed by PCR amplification of Ad5 E1A from bp 551 to 901 and HPV16 E7 from bp 604 to 858 (see Fig. 3A). The E1A forward primer (GTCGAATTCGTCGACGTCGGACTGAAAATGAGACAT ATT) contains engineered restriction sites EcoRI, SalI, and AatII and amplifies E1A from bp 551 at the 5⬘end. The E7-E1A reverse primer (AGTTGTCTCT GGTTGCAAGTTTGGCATAGAAACCGG) and the E1A-E7 forward primer (CCGGTTTCTATGCCAAACTTGCAACCAGAGACAACT) are complemen-tary and prime overlapping regions of E1A (bp 883 to 901) and E7 (bp 604 to 621). The E7 reverse primer (TGCGGATCCTTATGGTTTCTGAGAACA GAT) contains a BamHI restriction site and amplifies E7 from the 3⬘end (bp 858). The N-terminal E1A and C-terminal E7 PCR products were ligated by PCR with the overlapping primers (E1A reverse and E7 forward). The chimeric gene sequence was digested with EcoRI and BamHI and cloned into the com-plementary sites of pBActNeo (42). The integrity of the clonedE1A/E7gene was verified by DNA sequence analysis (data not shown). The resulting gene encodes a chimeric protein (E1A/E7) consisting of the N-terminal nonconserved region and CR1 of E1A fused to CR2 and the C-terminal portion of E7. The predicted protein translated from theE1A/E7chimeric gene would include amino acid residues 1 to 114 of E1A fused to residues 14 to 98 of E7.
Cell lines.The cells of the human NK cell line NKL (44) were cultured in RPMI 1640 medium supplemented with 10% serum, antibiotics, and interleu-kin-2 (1,000 U/ml). The NKL cell line was provided by M. J. Robertson (Dana-Farber Cancer Institute, Boston, Mass.). Cells of the rat NK cell line RNK-16 (43) were cultured in RPMI 1640 medium supplemented with 10% serum and antibiotics. RNK-16 was provided by M. Nakamura (University of California at San Francisco, San Francisco, Calif.).
H4 is a human fibrosarcoma cell line derived from HT1080, and H4-E1A (also known as P2AHT2A) is an Ad5 E1A-transfected H4 cell line (22) (provided by S. Frisch, La Jolla Cancer Research Institute, La Jolla, Calif.). H4-E1A-⌬p300, H4-E1A-⌬pRb, and H4-E1A-⌬p300-⌬pRb stably express E1A from the viral mutantsdl1104,dl1108, anddl04/08 (28), respectively (Fig. 1). The H4-E7 cell line expresses high levels of the HPV16 E7 oncoprotein, equivalent to levels expressed in the HPV16-transformed cervical cancer cell line Caski (49).
Stable cell lines were generated by transfection of the H4 cell line with mutant E1A- or chimeric E1A/E7-expressing plasmids (described above) with Superfect transfection reagent (QIAGEN, Valencia, Calif.), followed by selection in G418 (Sigma-Aldrich, St. Louis, Mo.). Clonally derived cell lines were screened for the expression of E1A, E7, or E1A/E7 by Western analysis as described below. The human cell lines were maintained in Dulbecco modified Eagle medium supple-mented with 5% calf serum, 15 mM glucose, and antibiotics. The absence of mycoplasma in all cell lines was confirmed with the Mycoplasma PCR assay kit (Stratagene, La Jolla, Calif.).
Mouse embryo fibroblasts (MEF) were isolated from C57BL/6 embryos after 16 days of gestation as described previously (58). MEF were cultured in RPMI medium supplemented with 10% fetal bovine serum, 15 mM glucose, and anti-biotics. MEF were transfected with plasmids that express the E1 region of Ad (p5XhoI-C) (2) or the chimeric E1A/E7 protein (pActin-neo-E1A/E7) with Superfect reagent (QIAGEN), followed by selection in G418 (Sigma-Aldrich).
[image:2.603.61.267.70.312.2]Western analysis.E1A protein expression was analyzed by Western analysis. Cells were lysed in radioimmunoprecipitation assay buffer (1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris [pH 7.4], 150 mM FIG. 1. Expression of wild-type and mutant E1A proteins.
(A) Schematic diagram of E1A protein showing p300- and pRb-bind-ing sites. The locations of the⌬p300 and ⌬pRb deletions, the RG2 point mutation, and the N- and C-terminal ends of the protein are indicated. Amino acid (aa) residue numbers are indicated at the top. (B) Cell lysates from wild-type (wt) and mutant E1A-expressing cell lines were analyzed for E1A protein expression by Western blotting with an E1A-specific antibody, M37, as described in Materials and
Methods. “#1” and “#2” indicate independently derived cell lines.
on November 8, 2019 by guest
http://jvi.asm.org/
NaCl), and protein concentrations were determined by the Lowry protein assay (Bio-Rad, Hercules, Calif.). Equal amounts of total cellular protein were sepa-rated on a 10 to 16% polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were probed with the anti-E1A monoclonal antibody M37 (26) (supplied by E. Harlow, Massachu-setts General Hospital, Charleston, Mass.), followed by a horseradish peroxi-dase-conjugated sheep anti-mouse antibody (Amersham, Piscataway, N.J.). M37 binds E1A between amino acids 15 and 50; therefore, it will bind the wild-type and mutant proteins used in these studies (26). Protein was visualized by using the Renaissance Chemiluminescence kit (DuPont-NEN, Boston, Mass.) accord-ing to the manufacturer’s recommendations. Membranes were stained with Pon-ceau red to ensure equal loading of proteins (data not shown).
