0022-538X/89/051907-09$02.00/0
Copyright C 1989, American Society for Microbiology
Molecular Analysis of the Function of Direct
Repeats
and
a
Polypurine Tract for Plus-Strand DNA Priming in
Woodchuck Hepatitis Virus
CHRISTOPH SEEGER*AND JOHNNAMARAGOS
Department of Microbiology, Immunology andParasitology, New York State Collegeof Veterinary Medicine,
Cornell University, Ithaca, New York14853-6401
Received 8 November1988/Accepted 23 January 1989
Thereplication of the hepadnavirus DNA genome is initiated by reversetranscription ofpregenomeRNA
intominus-strandDNAfollowedby plus-strand DNAsynthesis. Theprimingofplus-strandDNA requires the transfer ofanRNAprimer frompregenomeRNAtothe primer-bindingsiteonminus-strandDNA. Annealing
oftheprimertotheprimer-binding siteisfacilitatedby shortdirectrepeats,DR1 and DR2. To investigate the
mechanism of plus-strand primer formation, we have introducedspecific mutations into DRI and DR2 and measured theeffect ofthesemutantsoninitiation ofplus-strandDNAsynthesis.Tofacilitatesuchananalysis, wehave constructed a vectorfor the efficient expression of woodchuck hepatitis virus in cultured cells. Our
resultssuggestthat the3'endofthe RNA primer isdetermined priortoits transfertotheprimer-bindingsite and thatthe determination of the 3' endofthe primerdoesnot depend on aspecific sequence motifat the
cleavage site.In addition, wehave identifiedan alternative initiation siteforplus-strandDNAsynthesis ata purine-richsequencebetween DR1 and DR2.Initiationatthis siteoccursbyamechanism that isindependent
of thedirectrepeats and doesnotrequirethetransfer ofanRNAprimertotheprimer-binding site. Woodchuck hepatitis virus (WHV), a member of the
hepadnavirus family, has a small 3.3-kilobase (kb)-long
relaxed circular DNA genome that replicates by reverse transcription ofan RNA template, the pregenome (for re-views, see references 11and 19). Virion DNA is composed
ofacompleteminus strand covalentlylinked toaproteinat
its 5' end and ofan incomplete plus strand with heteroge-neous 3' ends(12). Thegenomeis held in acircular confor-mation by a cohesive overlap, 215 nucleotides in length, representing the distance between the 5' ends of plus- and
minus-strandDNA(23). Uponinfection, therelaxedcircular virion DNA is converted into a covalently closed circular (CCC)molecule present innuclei of virus-infected cells(18, 34; Fig.IA). CCCDNAis thetemplatefor thetranscription
ofa 3.6-kb terminallyredundant RNA initiatingnearthe 5'
end of the precore gene and terminating within the core antigen region (8; Fig. 1A and B). This transcript has
heterogeneous 5' ends bracketing the first AUG of the
precoregene(8).Theshortesttranscript, initiatingwithin the precoreregion, isthetemplateforreversetranscriptionand
is termed pregenome RNA (27). Reverse transcription of pregenomeRNAintominus-strand DNAoccursin immature
viruscoreparticlespresentin the cytoplasm of infectedcells
(32).
Theprimer forminus-strand DNAsynthesisismostlikely aprotein,thestructureandoriginof which havenotyetbeen determined (12, 20). The 5' end of minus-strand DNA lies withintheterminally redundant regionofpregenomeRNAat DR1 (16, 27). The primer forplus-strand DNA synthesis is
anRNAoligomerwhich is translocated from theDR1 region
ofpregenome RNA to the priming site at DR2 (15, 27). Annealingof this 18-nucleotide-long primeris facilitated by
the direct repeats, DR1 and DR2, 11 nucleotides in length (Fig. 1). Since thesequence motifofthe RNAprimermaps
totheterminally redundantportion ofpregenome RNA,the
* Correspondingauthor.
origin of the primer cannot unambiguously be determined from the sequences of cloned viral genomes (8, 27). The observation that the 5' end of the RNA primer of duck
hepatitis B virus (DHBV) is modified (capped) provides
strong circumstantial evidence that the primer is derived
from the5' endofpregenomeRNA(15).Furthersupportfor this model is derived from biochemical studies which
indi-cate that the position of the 5' end of the RNA primer
coincides with the position of the 3' end of minus-strand DNA(16, 27).The mechanism for the formation of theexact 3' end of this primerisnotknown. By analogyto retroviral systems, it has beenspeculatedthat the 3' end is formedby
the action ofan RNase H-likeactivity (27, 38).
Inthepast, models forthe replication strategy of
hepad-naviruses hadmainly been derived from the physical
analy-sis ofreplicativeintermediates isolated from livers and sera
of infected animals. Later, it became feasible to infect animals withcloned viralDNA, permittingageneticanalysis
of the hepadnavirus replication scheme (26, 39). However,
this system allows for only the genetic analysis of altered viralgenomes that remain competent through all stages of thereplication cycle. Mostrecently, it has becomepossible topropagate human hepatitis B virus (HBV)and DHBV in cell culturesystems,providingabasisforadetailed genetic analysisof this virus class(1, 5, 21, 33, 36, 40). To facilitate suchananalysis,weconstructedacytomegalovirus
(CMV)-basedexpression vectorfor the efficientproductionof
infec-tious WHV in cultured cells. An important feature of this systemis thepossibility togenetically manipulatethe termi-nalrepeatsofpregenomeRNAindependentlyof each other.
Using this methodology, we have investigated how the 3' end of the RNA primer for plus-strand DNA synthesis is determined. Specifically, we addressed the question of whether the3' end of the RNAprimeris determinedpriorto or after its transfer to DR2. Our results provide strong evidence that the 3' end ofthe RNA primeris determined before its transfertoDR2 andthat thegenesisofthe3'end
1907
on November 10, 2019 by guest
http://jvi.asm.org/
1908 SEEGER AND MARAGOS
A
DR2
5,
3,
B
R
3320 bp
IS11IS21SURI
POLYMERASE
PqLI|
1DR1
,Sp
DR2DRI|
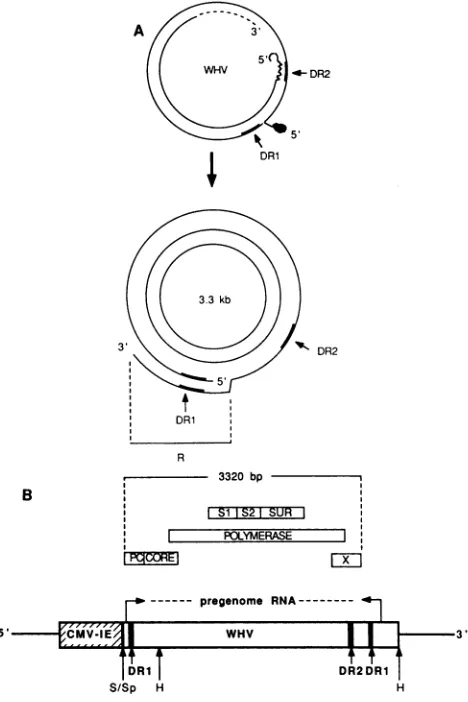
FIG. 1. Transcriptional and translational mapofpCMW82. (A)
The top halfof the figure shows the structure ofthe WHV virion DNA.The protein covalently linkedtothe 5' end of minus-strand DNA isindicatedwith the solidoblong symbol,and the RNAprimer attachedtothe5' end of theplusstrandisrepresented bythewavy
line(dashes imply variabilityatthe 3' endofplus-strand DNA).The
positions ofthe direct repeats, DR1 and DR2, are indicated with
heavy lines. The bottom halfof the figure shows the covalently closed circular(CCC) DNA and the 5' and 3' ends ofpregenome
RNA. R, Terminally redundant portion ofpregenome RNA. The
positionsofDR1and DR2areindicatedasdescribed before.(B)The
CMV-IE promoter-enhancer region is represented by the hatched
box,the WHVsequences arerepresented bythe dottedbox,and the fourWHVgenecoding regionsarerepresented byopenboxes(PC,
pre-core; Si, large surface antigen; S2, middle surface antigen;
SUR, majorsurface antigen).The solid bar above the translational
map of WHV marks the length of the CCC DNA, the natural
template for transcription ofpregenome RNA. DR1 and DR2 are
symbolized byvertical bars. The 5' and 3' ends ofpregenomeRNA
are symbolized by arrows. The restriction sites used for the
con-struction ofpCMW82 areindicated (for details, see Materials and
Methods): S, Sstl; Sp, Sphl; H, Hindlll. bp, Base pairs.
of this primer does not depend upon a specific sequence pattern at the cleavage site. Furthermore, we demonstrate
that priming of plus-strand DNA of WHV can initiate at a
purine-rich site by a mechanism independent of the
DRI-DR2sequence motif.
MATERIALS AND METHODS
Molecular clones. For the construction of pCMW8, the
precursorplasmid of pCMW82, anSstI-to-HindIII fragment
ofplasmid pBC12/CMV/IL-2 (a gift from Bryan Cullen
[71)
was replaced with anSphl-to-HindIII
fragment
frompWHV81-2 (14, 28). The correct fusion between the CMV
andWHVsequenceswasconfirmed
by
nucleotidesequenceanalysis. Subsequently, afull-lengthgenome of WHV clone
WHV2 was released from
pWHV81-2 by cleavage
withHindlIl and inserted into the Hindlll site of
pCMW8,
yielding pCMW82. The mutants
82DR12, 82DR1C,
and82DR1/2C were obtained
by
oligomer-directed
site-specific
mutagenesis
asdescribedby
Taylor
etal.(35)
by using
akitfrom Amersham
Corp.
The mutant genotypes were con-firmed by nucleotide sequenceanalysis.
Cell culture.
HepG2
cells(2)
were cultured in minimalessential medium
(GIBCO Laboratories) supplemented
with10% bovine calfserum
(HyClone
Laboratories, Inc.)
in the presence ofpenicillin
(90U/ml)
andstreptomycin (90
p.g/ml)
at
37°C
and3%CO,.
Thetissue culture mediumwaschanged
in 3- and
4-day
intervals. The transfection of cells withplasmid
DNA wasaccomplished by
the calciumphosphate
procedureas described
by
Chen andOkayama (6).
Isolationofvirion DNA.
Supernatant
from transfected cellswascollected in 3- and
4-day
intervalsover aperiod
of 7to30
days
after DNA transfection. The supernatant was clearedofcellulardebrisby
centrifugation
inaSorvallSS34rotorat9krpmfor10min. The cleared supernatant
(7.5
ml)
waslayered on3 ml ofa10% sucrose and on1 mlofa20%sucrose solution in 20 mM Tris
(pH 8)-150
mM NaCl. The virus was collected by centrifugation in a Beckman SW41rotorfor6 hat40
krpm
orfor 16 hat 33krpm
at10°C.
The supernatantsweredecanted,
and thepellets
weresuspended
in 100
RI
ofasolutioncontaining
10mMTris(pH 8),
100mMNaCl, and 0.1%
3-mercaptoethanol
for at least 6 h at4°C.
One-tenth volume of TM
(100
mM Tris[pH
8],
100 mMMgCI,)
and 5.g
of DNase I(Bethesda
Research Laborato-ries[BRL])
were added. After an incubation of 30 min at37°C,
50[LI
of a solutioncontaining
3% sodiumdodecyl
sulfate,
2 mg ofproteinase
K(BRL)
perml,
40mM EDTA(pH 8),and 200
jig
of yeast tRNA(BRL)
wasaddedand thesuspension
was incubated for 2 to 3 h at37°C.
DNA was extracted twice withphenol
followedby
oneextraction witha solution of 70% butanol and 30%
isopropanol
andethanolprecipitation
in the presence of LiCl(1
M finalconcentra-tion)
at-70°C.
Pellets were washed with 70%ethanol,
suspended
in 20 ,ul ofTE(10
mMTris[pH
8],
1 mMEDTA),
and stored at
-20°C.
Primer extension analysis. To 5
p[l
of a virion DNApreparation,
1 pl (200 pg)of 5'-end-labeled[32P]DNA
oligo-mer
primer
was added, and the mixture was denatured at95°C
for 2 min.Samples
were chilledimmediately
on ice. Theprimer
extension reaction wasperformed
under thefollowing
conditions. Five microliters of the virionDNA-primer
mixturewassupplemented
withdATP, dCTP,
dGTP,
and TTP
(100 FM
finalconcentration),
50mMTris(pH
8),
6 mMMgC1,,
40 mMKCI,
1mMdithiothreitol(Sigma
Chem-icalCo.),
0.1 mg ofacetylated
bovine serum albumin(Promega
Biotec)
and 2 U of avianmyeloblastosis
virusreverse
transcriptase (Seikagaku
AmericaInc.)
per ml. The final volume of the reaction was 10pL..
The reaction was incubated at42°C
for60minandstopped by
the addition of6
p.l
offormamide.Samples
weredenaturedat95°C
for3minprior to
electrophoresis through
a 7%polyacrylamide-8
M ureasequencing gel.
Gels weredried andexposed
toKodakXAR-5 film (Eastman Kodak
Co.)
with anintensifying
screenfor24to72 hat
-70°C.
Forthedigestion
of the RNAprimer prior
to the addition of avianmyeloblastosis
virusreverse
transcriptase,
RNase T1(BRL)
or RNase CL3J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.71.305.73.424.2]PLUS-STRAND DNA PRIMING IN WHV 1909
(BRL) was added to the reaction mixture prior to the
addition of avian myeloblastosis virusreverse transcriptase toyield afinal concentration of 140 or 0.8 U/u1l, respectively, and then samples were incubated for 30 min at 37°C. For treatment of virion DNA with sodium hydroxide, 10 ,lI of purified virion DNA was incubated for 5 min at 95°C in the presence of 200 mM sodium hydroxide. Tris hydrochloride wasadded to afinalconcentration of 333 mM, and DNA was precipitated with ethanol at -70°C. Pellets were suspended in 10 ,ulof TE. DNAoligomers (10 ng) were treatedattheir 5'endswithpolynucleotide kinase at 0.9 U/,ul(Pharmacia) in the presence of[y-32P]ATP (3 x
103
Ci/mmol; 1.7 ,uM), 40 mM Tris hydrochloride (pH 7.4), 10 mMMgCI2,
100 mMKCI, 5mMdithiothreitol (Sigma) inatotal volumeof10
RI.