For the detection of E7 and E1A/E7 proteins, cells were lysed in buffer containing 50 mM Tris-HCl, 120 mM NaCl, 0.5% NP-40, 4 mM NaF, and 2 mM sodium orthovanadate (E7 lysis buffer). Protein concentrations were determined by the Lowry protein assay (Bio-Rad). Equal amounts of total cellular protein were electrophoresed and blotted as described above. E7 and E1A/E7 were detected by using monoclonal antibody ED17 (Santa Cruz Biotechnology, Santa Cruz, Calif.), followed by rabbit anti-mouse antibody (Organon Teknika, West Chester, Pa.), followed by donkey anti-rabbit antibody linked to horseradish peroxidase (HRP; Amersham). Protein was visualized by using the Renaissance Chemiluminescence kit (DuPont-NEN) according to the manufacturer’s recom-mendations. Membranes were stained with Ponceau red to ensure equal loading of proteins (data not shown).
Coimmunoprecipitation of pRb and p300.The interactions of the wild-type or mutant E1A, E7, or E1A/E7 proteins with p300 or pRb were analyzed by coimmunoprecipitation with antibody to E1A or E7, followed by Western anal-ysis to detect p300 and pRb. Cells were lysed in E7 lanal-ysis buffer (described above), and E1A or E7 proteins were immunoprecipitated by using monoclonal antibody specific for Ad5 E1A (M37) (26) or HPV16 E7 (ED17; Santa Cruz Biotechnol-ogy). Antibody-bound proteins were precipitated on protein A-Sepharose beads (Amersham). Immunoprecipitated proteins were separated by electrophoresis on a 4% polyacrylamide gel and transferred to polyvinylidene difluoride mem-branes (Bio-Rad). Memmem-branes were probed with a polyclonal antibody to p300 (N-15; Santa Cruz Biotechnology), followed by HRP-conjugated anti-rabbit im-munoglobulin G (Amersham) or pRb (IF8; Santa Cruz Biotechnology), followed by HRP-conjugated anti-mouse immunoglobulin G (Amersham). Protein com-plexes were visualized as described for Western analysis.
Macrophage cytolysis assays.Bone marrow-derived macrophages extracted from femurs and tibias of C57BL/6 mice were grown in RPMI 1640 medium containing 10% serum and 20% granulocyte-macrophage colony-stimulating fac-tor (GM-CSF) for approximately 7 days prior to the assay. Flow cytometry indicated that over 90% of these cells expressed the macrophage marker F4/80 (Caltag, Burlingame, Calif.) (data not shown). Macrophages were induced to quiescence by culturing in GM-CSF-free medium for 24 h and were then acti-vated in lipopolysaccharide (1g/ml; Sigma-Aldrich) and gamma interferon (100 U/ml; R&D Systems, Minneapolis, Minn.) for 18 h prior to the assay. Target cells were labeled with [3H]thymidine, and standard 48-h cytolysis assays were
per-formed as described, with optimal effector-to-target ratios of 50:1 for the human cell lines and 30:1 for the murine target cells (16). The results shown represent the mean⫾the standard error of the mean for at least four separate experi-ments. The mean percentage of spontaneous release from all of the target cell lines was less than 20%. Repeated-measures analysis of variance was used with a mixed-effects model approach to compare percentages of killing between cell lines. APvalue of⬍0.05 was considered statistically significant.
NK cytolysis assays.Human target cells were labeled with Cr51and incubated
with human NKL cells at effector-to-target ratios of 6:1 to 100:1 for standard 18-h cytolysis assays as described previously (48). Assays using the murine target cells were done similarly except that target cells were incubated with RNK-16 cells for a 6-h assay. The results shown represent the mean⫾the standard error of the mean for at least three separate experiments. The mean percentage of spontaneous release from all of the target cell lines was less than 20%. Repeated-measures analysis of variance was used with a mixed-effects model approach to compare percentages of killing between cell lines at each effector-to-target ratio. APvalue of⬍0.05 was considered statistically significant.
RESULTS
Cell lines that stably express mutant E1A proteins.To de-fine the regions of E1A that are required to sensitize human tumor cells to killing by NK cells and macrophages, we gener-ated H4 fibrosarcoma cell lines that stably expressed
well-defined mutants of E1A (28). The mutant proteins have a deletion in CR1, which eliminates binding to p300 (E1A-⌬p300); a deletion in CR2, which eliminates binding to pRb (E1A-⌬pRb); or deletions in both regions (E1A-⌬p300-⌬pRb) (Fig. 1A). The expression of E1A proteins in the H4-E1A, H4-E1A-⌬p300, H4-E1A-⌬pRb, and H4-E1A-⌬p300-⌬pRb cell lines was confirmed by Western analysis (Fig. 1B). This analysis confirmed that the cell lines expressed E1A proteins of the expected sizes. The Western analysis also confirmed that the H4-E1A-⌬pRb cell line expressed only the product of the 12S form of E1A. Previous studies have shown that the expres-sion of either the 12S or the 13S form of E1A can sensitize cells to killing by NK cells and macrophages (9–11, 14).