The reactionwas incubated for 30 min at 37°C and stopped by incubation for 10 min at 65°C. The mixture was diluted 10-fold with TE. The sizes of the primer-extended products were determined with the help of a sequencing ladder derived from M13 clone MW2sh containing a Hindlll-to-SstII fragment of WHV2 (14). Primer extension and sequencing reactions were primed with the same DNA oligomer.DNA oligomers. Oligonucleotides for primer extension analysis correspond to position 3248 to 3229 (CS2) and to
position 26 to 3316 (DR17P) on the WHV genome.
Nomenclature. Tofacilitate a comparison of the presented data with previously published results, the nucleotide
se-quence numbering of the WHV2 genome (14) was arranged
tobeginwith thefirstATGof the precore openreading frame
(position 1931 [14]) in accord with the published sequences of HBV andground squirrel hepatitis virus (25, 37).
A r:. RNA PRIMfER r (+) DNA
UAUCUUUUUCACCUGUGCA 3
_--_--_ _ 5
A
I29 '3 27, NT 147
POS
A
3249'.8
CN C
1j2'3-
4
'516'--H7
_-04 r- ,C14O nt
r C, v-r_
\
~~146
nta
RESULTS
Construction ofan expression vector for theproductionof WHV incultured cells. Theproductionof HBV and DHBV in selected human hepatomacell lines transfected with cloned
viral DNA has previously been described (1, 5, 33, 40). To expressWHVinculturedcells,wefused WHVsequencesto
the CMV immediate-early (CMV-IE) promoter-enhancer
region (7, 31). The recombinant plasmid pCMW82 was designed to direct the synthesis of pregenome RNA, the template for reverse transcription (Fig. 1B). This strategy
wasbased ontwo considerations important forarapid and
detailed genetic analysis of the WHV genome: (i) the effi-cient production and secretion of virus particles for a
bio-chemicalanalysisof virion DNA and(ii)themanipulationof theterminallyredundant ends ofpregenome RNA indepen-dently of each other. We chose the CMV-IE
promoter-enhancer complex because it drives very high levels of
transcription and is relatively nonspecific for cell type (3). By deleting the WHV promoter and thereby reducing the length of the terminally redundant DNA template for
tran-scription, itwaspossibletominimize the chance for homol-ogous recombination of the redundant ends upon
transfec-tionofcultured cellswith cloned DNA.
ToinvestigatewhetherpCMW82 could direct the
produc-tionof virion DNA, HepG2 cells weretransfected with this
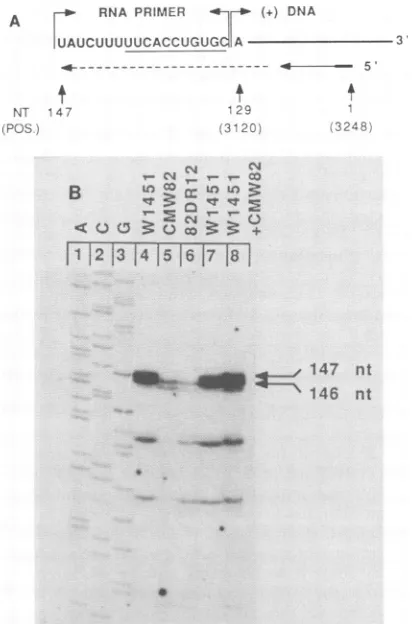
construct and virion DNA was purified from the culture supernatant as described in Materials and Methods. The ability of this system to lead to correct synthesis of virion DNAwasverifiedby mappingthe 5'end ofplus-strandDNA by primerextensionanalysisasshownschematicallyin Fig.
2A. Figure 2B shows a comparison of primer-extended products obtained from virion DNA isolated from an
in-fected woodchuck, W1451 (lanes 4 and 7), and fromvirion
FIG. 2. Comparison ofthe 5' end of plus-strand DNA from a WHV-infected animalandfromWHVproducedinHepG2cells.(A) The position of oligonucleotide CS2 used for primer extension analysis ofthe 5' endof virion DNAis indicated(position [POS.] 3248 to 3229). The position of the 5' end of plus-strand DNA (position 3120) and the sequence of the RNAprimer of WHV are shown as determined previously (27). The lengths ofthe primer-extendedproductsareindicated(NT).(B) Primer extensionanalysis was performed as described in Materials and Methods by using DNA oligomer CS2 as a primer. The lengths of the extended products are indicated. A nucleotide-sequencing ladder derived fromM13 cloneMWhs, primed with oligomerCS2, provided size markers(lanesI to 3). Lanes 4 and 7 show theextendedprimer from reactions with virion DNAisolated from woodchuck1451; lanes 5 and 6 show the extended primerfrom reactions with virionDNA isolated fromHepG2cellstransfected withpCMW82 (lane 5)orwith p82DR12, a derivative of pCMW82 (lane 6) (Fig. 3B). Lane 8 contains equalamounts of the reactionproducts shownin lanes 4 and 5. nt, Nucleotides.
DNA isolated from the supernatant ofHepG2 cells previ-ously transfected with pCMW82 (lane 5) or p82DR12 (lane 6),aderivative ofpCMW82 (seebelow). The DNA products
extended onvirion DNA derived from tissue culture cells are 146nucleotides in length, 1nucleotide short of the products
derived from virion DNA isolated from an infected wood-chuck. Mixing the extended products from those two
reac-tions priorto gel electrophoresis confirmed this conclusion
(Fig. 2B, lane8). The occurrence ofdoublets in the primer extension reactions ismost likely a consequenceof
prema-ture disengagement of the reverse transcriptase from the DNA template. Removal of the RNA primer with RNase
CL3 priortoprimerextension results in extensionproducts of thesamelength(seeFig. 4B,lanes3and4).This indicates that the position of the 5' ends of plus-strand DNA is
VOL. 63, 1989
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.330.537.70.382.2]1910 SEEGER AND MARAGOS
identical in virus propagated in tissue culture cells and in virus obtained from infected animals. We conclude from these results that the RNA primer producedin cultured cells is 1 nucleotide shorter (17 nucleotides in length) than the RNA primer made in vivo (27). On the basis of the distance between the TATA box and the cap site of the original CMV-IE transcript, the 5' end of pregenome RNA tran-scribed from the CMV-IE promoter maps to position 5 on the WHV genome, 1 nucleotide short of the transcript made by WHV in vivo (8, 27, 31). This result provides direct genetic evidence for the origin of the RNA primer for plus-strand DNA synthesis from the 5' end of pregenome RNA; the truncation of pregenome RNA by 1 nucleotide results in a 1-nucleotide-shorter RNA primer for plus-strand DNA synthesis. A comparison of the 5' ends of minus-strand DNA produced by pCMW82 in cultured cells with that of WHV made in vivo revealed no qualitative
difference.