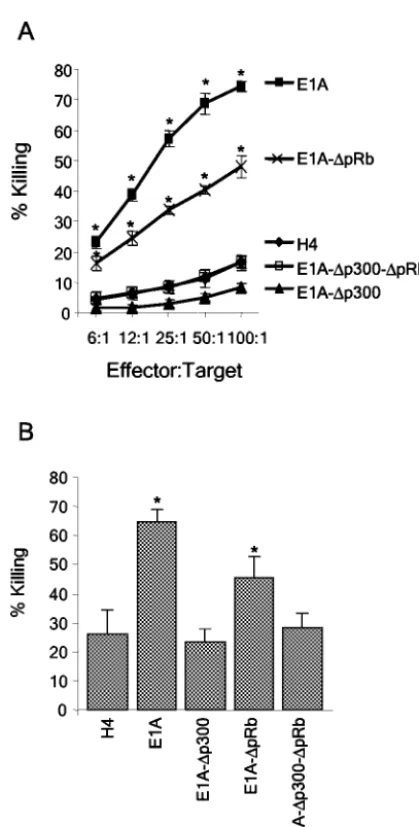
A deletion in CR1 eliminates E1A-induced sensitivity to killing by activated macrophages and NK cells, but a deletion in CR2 does not. Cytolysis assays involving activated macro-phages or NK cells were performed with the H4, H4-E1A, H4-E1A-⌬p300, H4-E1A-⌬pRb, and H4-E1A-⌬p300-⌬pRb cell lines (Fig. 2). We were particularly interested in determin-ing whether the p300-binddetermin-ing site in CR1 was required for E1A-induced sensitivity to killing, since prior data showed that a point mutation in the N-terminal, p300-binding site of E1A (E1A-RG2 [Fig. 1]) eliminated its ability to sensitize cells to killing by NK cells (15). The H4-E1A-⌬pRb cell line was sus-ceptible to killing by NK cells (P ⫽ 0.0001) and activated macrophages (P⫽0.0021) compared to the parental H4 cell line. In contrast, the H4-E1A-⌬p300 and H4-E1A-⌬ p300-⌬pRb cell lines were as resistant to lysis by NK cells and activated macrophages as H4 cells were. Equivalent results were seen when additional, independently derived clones of H4-E1A-⌬pRb, H4-E1A-⌬p300, and H4-E1A-⌬p300-⌬pRb were used in cytolysis assays (data not shown). These results indicate that the expression of the p300-binding region (CR1) of E1A was required to sensitize cells to killing by NK cells and activated macrophages but that the expression of the pRb-binding region (CR2) was not.
The E1A/E7 chimeric protein is expressed at high levels in stably transfected cell lines. Our initial experiments demon-strated that the expression of the p300-binding domain of E1A was required to sensitize tumor cells to killing by NK cells and activated macrophages. The E7 oncoprotein interacts with p300 through CR2 and the adjacent casein kinase II phosphor-ylation site (3). However, E7 lacks the N-terminal p300-bind-ing domain present in E1A (54). Therefore, we hypothesized that unique functions of the N terminus and CR1 of E1A, which encompasses the E1A-p300 binding domain, account for the difference in sensitivities of E1A- and E7-expressing cells to killing by NK cells and macrophages. The CR1s of E1A and E7 are interchangeable for the transformation function (6) but may not be interchangeable for binding to p300. For that reason, we constructed anE1A/E7chimeric gene that included the p300-binding region of E1A (amino acids 1 to 114) fused to the E7 gene in which CR1 was deleted (amino acids 14 to 98) (Fig. 3A). The chimeric gene expressed the p300-binding do-main of E1A (the N-terminal nonconserved region and CR1) and part of the nonconserved region between CR1 and CR2 that allowed for the detection of the translated protein with the E1A-specific monoclonal antibody M37 (26).
H4 cells that stably express the chimeric E1A/E7 protein were established, and the level of E1A/E7 protein was
on November 8, 2019 by guest
http://jvi.asm.org/
pared to the level of E7 expressed in the H4-E7 cell line. SiHa and Caski are cell lines derived from HPV16-induced cervical carcinomas that express small and large amounts of the HPV16 E7 oncoprotein, respectively (56). Previous studies demon-strated that the amount of HPV16 E7 expressed in H4-E7 cells was greater than that expressed in the SiHa cell line and approximately equivalent to that expressed in the Caski cell line (47, 49). Levels of the E1A/E7 protein expressed in H4-E1A/E7 cells were slightly higher than levels of the E7 protein expressed in H4-E7 cells (Fig. 3B). Thus, the expression of the E1A/E7 chimeric protein was comparable to the large amounts of E7 expressed in HPV16-transformed cervical carcinomas.
[image:4.603.59.269.62.476.2]The E1A/E7 chimeric protein binds p300 and pRb. Coim-munoprecipitation studies were performed to determine
FIG. 3. The E1A/E7 chimeric protein is expressed at high levels in stably transfected cell lines and binds p300 and pRb proteins. (A) Di-agram of the E1A/E7 chimeric protein that consists of the N terminus and CR1 of E1A fused to the E7 CR2 and C-terminal regions. Amino acid (aa) numbers are shown at the top, and nucleotide locations in base pairs are indicated at the bottom. The binding locations of p300 and pRb are indicated. (B) E1A/E7 and E7 protein expression were analyzed by Western blotting with stably expressing cell lines by using an E7-specific antibody, ED17, as described in Materials and Methods. Protein molecular weights in thousands are shown on the left. “#1” and “#2” indicate independently derived cell lines. E1A or E7 and associated proteins were coimmunoprecipitated from total cell lysates by using E1A-specific (M37) or E7-specific (ED17) antibodies. (C and D) Immunoprecipitated proteins were separated by electrophoresis, blotted, and probed with antibody to p300 (C) or pRb (D).
FIG. 2. The ability of E1A to bind p300 correlates with its ability to sensitize H4 cells to killing by NK cells and activated macrophages. Wild-type and mutant E1A-expressing H4 cell lines were tested for sensitivity to NK cells (A) or activated macrophages (B) as described in Materials and Methods. E1A, H4-E1A; E1A-⌬pRb, H4-E1A-⌬pRb; E1A-⌬p300, H4-E1A-⌬p300; E1A-⌬p300-⌬pRb, H4-E1A-⌬ p300-⌬pRb. An asterisk indicates a significant difference (P⬍0.05) from the value for the H4 parental cell line.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.603.340.493.107.573.2]whether the E1A/E7 chimeric protein is able to bind p300 and pRb (Fig. 3C and D). Consistent with published studies (28), the E1A-⌬p300 mutant, which contains a deletion in CR1, did not bind cellular p300 (Fig. 3C) but did bind pRb (Fig. 3D). The E1A-⌬pRb mutant, which contains a deletion in CR2, bound p300 (Fig. 3C) but did not bind pRb (Fig. 3D). As predicted based on the known sequence requirements for E1A-p300 and E7-pRb binding, the E1A/E7 chimeric protein bound both p300 (Fig. 3C) and pRb (Fig. 3D). We were unable to detect an interaction between E7 and p300 by using coim-munoprecipitation (Fig. 3C). The inability to detect an inter-action may be due to the insensitivity of the coimmunoprecipi-tation assay. Alternatively, the monoclonal antibody (ED17) that we used for immunoprecipitation may interfere with E7-p300 binding.