further confirming the competenceof this expression system to direct the synthesis of WHV in transfected cells. There-fore, we conclude that pCMW82 is a suitable vector for the genetic analysis of the mechanism for plus-strand DNA synthesis.Is the 3' end of the RNA primer for plus-strand DNA synthesis determined prior to its transfer to DR2? Previous studies have shown that the 5' ends of plus-strand DNA map to position 3120, the first nucleotide 3' of DR2 (15, 27). The accurate initiation of plus-strand DNA thus depends upon the precise formation of the 3' end of the RNA primer. At least two different mechanisms can be envisaged which could lead to the formation of an exact 3' end of the RNA primer; models for these mechanisms are shown in Fig. 3A. (i) If the 3' end of the primer is determined before its transfer to DR2, cleavage could occur at
DR1
by a sequence-specific RNase H-like endonuclease activity, followed by the trans-fer of the primer to DR2. (ii) Alternatively, if the 3' end is determined after the transfer, primers with heterogenous 3' ends could anneal to DR2 and the exact 3' ends would be determined by a nuclease activity, in the simplest model an exonuclease, that trims the overhanging 3' ends to the position where the homology betweenDR1
and DR2 begins. The observation that the sequence motif dCCT(T/A), with the first dC (deoxycytidine) residue at position 21 specifying the last nucleotide ofDR1,
is present in the sequenced genomes of mammalian and avian hepadnaviruses favors the first of the two models (10, 14, 17, 25, 30, 37). According to this model, an RNase H-mediated cleavage occurs between the two rC residues at position 21 and 22, respectively (Fig.3B,
line a). To test this hypothesis, we have changed the nucleotide sequence flankingDR1
in pCMW82, shown in Fig. 3B (line a), and measured its effect on plus-strand DNA priming. The sequence motif 5'-dCCTT-3' was moved in the 5' direction by 1 nucleotide. To assure proper annealing of the primer to DR2, the penultimate nucleotide of DR2 (position 3118) had to be changed from dG to dC. The DR1and DR2 sequences of the resulting mutant p82DR1/2C are shown in Fig.
3B,
line b. If the rCCUU sequence pattern served as a recognition sequence for an endonucleolyticcleavage, the length of the RNA primer should be reduced by 1 nucleotide at its 3' end, thus moving the initiation site for plus-strand DNA synthesis in the 5' direction by1 nucleotide, from position 3120 to 3119.
HepG2 cells were transfected with
p82DR1/2C,
and virion DNA was purified from culture supernatants and subjected to primer extension analysis. To determine the exact posi-tion of the 5' end of plus-strand DNA, the RNA primer was removed by treatment with sodium hydroxide. As shown inA RNaseH
t_
+ ;4_
5
L-
//l
DRI DR2
5' 5' ---_ ,
-//
7//
5' Exonuclease
5'
i)
2
ii)
2
3
B
a) 5' ACTGGCTT // TTCACCTGTG A -pCMW82
_._+)
b)
5'-TTECiA
TT //ITTCACCTGTC5A
- p82DR1/2Cc)
5'TTCACCTGTCTT
// TTCACCTGTG _p82DR1Cd) 5' T TT // TTCACCTGTGC p82DR12
position 1 1 21 3109 3119
FIG. 3. Mutations for the determination of the mechanism for the creation of the 3'end of the RNA primer.(A) Thefigure shows two possible mechanismsfor theformation ofthe3' endof theRNA primer, dependingonwhetherthe 3' end is determinedbefore(i)or after (ii) its transfer to DR2. The protein covalently attachedto the 5'end of minus-strand DNA isindicatedwiththesolidoval,and the
5' end of the RNA primeris tagged with the symbol 5'. The small arrows in lane 1 indicate random RNase H cleavage sites on pregenome RNA. The large arrow in part i, lane 1, marks the specific RNase Hcleavagesite for thedetermination ofthe 3'end of the RNA primer. (B)Thenucleotide sequence ofDR1 andDR2are
shown, and their positions are indicated asdescribed in Materials and Methods. *,Mutationsgenerated bysite-directed
mutagenesis:
(a) the wild-type sequence ofpCMW82; (b) p82DR1/2C with base changes at positions 20,22,24, and 3118 (aT-to-Gchangeatposition
24 was introduced to preserve the amino acid composition ofthe precore gene): (c) p82DR1C, which is the same as p82DR1/2C except for position 3118, whichremainedunchanged; (d)p82DR12,which has a single base change at position 3120from dAtodC.+, Positionsofthe5' ends ofplus-strand DNA.
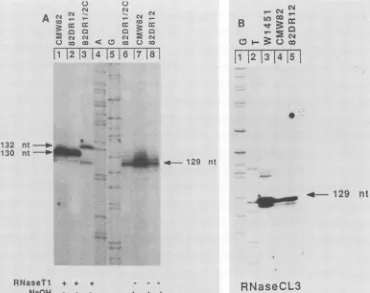
Fig. 4A (lanes 6 and 7), the length of the extended
product
from mutant p82DR1/2C is identical tothat of the
wild-type
pCMW82. The authenticity of the mutant clone was
con-firmed by treatment ofvirion DNA with RNase T1
prior
to primerextension analysis (Fig. 4A, lanes1to3).
Thechange
of the penultimate nucleotide of DRI from dG to dC
(posi-tion 20) removes an RNase T1 recognition site and
conse-quently leads to an extension product 2 nucleotides
longer
than the products obtained with wild-type DR1 sequence
motifs (Fig. 3B). In summary, the introduced base
changes
do not affect the position of the 5' end of the
plus-strand
DNA, indicatingthatthe 3'end ofthe RNA
primer
iscreatedby a mechanism independentof the exact sequence atthe3' end ofDR1.
A possible explanation of theforegoingresult is that the 3'
end of the RNA primerisformed after its transfertoDR2at
the last nucleotidewhere DR1 andDR2 are
homologous.
To test this possibility, thefollowing experiment wasdesigned.
Thehomologybetween DR1 and DR2 wasextended from11 to 12 nucleotides atDR2 by introducing asingle
nucleotidechange at position 3120, converting a dA to a dC residue
(Fig.3B,lined). Ifthe 3'end of the RNAprimerwasdefined
J. VIROL.
1
1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.327.557.72.310.2]PLUS-STRAND DNA PRIMING IN WHV 1911
A
-31 a
E co
1 a:> 0
r'
0
co
<5
13 14
15
m
v-cc 0
00
T6-C.,'
co G
E CM Q CO
TT8
1302 nt
130 nt---10..
B
't3:
0- 2
cN
a[i-
2W T
co [image:5.612.117.487.70.363.2]ji
12
1314
151
...
_M_"_ 129 nt
w___
_low -*- 129 nt
RNaseTl
+ +
+
;
RNaseCL3
NaOH
- - - + + +FIG. 4. Mappingthe 5'ends of plus-strandDNAofmutantsp82DR1/2Candp82DR12. Primer extensionanalysiswasdone asdescribed in thelegendtoFig.2B, except that virionDNA wastreated with eitherRNaseT1,sodiumhydroxide, orRNase CL3 priortotheprimer
extension.(A)Reactionproducts obtained aftertreatmentwith RNase
T,
(lanes1 to3)and sodiumhydroxide (lanes6 to8).Size markerswereprovidedby thenucleotidesequence ladder shown in lanes 4 and 5. Other lanes show theextension products from virionDNAobtained from DNAtransfections with pCMW82 (lanes1and7),withp82DR12 (lanes 2and8),and withp82DR1/2C (lanes3and 6).(B)Primerextension products aftertreatmentof virionDNAwithRNaseCL3(lanes3 to5). Lanesshowthereactionproducts from virionDNAfromwoodchuck W1451(lane3) and DNAtransfection with pCMW82 (lane4) and p82DR12 (lane 5).Thenucleotidesequenceladder in lanes1and2provided size markers.Thelengths of the extended products areindicated (nt,nucleotides).
by the relative length of the directrepeats,the 5' endof the plus-strandDNA wouldbe shifted by 1 nucleotide in the 3'
direction, initiatingatposition3121 rather thanat3120.The
analysis of virion DNA isolated from the supernatant of
HepG2cells transfected withp82DR12was performedafter removal of the RNAprimer with either RNase C13 (Fig. 4B,
lane5)orsodium hydroxide (Fig. 4A, lane 8). The data show
that inboth cases thelengths of the extended products are
identical with thoseobtainedwith virion DNA fromCMW82 (Fig.4B, lane 4, and Fig. 4A, lane 7). This result indicates that an extension ofthe homology between DR1 and DR2 has no effect on the initiation site of plus-strand DNA synthesis.