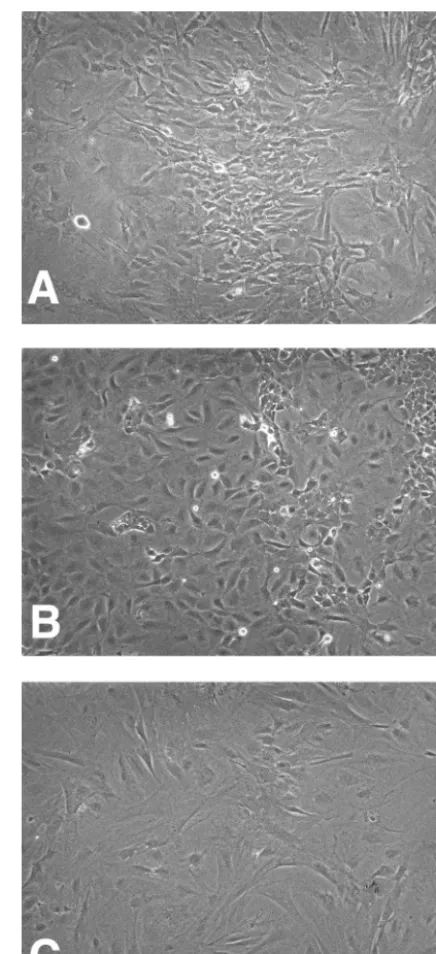
Expression of the E1A/E7 chimeric protein immortalizes primary murine fibroblasts.Next, we determined whether the E1A/E7 chimeric protein was able to immortalize primary cells. Primary MEF cells were isolated from C57BL/6 embryos and transfected with plasmids that expressed the E1 region of Ad5 or the E1A/E7 chimeric protein. MEF cell lines that stably express E1 or E1A/E7 proteins were established. There were no observable differences in growth rates between the MEF immortalized by E1 and those immortalized by E1A/E7 (data not shown). In contrast to primary MEF, MEF immortalized with E1 or E1A/E7 were continually passaged for months with-out evidence of senescence. The E1A/E7-immortalized MEF cell line retained a fibroblastic morphology similar to that of primary MEF cells, whereas the E1-immortalized cells exhib-ited an epithelial cell morphology (Fig. 4). In summary, the
E1A/E7 gene encoded a functional chimeric protein that
bound p300 and pRb and immortalized primary MEF. The expression of this chimeric protein allowed us to determine whether the expression of the E1A p300-binding domain was sufficient to sensitize cells to killing in the context of a fully functional protein.
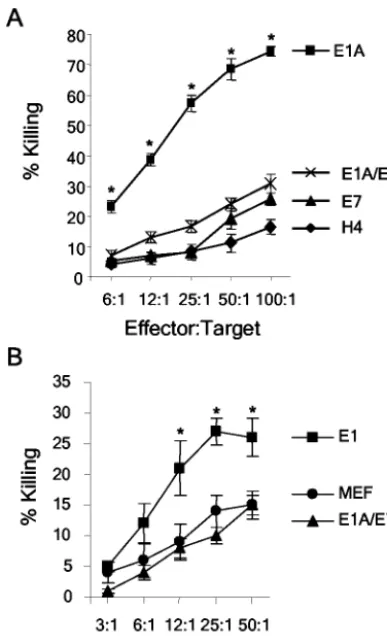
Expression of E1A/E7 sensitizes human and murine cells to killing by activated macrophages. We used the E1A/E7-ex-pressing cell lines to determine whether the expression of CR2 and the C terminus of E7 could complement the p300-binding domain of E1A in sensitizing cells to killing by activated mac-rophages. The human (H4, H4-E1A, H4-E7, and H4-E1A/E7) and murine (MEF, MEF-E1, and MEF-E1A/E7) cell lines were tested in cytolysis assays by using activated macrophages. It was possible to test the sensitivity of murine and human cells with activated murine macrophages because macrophage-me-diated killing of E1A-expressing target cells is effective across species barriers (12, 16, 36). The H4-E1A/E7 cell line was significantly more sensitive to killing by macrophages than were the parental H4 (P⫽0.0004) and H4-E7 (P⫽0.0001) cell lines (Fig. 5A). Similar results were seen when a second, independently derived clone of H4-E1A/E7 was used in mac-rophage cytolysis assays (data not shown). E1A/E7-immortal-ized MEF were significantly more sensitive than primary MEF cells (P⫽ 0.0187) (Fig. 5B) but less sensitive than MEF-E1 cells (P⫽0.0005) to killing by macrophages. Because H4-E1A and H4-E1A/E7 were killed equivalently by macrophages, the significance of the intermediate phenotype of MEF-E1A/E7 cells is unclear. Regardless, these data demonstrate that ex-pression of the first 114 amino acids of E1A, which
encom-passes the E1A-p300 binding domain, was sufficient to sensi-tize cells to lysis by activated macrophages. These data also established that the N-terminal p300-binding domain present in E1A but lacking in E7 accounted for the inability of E7 to sensitize cells to lysis by macrophages.
Expression of E1A/E7 does not sensitize human and murine cells to killing by NK cells.The human (H4, H4-E1A, H4-E7, and H4-E1A/E7) and murine (MEF, MEF-E1, and MEF-E1A/ E7) cell lines were tested in cytolysis assays involving human (NKL) or rat (RNK) cell lines, respectively. In agreement with
FIG. 4. E1A/E7-immortalized MEF. MEF were transfected with plasmids that express the E1 region (E1A and E1B) of Ad5 or the E1A/E7 chimeric protein and selected for resistance to G418. The cell lines were photographed at a magnification of ⫻100. (A) MEF; (B) MEF-E1; (C) MEF-E1A/E7.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.603.312.530.67.544.2]prior studies, the expression of E1A sensitized H4 (Fig. 6A) and MEF (Fig. 6B) cells to lysis by NK cells. In contrast, H4 and MEF cells that expressed E1A/E7 were as resistant as parental or E7-expressing cells to lysis by NK cells (Fig. 6). Similar results were seen when a second, independently de-rived clone of H4-E1A/E7 was used in NK cytolysis assays (data not shown). These data establish that expression of the region of E1A that encompasses the p300-binding domain is not sufficient to sensitize cells to lysis by NK cells. The E7 portion of the chimeric protein did not functionally replace the additional region of E1A that is required to sensitize cells to killing by NK cells.
DISCUSSION
The purpose of this study was to define functional differ-ences between the E1A and E7 oncoproteins that account for the ability of E1A and the inability of E7 to sensitize cells to killing by activated macrophages and NK cells. Using H4 cells that stably expressed mutant E1A proteins, we demonstrated that the expression of the CR1 (p300-binding) region of E1A is required to sensitize tumor cells to lysis by NK cells and activated macrophages (Fig. 2). In contrast, the correlation between binding to pRb by E1A and sensitivity to killing by macrophages or NK cells was more complex. H4-E1A-⌬pRb was less sensitive than H4-E1A to killing by NK cells (Fig. 2A) and macrophages (Fig. 2B). It is possible that this intermediate sensitivity is due to lower levels of E1A being expressed in the H4-E1A-⌬pRb cell line (Fig. 1B), since there is a threshold level of E1A that needs to be expressed in order to induce sensitivity (10, 11, 18). Previous studies showed that an intact CR2 (pRb-binding) region of E1A is required to sensitize cells to TNF-␣-dependent killing (17). Therefore, an alternative explanation is that an E1A-pRb interaction is necessary to fully sensitize cells to killing by macrophages and NK cells.
In contrast to a previously reported observation (1), our laboratory recently found that the expression of E7 in murine (3T3 and MCA-102) and human (H4) cells does not sensitize cells to killing by macrophages (36). The E7 oncoprotein also does not sensitize cells to killing by NK cells (47, 49). E7 lacks the N-terminal p300-binding domain of E1A required for NK cell and macrophage sensitivity (3). Therefore, we constructed a chimericE1A/E7gene that included the N terminus and CR1 of E1A fused to CR2 and the C-terminal sequences of E7 (Fig. 3A). TheE1A/E7gene encoded a chimeric protein that bound p300 and pRb (Fig. 3C and D) and immortalized primary MEF (Fig. 4). The expression of the E1A/E7 protein sensitized both H4 and MEF cells to killing by activated macrophages (Fig. 5) but not to killing by NK cells (Fig. 6). Thus, functional differ-ences within the N-terminal regions of E1A and E7 accounted for the ability of E1A to sensitize cells to killing by macro-phages but not to killing by NK cells.
Prior studies established that a point mutation in E1A at amino acid residue 2 (E1A-RG2) that abolishes the ability of E1A to bind p300 also abolishes E1A-induced sensitivity of H4 cells to lysis by NK cells (15). In our study, the expression of E1A-⌬p300, which contains a deletion in the p300-binding site within CR1, also did not sensitize H4 cells to lysis by NK cells. Taken together, these observations indicate that the expression of the p300-binding domain of E1A is required to sensitize
cells to lysis by NK cells. However, the expression of E1A/E7 did not sensitize cells to lysis by NK cells. Therefore, regions of E1A in addition to the N-terminal p300-binding site are re-quired to sensitize cells to lysis by NK cells.
[image:6.603.342.490.71.466.2]Prior studies indicated that the expression of any of several nonoverlapping regions of exon 2, in addition to the p300-binding domain in exon 1, is required to sensitize E1A-trans-fected or Ad-inE1A-trans-fected rodent cells to killing by NK cells (9, 33). Those results raised the possibility that tethering the p300-binding domain of E1A to a short peptide of random sequence may be sufficient to sensitize cells to killing by NK cells. Our study using cells that express the E1A/E7 protein established that this hypothesis was not correct. Furthermore, the failure of the E1A/E7 chimeric protein to sensitize tumor cells to NK cell-mediated killing demonstrated that functions encoded
FIG. 5. Expression of E1A/E7 sensitizes human and murine cells to killing by activated macrophages. Macrophage cytolysis assays were performed with H4, H4-E1A, H4-E7, and H4-E1A/E7 (A) or MEF, MEF-E1, and MEF-E1A/E7 (B) cell lines as described in Materials and Methods. An asterisk indicates a significant difference (P⬍0.05) from the value for the parental cell line (H4 or MEF).
on November 8, 2019 by guest
http://jvi.asm.org/
within exon 2 of E1A could not be complemented by amino acids 14 to 98 of E7.
Exon 2 of E1A has unique functions that may be required, in addition to p300- binding, to sensitize cells to killing by NK cells. Exon 2 interacts with C-terminal binding protein, which is a transcriptional repressor protein (8). Exon 2 also contains a histone acetyltransferase (HAT) inhibitory domain, which inhibits the HAT activity of p300 (7). E1A inhibits p300-de-pendent transcription by disruption of coactivation complexes (34, 55) as well as by inhibition of HAT activity. Thus, it is possible that the exon 2-mediated inhibition of transcription by either HAT inhibition or C-terminal binding protein binding may act in concert with exon 1-p300 binding to sensitize cells to killing by NK cells.