To confirm this result, we have constructed p82DR1C (Fig. 3B,linec). Thismutantis identical with thepreviously
describedp82DR1/2C, except that the sequence pattern in DR2remained unchanged. This mutant was assayed for its capacity to direct the synthesis ofplus-strand DNA. Ifthe RNAprimer was determined priorto its transferat DR1, it would notbase pair properly atDR2as a consequence ofa nucleotide mismatchat the penultimate nucleotide between DR1 and DR2. In the absence ofa nuclease activity that
couldremovethe last 2 nucleotides of theprimer,initiation ofplus-strand DNA thus would beunlikelytooccur.
Trans-fection ofHepG2 cells withp82DR1Cledtotheproduction
ofvirion DNAasobservedbydot blotanalysisandagarose gel electrophoresis. However, primer extension analysis
with purified virion DNA did not reveal any extended products characteristic of initiation ofplus-strand DNA at
DR2(datanotshown).This observation confirmsour
previ-ousresult that the 3' end of the RNAprimerisnotmodified after its transfertoDR2.
Thusfar, wehavedemonstrated that theformation ofthe 3' end of the RNA primer is not dependent either on a
distinct sequence motifatthe 3' endof DR1or ontheextent
of the homology between DR1 and DR2. In addition, our
dataindicate thatcorrectannealing oftheprimeratits 3' end isarequirementfor initiation ofplus-strandDNAsynthesis
at DR2.
Identification of an alternative site for initiation of
plus-strand DNAsynthesis. The failuretomapthe 5'endofvirion DNA from mutant p82DR1C could have resulted from the
lack ofplus-strandDNAsynthesizedorfromthe presence of
anovel 5' endlocated 3' of the normal 5' end. In the latter case,primer extension
analysis
with theoligonucleotide
CS2 usedpreviouslyin the primerextensionreaction would nothavegivenanysignal. Since dot blotanalysisof virionDNA
purified from the supernatant of cells transfected with
p82DR1C revealed the presence ofplus-strand DNA (data
not shown), we attempted to identify the novel 5' end by primer extension with a different DNA oligomer (DR17P),
derived from position 26 to 3316 (Fig. SB). A
comparison
betweenprimer-extended productsfrompCMW82
(Fig.
5A,lane1) andmutantp82DR1C (lane 2)reveals the presenceof VOL.63, 1989
on November 10, 2019 by guest
http://jvi.asm.org/
1912 SEEGER AND MARAGOS
r--- e -~
1 2r 13 4 15 1o7iS
~ ~ 10
* _ t:>'SK 5
RNaseCL3 RNaseTl NaOH
B DR2 PPT
I1
5' // GGGAGGAGGGCAGC 3
5' A 5A
--_ 5' C
I ~~~~~~-5' D
NT 227
(POS.) (31 20) (3239)
97 95
(3252)
FIG. 5. Mapping the 5' end ofplus-strand DNAof virion DNA from p82DR1C. (A)PrimerextensionwasperformedonvirionDNA
isolated from HepG2 cells transfected with pCMW82 (lane 1) and
p82DR1C (lanes 2, 6, 7,and 8)asdescribed in the legend to Fig. 4,
exceptthat DNAoligomer DR17P(position 26to3316)wasusedas aprimer.Virion DNAwastreated withRNaseCL3 (lane6), RNase
T1(lane7), orsodium hydroxide (lane 8) priorto primer extension.
Thesizes of the extendedproductswerededuced from the
sequenc-ing ladder of M13 clone MWhs shown in lanes 3 tb 5. The
sequencing reaction was primed with oligonucleotide DR17P. nt,
Nucleotides. (B) Depiction of theprimerextensionresultsobtained
asshownin panel A. The majorextensionproductsobserved before
and after removal of the RNA primer are lined up with the
nucleotide sequence of WHV (14). Lane A shows the extended productaftertreatmentofvirion DNAwith RNase
T,
oralkali,laneBshows the productafter RNase CL3treatment, and lane C shows
the product without any treatment. Lane Dindicates the length of
the extended product characteristic for initiation of plus-strand
DNA at DR2. The length of the extended products and the
corre-sponding positions of their 3' ends on the WHV genome are
indicated (NT, nucleotides; POS., position).
atleast six additional bands 108 to 114 nucleotides in length with virion DNA from p82DR1C. This could indicate the
presence of heterogenous 5' ends ofplus-strand DNA
map-pingtoposition 3233 to3239,approximately 110 nucleotides 3' of the original 5' end at DR2. The bands routinely
observed in primer extension analysis of virion DNA that has notbeen treated with ribonuclease or alkali most likely
representextension products from residual pregenome RNA
speciespresentin virion DNA preparations. Upontreatment
of virion DNA with RNase CL3, RNase T1, or sodium
hydroxide prior to primer extension (lanes 6 to 8, respec-tively), the sizes ofthe extended products werereduced to
97 and 95 nucleotides depending on the treatment. A
sum-mary of these results is shown in Fig. 5B.
On the basis of the results obtained after treatment of
virion DNAeither with RNase T1 orwithalkali,the 5' end of
the plus DNA strand starts withdC at position 3252 onthe WHV map. Since this dC residue is preceded by a G
(guanosine),treatment with RNaseT1and alkali showedthe
same band, provided that the origin of the RNA primer coincides with thepriming site. This assumption is directly
confirmed by the pattern obtained upon digestion with
RNase CL3. Thisribonuclease cleaves after the rC residues at position 3249 and hence leaves the two terminal ribonu-cleotides rArG of the RNA primer intact, resulting in a
primerextension product 2 nucleotides longer(lane6) than the reaction products obtained after treatment with RNase T1and alkali (lanes 7and8,respectively). On the basis of the most prominent band corresponding to position 3239, the RNA primer is 13 nucleotides in length. However, longer
primers up to 19 nucleotides in length could be present, as
judged fromadditional bandsobserved inlarne2. The nucle-otide sequence of the RNA primer is 5'-GGGAGGAGG GCAG-3'. This purine-rich sequence motif is reminiscentof
polypurine tracts (PPT) found at the priming sites for plus-strand DNA synthesis in retroviruses and other retroid elements (22, 29, 38). Evidence based upon in vitro experi-ments suggests that in retroviruses polypurine sequences cannot be cleaved internally by RNase H or other ribonu-cleases and thus can serve as primers for DNA synthesis (29). In summary, our results suggest that a PPT-like
se-quence at position 3239to3251 cansubstituteforadefective priming site for plus-strand DNA synthesis atDR2 and can
direct the synthesis of plus-strand DNA with a discrete 5' end at position 3252. Initiation of plus-strand DNA at this PPT reduces the length of the cohesive overlap region in virion DNA from p82DR1C from 215 to83 nucleotides.
Initiation of plus-strand DNA synthesis from polypurine tracts. We have shown that priming ofplus-strand DNA at
the PPT occurs in a mutant that is defective for priming of plus-strand DNA at DR2. To investigate whether the PPT
can serve as a priming site in the presence ofa functional DR1-DR2 sequence motif, primer extension was performed
on virion DNA isolated from a chronically infected wood-chuck, W1451. As evident from Fig. 6 (lane 7), the vast
majority of extended products are 227 nucleotides long,
positioning the 5' end of plus-strand DNAto position 3120 (27), adjacenttoDR2,asobserved with p82DR1/2C (lane6).
However,a minor fraction(about 1%)of extension products
are shorter, 97 or 95 nucleotides in length,
depending
onwhether RNase CL3 (lane 7) or RNaseT1 (lane8) was used for the removal ofthe RNA oligomeron plus-strand DNA.
Identical extension products are obtained upon RNase CL3 digestion of virion DNA from woodchuck W1451 and from virion DNA from HepG2 cells transfected with p82DR1C
(Fig. 6, lanes 5 and 7). Thus, the 5' end of the shorter
plus-strand DNA clustercoincides with thepositionof the5'
end of plus-strand DNA mapped from p82DR1/C. These
resultsdemonstrate that eveninwild-type virus,initiationof plus-strand DNA synthesis can occur atthe PPT, albeit at a
low level compared with initiation at DR2.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.68.300.70.460.2]c)N
0--N "
11 12
131415
617 181d" 227 nt
a.
. .1 i:'
x~~~~~~~~~~~~~~9.n.
95.n.
[image:7.612.99.252.69.373.2]RNaseCL3 RNaseTl
FIG. 6. Initiationofplus-strandDNAatthe PPT. Primer exten-sion analysis was performed as described in the legend to Fig. 5.
Virion DNA was purified from the supernatant of HepG2 cells transfected with p82DR1C (lane 5)orp82DR1/2C (lane 6) andfrom
virionDNAisolated from woodchuckW1451(lanes7and8).Virion
DNAwastreated with either RNase CL3(lanes5to7)orRNaseT1
(lane 8) priortotheprimer extension. Lanes 1to4,A, C, G, andT
tracks of the sequencing reaction performed withclone MWhs by using oligomer DR17P as a primer. The lengths of the extended
products are indicated. To visualize the 227-nucleotide (nt)-long
extensionproductinlane 6 theportionof the driedgel containingthe
corresponding extension products inlanes 7 and 8 was physically
separated from its original positionafter initial exposure. DISCUSSION
Efficientexpressionof WHV inHepG2cells. The develop-mentof cell culturesystemsfor theexpressionof HBV from cloned DNA createdan exciting path fora detailedgenetic analysis of the hepadnavirus genome (1, 5, 13, 21, 33, 40). Genetically altered genomes can be assayed in a transient
expression system for their ability to execute individual steps important for replication. A requirement for such a
system is that sufficient virus is secreted into the culture medium to allow for abiochemical analysis ofvirion DNA without extensivepurification steps.
The synthesis of pregenome RNA, the template for
re-verse transcription, occursforma closed circular molecule
and results ina RNA with terminally redundant ends. This terminally redundant portion contains several important
functions forreplication; amongthem are the initiation site
for minus-strand DNA synthesis, the origin of the RNA
primer for plus-strand DNA synthesis, and most likely signals important forpackagingof the viralgenome (9, 27). Therefore, we attempted to resolve this portion of prege-nome RNA on the DNA level, in order to be able to
manipulateoneelementindependentlyof the other. We have
fused WHV sequences to the CMV-IE promoter unit such that the synthesis of WHV pregenome RNA is under the control of this regulatory element. The 3.6-kb RNA of
hepadnaviruses has heterogenous 5' ends, separated by
approximately 30nucleotides, bracketing the precore AUG codon (8). Previous reports have demonstrated that the
shortest RNA species initiating 3' to the precore AUG is
packaged into viral core particles and thus serves as the
template for reverse transcription (9, 27). Furthermore,
experiments conducted with DHBV showed that a func-tional precore open readingframe is not required for virus
replication and infectivity (4, 24). Therefore, we designed
the CMV-IE-WHV vector for the expression of a WHV
transcript correspondingto pregenome RNA.
Our results demonstratethat sufficient virion DNAcanbe
obtained from as little as 3 ml of culture medium for the
biochemicalanalysisof plus-strandDNA.Comparisonof the viral DNA produced in vitro with virion DNA produced in the animal revealed identical initiation sites of plus- and minus-strand DNA (Fig.
4W;
J. Maragos and C. Seeger, unpublished results). In contrast, the RNA primerforplus-strand DNA synthesis made in vitro is shorterby 1 nucleo-tide than theprimermade in vivo. This is the consequenceof a 1-nucleotide-shorter pregenome RNA made from the
CMV-IE promoter (Fig. 2B) compared with pregenome
RNA made from the WHV promoter. This observation
provides direct genetic evidence for the origin of the RNA
primerfrom the5' endof pregenomeRNAand is in accord with previous results obtained from the biochemical and
genetic analysis of this RNA primer (15, 16, 27). In
agree-ment with this result is the observation that a mutation introduced intothe5' copyof DR1 (p82DR1C) caninterfere with the primingofplus-strandDNA at DR2. Inoculation of woodchucks with culture supernatant from
pCMW82-trans-fectedHepG2cells, aswellassomecell lines of
nonhepatic
origin,
can lead toproductive
WHV infection in theseanimals (J. Maragos, B. Baldwin, B. C. Tennant, and C.
Seeger,
unpublished results),
emphasizing
theutility
of this system for a carefulgenetic analysis
of thehepadnavirus
replication strategy in vitro and in vivo.
Determination of the 3' end of the RNA primer. The primaryobjectiveof thisstudywas toinvestigatethe mech-anism
by
which the RNA primer forplus-strand
DNAsynthesis of
hepadnaviruses
is generated; inparticular,
weaddressed thequestion ofwhetherthe primeris determined
prior to orafter its transfer to DR2. In resemblance tothe retrovirus model(38), it would seem
likely
that the 3' endof the RNA primer is created by an RNase H-likeactivity.
However, unlike the sequence of retroviruses and other retroidelements, thenucleotide sequence ofthe
hepadnavi-rus RNA primer does not include the PPT involved in retrovirus primer formation. Instead, a sequence pattern,5'-CCU(U/A)-3',
flanking the cleavage site at the 3' end of the RNAprimer
isconserved inhepadnaviruses.
We tested thehypothesis
that this sequence motifserves as arecogni-tion siteforRNaseH,
by
shifting
this patternby
1nucleotidein the 5' direction. This change did not affect the relative
length ofthe RNA
primer, implying
that cleavage occurred atthe same position but betweentwodifferentnucleotides. This result is not consistent with thehypothesis
that theconserved sequence pattern5'-CCUU-3' governs the
deter-mination oftheexact 3' endofthe RNA
primer. Therefore,
we tested whether the exact 3' end of the primer could begenerated after its transfer to DR2, perhaps
by
an exonu-clease-likeactivity.
The observation that the extension ofon November 10, 2019 by guest
http://jvi.asm.org/
1914 SEEGER AND MARAGOS J it~ the
homology
between the two repeats did not affect theposition
of the 3' end of the RNAprimer strongly
arguesagainst
theinvolvement of such anactivity
inprimer
forma-tion. This result was confirmed with mutantp82DR1C (Fig.