Recent studies demonstrated that the E7 oncoprotein inter-acts with p300 (3). However, E7 lacks a region analogous to the N terminus of E1A and interacts with p300 through a region in CR2 and the adjacent casein kinase II phosphorylation site (3). E7 interacts predominantly with the C/H1 domain of p300, while E1A interacts primarily with the C/H3 (TRAM) domain. Based on the known functional regions of p300, the interaction of E1A and E7 with p300 in distinct locations would likely
result in different effects on p300-mediated gene expression (3). For example, the human T-cell leukemia virus type 1 Tax protein interacts with p300 at the KIX domain and activates p300-dependent transcription. Conversely, E1A interacts with the C/H3 domain and inhibits p300-dependent transcription (23). We hypothesize that the interaction of E1A and E7 at distinct sites within p300 results in the ability of E1A and the inability of E7 to sensitize cells to killing by macrophages and NK cells.
There are additional complexities regarding the functional consequences of the interaction of viral oncoproteins with p300 and CBP that may explain our results. First, p300 and CBP have many overlapping functions but also exhibit unique functions (4, 31, 45, 51, 57). Therefore, the interaction of viral oncoproteins with p300 and CBP can have opposing effects on transcription. For example, Kaposi’s sarcoma-associated her-pesvirus v-IRF and Epstein-Barr virus EBNA-2 transactivate theMYCpromoter through interaction with CBP (29). How-ever, overexpression of p300 suppresses v-IRF- and EBNA-2-mediated transactivation ofMYC(29). Second, even when viral oncoproteins interact with p300 at the same domain, the con-sequences of this interaction may be different. For example, although both E1A and simian virus 40 large T interact with p300 at the C/H3 domain, large T inhibits, whereas E1A en-hances, the phosphorylation of p300 (21).
There are several possible explanations, which are not mu-tually exclusive, for the ability of the E1A/E7 chimeric protein to sensitize cells to killing by macrophages but not to killing by NK cells. One possible explanation is that the molecular re-quirements that mediate the E1A-induced sensitivity of tumor cells to killing by macrophages are different from those that mediate such sensitivity to killing by NK cells. Our results, along with previous reports, indicate that E1A-induced sensi-tivity to killing by NK cells requires both the p300-binding region and portions of exon 2. However, E1A-induced sensi-tivity to killing by macrophages required only the p300-binding region. Activated macrophages kill E1A-expressing cells through the production of TNF-␣and NO (11, 19, 36), whereas NK cells kill E1A-expressing cells through perforin/granzyme-and Fas-dependent mechanisms (14, 32). E1A-induced sensi-tivities of a tumor cell to killing by these various molecules may be mediated by different molecular mechanisms, reflecting a requirement for different regions of E1A. Alternatively, recep-tors on NK cells and macrophages may be triggered by differ-ent ligands on E1A-expressing cells. The regions of E1A re-quired to increase the expression of these ligands may also differ.
[image:7.603.67.261.69.391.2]Despite the strong functional similarities between the Ad E1A and HPV E7 oncoproteins, the ability of E1A and the inability of E7 to sensitize transformed cells to killing by immunological mediators is striking. This study demon-strates that functional differences in the N terminus of E1A and E7 partly account for disparities in the sensitivities of E1A- and E7-expressing human and murine tumor cells to lysis by innate effector cells. Furthermore, our results sug-gest that the expression of E1A sensitizes cells to killing by macrophages and NK cells through distinct molecular mech-anisms.
FIG. 6. Expression of E1A/E7 does not sensitize human or murine cells to killing by NK cells. NK cell cytolysis assays were performed with H4, H4-E1A (E1A), H4-E7 (E7), and H4-E1A/E7 (E1A/E7) cell lines (A) or MEF, MEF-E1 (E1), and MEF-E1A/E7 (E1A/E7) cell lines (B) as described in Materials and Methods. An asterisk indicates a significant difference (P⬍0.05) from the value for H4-E7 (A) or MEF (B).
on November 8, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
This work was supported by Public Health Services grant RO1-CA76491 and seed grant support funded by the University of Colorado Cancer Center (to J.M.R); Public Health Services grant RO1-CA86727 and Department of the Army grant DAMD-17-98-1-8324 (to J.L.C.); the Cancer League of Colorado, NIH training grant T32-AI00048, and a Great West Life Assurance endowed fellowship (to T.A.M); and the Cancer Research Institute Predoctoral Emphasis Pathway in Tumor Immunology (grant to K.M.).
We thank S. Bayley, A. van der Eb, M. J. Robertson, M. Nakamura, S. Frisch, and E. Harlow for reagents, M. Ellison for statistical analy-ses, and G. Cheatham for secretarial assistance.
REFERENCES
1. Banks, L., F. Moreau, K. Vousden, D. Pim, and G. Matlashewski.1991. Expression of the human papillomavirus E7 oncogene during cell transfor-mation is sufficient to induce susceptibility to lysis by activated macrophages. J. Immunol.146:2037–2042.
2. Bernards, R., A. Houweling, P. I. Schrier, J. L. Bos, and A. J. van der Eb. 1982. Characterization of cells transformed by Ad5/Ad12 hybrid early region I plasmids. Virology120:422–432.
3. Bernat, A., N. Avvakumov, J. S. Mymryk, and L. Banks.2003. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene22:5927–5937.
4. Bordoli, L., S. Husser, U. Luthi, M. Netsch, H. Osmani, and R. Eckner.2001. Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic Acids Res. 29:4462–4471.
5. Bosch, F. X., M. M. Manos, N. Mun˜oz, M. Sherman, A. M. Jansen, J. Peto, M. H. Schiffman, V. Moreno, R. Kurman, and K. V. Shah.1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. J. Natl. Cancer Inst.87:796–802.
6. Brokaw, J. L., C. L. Yee, and K. Munger.1994. A mutational analysis of the amino terminal domain of the human papillomavirus type 16 E7 oncopro-tein. Virology205:603–607.
7. Chakravarti, D., V. Ogryzko, H. Y. Kao, A. Nash, H. Chen, Y. Nakatani, and R. M. Evans.1999. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell96:393–403.