3B),
in which a mismatch at thepenultimate
nucleotide between theprimer
and theprimer-binding
site abolishedpriming
from DR2. We conclude that properannealing
of theprimer
at its 3' end ismandatory
for thepriming
event and that the virus lacks the necessaryenzymatic
activities torepair
suchamismatch. In this context, it isnoteworthy
that a mutationchanging
nucleotide 4 from the 3' end ofDRi(position
18) doesnotimpede priming
at DR2 (27).The data
presented
heresupportamodel where the 3' end of the RNAprimer
is determinedby
anendonucleolytic
ribonucleaseactivity prior
toitstransfer toDR2. Itappearsthat the
cleavage
site for the creation of the 3' end doesnotcoincide with the actual
recognition
site of the ribonucleaseengaged
in this process. It is conceivable that sequences 5' or 3' to thecleavage
site serve asrecognition
sites for the ribonuclease. Apossible recognition
sequence could be theDRi sequence motif itself. However,recentobservations in
our
laboratory
indicate that substantial nucleotide substitu-tions inDRidonot interferewith thecorrectcleavage
of theprimer (A.
Glaser and C.Seeger,
unpublished
results).These observations
would
argueagainst
an involvement of the DRi sequence pattern inprimer
formation. While thepresented
models consider theprimary
nucleotide sequence asthedetermining
factor forprimer
formation,analternativepossibility
is that thesecondary
structureof the RNA in thecore
particle
is thedetermining
factor. In this model, thecleavage
site would be located in anaccessibleposition
forrecognition
andcleavage by
RNase Handcleavage
would beindependent
ofaprimary
nucleotide sequence pattern.A polypurine tract can serve as an additional plus-strand DNA
priming
site.Anunexpected finding
ofourinvestigation
is thatplus-strand
DNAsynthesis
can initiate at a purine-richsequenceindependently
of the DR1-DR2priming
mech-anism. We have observedpriming
at this site in a mutant(p82DR1C)
defective for initiation ofplus-strand
DNA atDR2,
as well as in virion DNA isolated from an infected animal. In the latter DNA, about 1% ofplus-strand
DNA initiatedat the PPT. Thus thepool
of WHV DNApresentinseraof infected animals consists ofatleast two
populations,
which differatthe 5'ends of
plus-strand
DNA. SincesimilarPPTs are present on the genomes of the
closely
relatedground
squirrel hepatitis
virus andHBV, itislikely
thatthis observationcanbegeneralized
for the mammalianhepadna-viruses(25, 37). Sincethe PPT maps to thecohesive
overlap
region
ofplus-
and minus-strand DNA, virion DNA withplus-strand
DNAinitiating
at this site should still form a relaxed circle and could be converted to CCC DNA uponinfection
(Fig.
7).Thusfar,wehaveidentifiedonePPTas an initiation site for DNAreplication.
However,scanning
of thehepadnavirus
genomesfor similar sequence motifs revealedseveraladditional sites scatteredovertheviralgenomes, any
ofwhich couldserve as atarget for
plus-strand
DNApriming
(10, 17, 25, 37).
The formation of the RNA
primer
by RNase H and thepriming
ofplus-strand DNAatDR2 canoccuronly after thecompletion
of minus-strand DNAsynthesis.
This is acon-sequenceofthe
identity
ofthe RNAprimer
with the 5' endofpregenome RNA. This mechanism differs fromthe strat-egy
employed by
other retroid elements in whichtheorigin
of theRNA
primer
forplus-strand
DNAsynthesis coincides with thepriming
site andinwhichplus-strandDNAsynthe-sis commences
prior
tocompletion
of minus-strand DNA50
---I
r
5
.-POSITIN 3120 3252
40(-)
DNA A (+) DNA4W(-)
DNA(+) DNA B
t
1 4
FIG. 7. Initiation of plus-strand DNA at DR2 and at the PPT. The positions of the 5' ends ofplus-strandDNA initiating at DR2
(position 3120) and at the PPT (position 3252) are indicated. The
positionof the 5' end ofminus-strand DNAatposition14is marked with the solid oblong symbol (27). Line A depicts the condition where primingofplus-strand DNAatDR2is blocked (symbolized
by dr2), and line B shows initiation of plus-strand DNA in the presence ofafunctional DR2 atboth DR2 and the PPT.
synthesis (22, 29, 38). An important consequence of the
priming
event atthePPT inhepadnaviruses
is thatplus-
and minus-strand DNAsynthesis
couldoccursimultaneously.
Itisnotclear
why only
aminor fraction ofplus
strandsinitiatesat the PPT. Itis conceivable that the formation of theRNA
primer
is inefficient or,alternatively,
that eventhough
aprimer
is available, steric constraints prevent efficient initi-ationat this site. It will beinteresting
to determine whether theWH-V withplus
strandsinitiating exclusively
atthe PPTis infectious in the animal. Infection of woodchucks with virus
originating
from tissue culture cells transfected with the mutantp82DR1C
shouldprovide
an answer to thisquestion.
ACKNOWLEDGMENTS
We thank Bryan Cullen for the generous gift ofplasmid pBC/ CMV/1L2; Mary Ann Sells, PeterPrice, and George Acs forhelp
with themaintenance ofHepG2 cells; Bud Tennantfor woodchuck
serumsamples; WilliamMason,JohnTaylor, andVolkerVogtfor
helpfuldiscussions andencouragement; and JimCaseyandVolker
Vogtfor valuable comments tothe manuscript.
This workwassupported byPublicHealth ServicegrantAI-24972 from the National Institutes of Health.
LITERATURE CITED
1. Acs, G., M. A. Sells, R. H. Purcell, P. Price, R. Engle, M.
Shapiro, and H. Popper. 1987. Hepatitis B virus produced by
transfectedHepG2 cellscauseshepatitisinchimpanzees. Proc. Natl. Acad. Sci. USA 84:4641-4644.
2. Aden,D.P.,A.Fogel,S.Plotkin,I.Damjanov,and B.Knowles. 1979. Controlledsynthesis ofHBsAginadifferentiated human liver carcinoma-derived cell line. Nature(London)282:615-616. 3. Boshart, M., F. Weber, G.Jahn, K. Dorsch-Haesler, B. Fleck-enstein, and W. Schaffner. 1985. A very strong enhancer is
located upstream ofan immediate early gene of human cyto-megalovirus.Cell 41:521-530.
4. Chang, C., G. Enders, R. Sprengel,N. Peters, H. E. Varmus, and D. Ganem. 1987. Expression of the precore region ofan avian hepatitis B virus is not requiredfor viral replication. J. Virol. 61:3322-3325.
5. Chang,C.M.,K.S.Jeng,C. P.Hu,S.J.Lo,T.Su,L.-P.Ting, C. K.Chou,S. Han,E. Pfaff,J.Salfeld,andH.Schaller. 1987. Production ofhepatitis Bvirus in vitro bytransientexpression
of cloned HBV DNA in a hepatoma cell line. EMBO J. 6:675-680.
6. Chen, C., and H. Okayama. 1987. High-efficiency transforma-tion of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745-2752.
7. Cullen, B. R. 1987. Trans-activation of human
immunodefi-ciency virusoccursviaabimodalmechanism. Cell 46:972-982. J. VIROL.