8. Chinnadurai, G.2002. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell9:213–224.
9. Cook, J. L., C. K. Krantz, and B. A. Routes.1996. Role of p300-family proteins in E1A oncogene induction of cytolytic susceptibility and tumor cell rejection. Proc. Natl. Acad. Sci. USA93:13985–13990.
10. Cook, J. L., D. L. May, A. Lewis, Jr., and T. A. Walker.1987. Adenovirus E1A gene induction of susceptibility to lysis by natural killer cells and activated macrophages in infected rodent cells. J. Virol.61:3510–3520. 11. Cook, J. L., D. L. May, B. A. Wilson, B. Holskin, M. J. Chen, D. Shalloway,
and T. A. Walker.1989. Role of tumor necrosis factor-alpha in E1A onco-gene-induced susceptibility of neoplastic cells to lysis by natural killer cells and activated macrophages. J. Immunol.142:4527–4534.
12. Cook, J. L., D. L. May, B. A. Wilson, and T. A. Walker.1989. Differential induction of cytolytic susceptibility by E1A,myc, andrasoncogenes in im-mortalized cells. J. Virol.63:3408–3415.
13. Cook, J. L., T. A. Miura, D. N. Ikle, J. A. M. Lewis, and J. M. Routes.2003. E1A oncogene-induced sensitization of human tumor cells to innate immune defenses and chemotherapy-induced apoptosis in vitro and in vivo. Cancer Res.63:3435–3443.
14. Cook, J. L., T. A. Potter, D. Bellgrau, and B. A. Routes.1996. E1A oncogene expression in target cells induces cytolytic susceptibility at a post-recognition stage in the interaction with killer lymphocytes. Oncogene13:833–842. 15. Cook, J. L., B. A. Routes, T. A. Walker, K. L. Colvin, and J. M. Routes.1999.
E1A oncogene induction of cellular susceptibility to killing by cytolytic lym-phocytes through target cell sensitization to apoptotic injury. Exp. Cell Res. 251:414–423.
16. Cook, J. L., T. A. Walker, A. M. Lewis, Jr., H. E. Ruley, F. L. Graham, and S. H. Pilder.1986. Expression of the adenovirus E1A oncogene during cell transformation is sufficient to induce susceptibility to lysis by host inflam-matory cells. Proc. Natl. Acad. Sci. USA83:6965–6969.
17. Cook, J. L., T. A. Walker, G. S. Worthen, and J. R. Radke.2002. Role of the E1A Rb-binding domain in repression of the NF-kB-dependent defense against tumor necrosis factor-alpha. Proc. Natl. Acad. Sci. USA99:9966– 9971.
18. Cook, J. L., B. A. Wilson, L. A. Wolf, and T. A. Walker.1993. E1A oncogene expression level in sarcoma cells: an independent determinant of cytolytic susceptibility and tumor rejection. Oncogene8:625–635.
19. Day, D. B., N. A. Zachariades, and L. R. Gooding.1994. Cytolysis of ade-novirus-infected murine fibroblasts by IFN-␥-primed macrophages is TNF-and contact-dependent. Cell. Immunol.157:223–238.
20. Dyson, N., P. Guida, K. Munger, and E. Harlow.1992. Homologous
se-quences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol.66:6893–6902. 21. Eckner, R., J. Ludlow, N. Lill, E. Oldread, Z. Arany, N. Modjtahedi, J.
DeCaprio, D. Livingston, and J. Morgan.1996. Association of p300 and CBP with simian virus 40 large T antigen. Mol. Cell. Biol.16:3454–3464. 22. Frisch, S.1991. Antioncogenic effect of adenovirus E1A in human tumor
cells. Proc. Natl. Acad. Sci. USA88:9077–9081.
23. Goodman, R. H., and S. Smolik.2000. CBP/p300 in cell growth, transfor-mation, and development. Genes Dev.14:1553–1577.
24. Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn.1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol.36:59–72.
25. Green, M., W. Wold, J. Mackey, and P. Rigden.1979. Analysis of human tonsil and cancer DNAs and RNAs for DNA sequences of group C (sero-types 1, 2, 5 and 6) human adenovirus. Proc. Natl. Acad. Sci. USA76:6606– 6610.
26. Harlow, E., R. Franza, Jr., and C. Schley.1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol.55:533–546.
27. Helt, A.-M., and D. A. Galloway.2003. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24:159–169.
28. Howe, J. A., J. S. Mymryk, C. Egan, P. E. Branton, and S. T. Bayley.1990. Retinoblastoma growth suppressor and a 300-kDa protein appear to regulate cellular DNA synthesis. Proc. Natl. Acad. Sci. USA87:5883–5887. 29. Jayachandra, S., K. G. Low, A.-E. Thlick, J. Yu, P. D. Ling, Y. Chang, and
P. S. Moore. 1999. Three unrelated viral transforming proteins (vIRF, EBNA2, and E1A) induce the MYC oncogene through the interferon-re-sponsive PRF element by using different transcription coadaptors. Proc. Natl. Acad. Sci. USA96:11566–11571.
30. Jelsma, T. N., J. A. Howe, C. M. Evelegh, N. F. Cunniff, M. H. Skiadopoulos, M. R. Floroff, J. E. Denman, and S. T. Bayley.1988. Use of deletion and point mutants spanning the coding region of the adenovirus 5 E1A gene to define a domain that is essential for transcriptional activation. Virology 163:494–502.
31. Kawasaki, H., R. Eckner, T. P. Yao, K. Taira, R. Chiu, D. M. Livingston, and K. K. Yokoyama.1998. Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature393:284–289. 32. Klefstrom, J., P. E. Kovanen, K. Somersalo, A. O. Hueber, T. Littlewood,
G. I. Evan, A. H. Greenberg, E. Saksela, T. Timonen, and K. Alitalo.1999. c-Myc and E1A induced cellular sensitivity to activated NK cells involves cytotoxic granules as death effectors. Oncogene18:2181–2188.