---l
dr2 PPT
F
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.339.542.68.160.2]8. Enders, G. H., D. Ganem, and H. E. Varmus. 1985. Mapping the major transcripts of ground squirrel hepatitis virus: the pre-sumptive template for reverse transcriptase is terminally redun-dant. Cell 42:297-308.
9. Enders, G. H., D. Ganem, and H. E. Varmus. 1987. 5'-Terminal sequences influence thesegregation of ground squirrel hepatitis virus RNA into polyribosomesand viral core particles. J. Virol. 61:35-41.
10. Galibert, F., T. N. Chen, and E. Mandart. 1982. Nucleotide sequenceof a cloned woodchuck hepatitis virus genome: com-parison with the hepatitis B virus sequence. J. Virol. 41:51-65. 11. Ganem, D., and H. E. Varmus. 1987. The molecular biology of
the hepatitis B viruses. Annu. Rev. Biochem. 56:651-693. 12. Gerlich, W. H., and W. S. Robinson. 1980. Hepatitis B virus
contains protein attached to the 5' terminus of its complete DNA strand. Cell 21:801-809.
13. Junker, M., P. Galle, and H. Schaller. 1987. Expression and replication of the hepatitis B virus genome under foreign pro-moter control. Nucleic Acids Res. 15:10117-10132.
14. Kodama, K., N. Ogasawara, H. Yoshikawa, and S. Murakami. 1985. Nucleotide sequence of a cloned woodchuck hepatitis virus genome: evolutional relationship between hepadnavi-ruses. J. Virol. 56:978-986.
15. Lien, J.-M., C. E. Aldrich, and W. S. Mason. 1986. Evidence that acappedoligoribonucleotide is the primer for duck hepa-titis B virus plus-strand DNAsynthesis. J. Virol. 57:229-236. 16. Lien, J.-M., D. J. Petcu, C. E. Aldrich, andW.S. Mason. 1987.
Initiation and termination of duck hepatitis B virus DNA synthesisduringvirus maturation. J. Virol. 61:3832-3840. 17. Mandart, E., A. Kay, and F. Galibert. 1984. Nucleotide
se-quenceof a cloned duck hepatitis B virus genome: comparison with woodchuck and human hepatitis B virus sequences. J. Virol. 49:782-792.
18. Mason, W. S., M. S. Halpern, J. M. England, G. Seal, J. Egan, L. Coates, C. Aldrich, and J. Summers. 1983. Experimental transmission of duckhepatitis B virus. Virology 131:375-384. 19. Mason, W. S., J. M. Taylor, and R. Hull. 1987. Retroid virus
genomereplication. Adv. Virus Res. 32:35-96.
20. Molnar-Kimber, K. L., J. W. Summers, J. M.Taylor,andW.S. Mason. 1983. Protein covalently bound to minus-strand DNA intermediates ofduckhepatitis B virus. J. Virol. 45:165-172. 21. Pugh, J. C., K. Yaginuma, K. Koike, and J. Summers. 1988.
Duck hepatitisBvirus(DHBV)particles produced bytransient
expression of DHBV DNA in a humanhepatomacell line are
infectious in vitro. J. Virol. 62:3513-3616.
22. Resnick, R., C.A.Omer, andA.J. Faras. 1984. Involvementof retrovirus reverse transcriptase-associated RNase H in the initiation of strong-stop (+) DNA synthesis and the generation ofthe longterminal repeat. J. Virol. 51:813-821.
23. Sattler, F., and W. S. Robinson. 1979. Hepatitis B DNA has cohesive ends. J. Virol. 32:226-233.
24. Schlicht, H. J., J. Salfeld, and H. Schaller. 1987. The duck hepatitisBviruspre-C regionencodesasignalsequencewhich is essential for synthesis and secretion ofprocessed core pro-teins but not for virus formation. J. Virol. 61:3701-3709.
25. Seeger, C., D. Ganem, and H. E. Varmus. 1984. The nucleotide sequence of an infectious molecularlycloned genome of ground squirrel hepatitis virus. J. Virol. 51:367-375.
26. Seeger, C., D. Ganem, and H. E. Varmus. 1984. The cloned genome of ground squirrel hepatitis virus is infectious in the animal. Proc. Natl. Acad. Sci. USA 81:5849-5852.
27. Seeger, C., D. Ganem, and H. E. Varmus. 1986. Biochemical and geneticevidence for the hepatitis B virus replication strat-egy. Science 232:477-484.
28. Seeger, C., D. Ganem, and H. E. Varmus. 1987. In vitro recombinants of ground squirrel and woodchuck hepatitisviral DNA produce infectious virus in squirrels. J. Virol. 61:3241-3247.
29. Smith, J. K., A. Cywinski, and J. M.Taylor. 1984. Specificityof initiation of plus-strand DNA by Roussarcoma virus. J. Virol. 52:314-319.
30. Sprengel, R., C. Kuhn, H. Will, and H. Schaller. 1985. Compar-ative sequence analysis ofduck and human hepatitis B virus genomes. J. Med. Virol. 15:323-333.
31. Stenberg, R. M., D. R. Thomsen, and M. F. Stinski. 1984. Structuralanalysis of the major immediate early geneofhuman cytomegalovirus. J. Virol. 49:190-199.
32. Summers, J., andW.S.Mason.1982.Replicationof the genome of a hepatitis B-like virus by reverse transcription ofan RNA intermediate. Cell 29:403-415.
33. Sureau, C., J. Romet-Lemonne, J. I. Mullins, and M. Essex. 1987. Production ofhepatitis Bvirusbyadifferentiated human hepatomacell line after transfection withcloned circular HBV DNA. Cell 47:37-47.
34. Tagawa, M., M. Omata, and K. Okuda. 1986. Appearance of viral RNA transcripts in the early stageof duck hepatitis B virus infection. Virology 152:477-482.
35. Taylor, J. W., J. Ott, and F.Eckstein. 1985.The rapid genera-tion of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res. 13:8765-8784.
36. Tuttleman, J. S., J. C. Pugh, and J. Summers. 1986. In vitro experimental infectionofprimaryduckhepatocytecultures with duckhepatitisB virus. J. Virol. 58:17-25.
37. Valenzuela, P., M. Quiroga, J. Zaldivar, P. Gray, and W. J. Rutter. 1980. The nucleotide sequence of the hepatitis B viral genomeand theidentification ofthemajorviral genes, p. 57-70. In B. Fields. R. Jaenisch, andC. F. Fox (ed.). Animal virus genetics. AcademicPress, Inc., New York.
38. Varmus,H. E.,and R. Swanstrom. 1985. Replication of
retro-viruses, p. 75-134. In R.Weiss,N.Teich, H. E. Varmus, and J. Coffin (ed.), RNAtumor viruses. ColdSpring Harbor Labora-tory, Cold Spring Harbor,N.Y.
39. Will, H., R. Cattaneo, H. Koch, G. Darai, H. Schaller, H.
Schellekens,P. VanEerd,andS. Deinhardt. 1982. Cloned HBV
DNA causes hepatitis in chimpanzees. Nature (London) 299:
740-742.
40. Yaginuma, K., Y.Shirakata,M.Kobayashi, and K. Koike. 1987. HepatitisBvirus(HBV)particlesareproducedinacell culture systembytransientexpressionof transfected HBV DNA. Proc. Natl. Acad. Sci. USA84:2678-2682.