33. Krantz, C. K., B. A. Routes, M. P. Quinlan, and J. L. Cook.1996. E1A second exon requirements for induction of target cell susceptibility to lysis by natural killer cells: implications for the mechanism of action. Virology217: 23–32.
34. Kurokawa, R., D. Kalafus, M.-H. Ogliastro, C. Kioussi, L. Xu, J. Torchia, M. G. Rosenfeld, and C. K. Glass.1998. Differential use of CREB binding protein-coactivator complexes. Science279:700–703.
35. Miller, A. D., D. G. Miller, J. V. Garcia, and C. M. Lynch.1993. Use of retroviral vectors for gene transfer and expression. Methods Enzymol.217: 581.
36. Miura, T. A., K. Morris, S. Ryan, J. L. Cook, and J. M. Routes.2003. Adenovirus E1A, not human papillomavirus E7, sensitizes tumor cells to lysis by macrophages through nitric oxide- and TNF-a-dependent mecha-nisms despite up-regulation of 70-kDa heat shock protein. J. Immunol. 170:4119–4126.
37. Munger, K., J. R. Basile, S. Duensing, A. Eichten, S. L. Gonzalez, M. Grace, and V. L. Zacny.2001. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene20:7888–7898. 38. Phelps, W. C., S. Bagchi, J. A. Barnes, P. Raychaudhuri, V. Kraus, K.
Mu¨nger, P. M. Howley, and J. R. Nevins.1991. Analysis oftransactivation by human papillomavirus type 16 E7 and adenovirus 12S E1A suggests a com-mon mechanism. J. Virol.65:6922–6930.
39. Phelps, W. C., C. L. Yee, K. Munger, and P. M. Howley.1989. Functional and sequence similarities between HPV16 E7 and adenovirus E1A. Curr. Top. Microbiol. Immunol.144:153–166.
40. Phelps, W. C., C. L. Yee, K. Munger, and P. M. Howley.1988. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell53:539–547.
41. Raska, K., Jr., and P. H. Gallimore.1982. An inverse relation of the onco-genic potential of adenovirus-transformed cells and their sensitivity to killing by syngeneic natural killer cells. Virology123:8–18.
42. Reis, L. F., H. Harada, J. D. Wolchok, T. Taniguchi, and J. Vilcek.1992. Critical role of a common transcription factor, IRF-1, in the regulation of IFN-beta and IFN-inducible genes. EMBO J.11:185–193.
43. Reynolds, C., E. Bere, Jr., and J. Ward.1984. Natural killer activity in the rat. III. Characterization of transplantable large granular lymphocyte (LGL) leukemias in the F344 rat. J. Immunol.132:534–540.
44. Robertson, M. J., K. J. Cochran, C. Cameron, J. M. Le, R. Tantravahi, and J. Ritz.1996. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol.24:406–415.
on November 8, 2019 by guest
http://jvi.asm.org/
45. Roth, J. F., N. Shikama, C. Henzen, I. Desbaillets, W. Lutz, S. Marino, J. Wittwer, H. Schorle, M. Gassmann, and R. Eckner.2003. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J.22:5186–5196.
46. Routes, J., S. Ryan, A. Clase, T. Miura, A. Kuhl, T. Potter, and J. Cook.2000. Adenovirus E1A oncogene expression in tumor cells enhances killing by TNF-related apoptosis-inducing ligand (TRAIL). J. Immunol.165:4522– 4527.
47. Routes, J., S. Ryan, H. Li, J. Steinke, and J. Cook.2000. Dissimilar immu-nogenicities of human papillomavirus E7 and adenovirus E1A proteins in-fluence primary tumor development. Virology277:48–57.
48. Routes, J. M., and J. L. Cook.1995. E1A gene expression induces suscep-tibility to killing by NK cells following immortalization but not adenovirus infection of human cells. Virology210:421–428.
49. Routes, J. M., and S. Ryan.1995. Oncogenicity of human papillomavirus- or adenovirus-transformed cells correlates with resistance to lysis by natural killer cells. J. Virol.69:7639–7647.
50. Sawada, Y., B. Fohring, T. E. Shenk, and K. Raska, Jr.1985. Tumorigenicity of adenovirus-transformed cells; region E1A of adenovirus 12 confers resis-tance to natural killer cells. Virology147:413–421.
51. Vo, N., and R. H. Goodman.2001. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem.276:13505–13508.
52. Vousden, K. H., and P. S. Jat. 1989. Functional similarity between HPV16E7, SV40 large T and adenovirus E1a proteins. Oncogene4:153–158. 53. Walker, T. A., B. A. Wilson, J. A. M. Lewis, and J. L. Cook.1991. E1A oncogene induction of cytolytic susceptibility eliminates sarcoma cell tumor-igenicity. Proc. Natl. Acad. Sci. USA88:6491–6495.
54. Wang, H.-G. H., Y. Rikitake, M. C. Carter, P. Yaciuk, S. E. Abraham, B. Zerler, and E. Moran.1993. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol.67:476–488.
55. Yang, X.-J., V. V. Ogryzko, J. Nishikawa, B. Howard, and Y. Nakatani.1996. A p300/CBP-associated factor that competes with the adenoviral protein E1A. Nature382:319–324.
56. Yee, C., I. Krishnan-Hewlett, C. Baker, R. Schlegel, and P. Howley.1985. Presence and expression of human papillomavirus sequences in human cer-vical carcinoma cell lines. Am. J. Pathol.119:361–366.
57. Yuan, Z. M., Y. Huang, T. Ishiko, S. Nakada, T. Utsugisawa, H. Shioya, Y. Utsugisawa, Y. Shi, R. Weichselbaum, and D. Kufe.1999. Function for p300 and not CBP in the apoptotic response to DNA damage. Oncogene18:5714– 5717.
58. Zucker, M. L., and S. J. Flint.1985. Infection and transformation of mouse cells by human adenovirus type 2. Virology147:126–141.