Copyright C) 1986, AmericanSociety for Microbiology
Coronavirus Minus-Strand
RNA
Synthesis and Effect of
Cycloheximide
on
Coronavirus
RNA
Synthesis
STANLEY G. SAWICKI* ANDDOROTHEA L. SAWICKI DepartmentofMicrobiology, Medical College of Ohio, Toledo, Ohio 43614
Received 20 June 1985/Accepted 6 September 1985
The temporal sequence of coronavirus plus-strand and minus-strand RNA synthesis was determined in
17CL1cells infected with the A59 strain ofmousehepatitis virus (MHV). MHV-induced fusionwasprevented
by keeping the pH of the medium below pH 6.8. This hadnoeffectontheMHVreplicationcycle, butgave
5-to 10-fold-greater titers of infectious virus and delayed the detachmentofcells from the monolayer which
permitted viralRNA synthesis tobe studied conveniently until atleast 10 h postinfection. Seven species of
poly(A)-containingviral RNAsweresynthesizedatearlyand latetimes afterinfection,innonequal butconstant
ratios. MHV minus-strand RNA synthesis was first detected at about 3 h after infection and was found
exclusivelyintheviralreplicative intermediatesandwasnotdetected in60Ssingle-stranded formininfected
cells.Earlyinthereplication cycle,from 45to65% ofthe[3H]uridine pulse-labeledRFcoreofpurifiedMHV
replicative intermediates was inminus-strand RNA. The rateof minus-strand synthesis peakedat 5 to6 h
postinfectionand then declinedtoabout 20% of the maximum rate. Theaddition ofcycloheximide before 3h
postinfection preventedviral RNAsynthesis,whereas theaddition ofcycloheximideafterviralRNAsynthesis
had begun resulted inthe inhibition of viral RNAsynthesis. Thesynthesisof both genome and subgenomic
mRNAsandof viral minus strands required continued protein synthesis,andminus-strand RNAsynthesiswas
three- tofourfoldmore sensitive to inhibitionby cycloheximide thanwasplus-strand synthesis.
Coronaviruses areenveloped RNAviruses thatreplicate
in the cytoplasmof infectedcells. Thebest-studied member
is mousehepatitis virus (MHV). Coronavirusassembly (9),
molecularbiology, andpathogenesis(23) have recentlybeen reviewed. The coronaviruses possess a continuous
plus-strand RNA genome of 5.4 x
106
to6.9 x106
daltons that iscappedatits5'end andcontainsapoly(A) sequenceatits3'
end (12, 13, 22, 27). Coronaviruses resemble two other
families of plus-strand RNA viruses, thepicornaviruses and the alphaviruses, in their intracellular synthesis of virus-encoded, RNA-dependent RNApolymerases (5, 8), which
arenotfound inthe virion-associated form(7).
Thecoronavirus plus-strandgenomeserves as atemplate forthesynthesis ofanegative-strand RNA(12), which then serves as a template for transcription of progeny genome RNA and, depending on whether the virus is an avian or
mammalian coronavirus, for transcription of five or six
subgenomic mRNAs (25-27, 30). The subgenomic mRNAs
formanestedsetof molecules whichare3'coterminal(6,27,
31). Several models (11, 24) for transcription of these
mRNAs havebeenproposed toexplainthefindingsthat the
UV target size is proportional to thelength of each mRNA
(10) and that there is present in each mRNA a common 5'
leadersequence (12, 26).
Wedetermined thetemporalsequence andprotein
synthe-sisrequirementsfor thesynthesis of coronavirus plus-strand
and minus-strand RNA. Braytonetal. (5)reportedthatonly
minus-strand synthesiswasdetectable in vitro withextracts
ofMHV (strain A59)-infectedcells harvested at 1 h postin-fection (p.i.),andonlyplus-strand synthesiswasdetectable
in extractsof cellsharvested at6h p.i. Theyconcluded that
coronavirus minus-strand synthesiswas temporally distinct
from that of plus-strandsynthesis. Ifconfirmed, this pattern
would be unique among the plus-strand viruses.
*Correspondingauthor.
MATERIALS ANDMETHODS
Cellsand virus.Seventeen cloneone(17CL1)mousecells
and the A59 strain of MHV (28) were kindly provided by
LawrenceS. Sturman. 17CL1 cellsweregrowninDulbecco
modified Eagle mediumcontaining 5%fetalbovineserum,5%
tryptosephosphate broth, 500 Uof penicillinperml, and 100
Uofstreptomycinperml. MHVwastwiceplaquepurifiedin
17CL1 cells and grown in 17CL1 cells. High-titer stocks of
MHV were prepared by infecting confluent monolayers of
17CL1 cells with MHVdiluted inpH 6.8 DMEM [Dulbecco
modified Eagle medium containing 740 mg of sodium bicarbonate per liter, 10 mM N-1-hydroxyethylpiperazine-N'-2-ethanesulfonic acid, 10 mM 2-(N-morpholino)ethane-sulfonic acid] containing 5% fetal bovine serum and 5%
tryptose phosphate broth. The cellswere not rinsed before
infectingthem.Afteranadsorption period of1hat37°C,the
inoculum was removed, and the cells were fed withpH6.8
DMEM supplemented with 5% fetal bovine serumand 5%
tryptose phosphate broth. One day after infection the
supernatant wasclarifiedbycentrifugationat10,000 x gfor
10 min at 4°C and stored on ice or at -80°C. Titers were
determinedby plaqueassayonmonolayers of 17CL1cells in
60-mm petri dishes overlaid with medium containing 0.1%
Gelrite(Kelco, SanDiego, Calif.).Titers of S x 108PFU/ml were routinely obtained.
Radiolabeling and isolation ofMHV RNA. 17CL1 cells in
35-,60-, or100-mmpetridisheswere infected with MHVas
described above with amultiplicityof infectionof50to 100
PFU per cell. After theadsorption period,theinfected cells
were fed with pH 6.8 DMEM supplemented with 5% fetal
bovine serum. The cells were fed with medium that
con-tained2,ugofactinomycinD(a generousgiftof MerckSharp
&Dohme, West Point, Pa.) perml 1 or 2h before
labeling
with
[3H]uridine
(34Ci/mmole; ICNRadiochemicals, Irvine, Calif.)in the continued presence ofactinomycin
D. Attheend of the labeling period, the
[3H]uridine-containing
me-diumwas removed, themonolayer waswashed with
phos-328
on November 10, 2019 by guest
http://jvi.asm.org/
phate-buffered saline, and the cells were solubilized in a
small volume ofNETbuffer (0.1 M NaCl,0.001 M EDTA,
and 0.01 M Tris hydrochloride, pH 7.2) containing 5%
sodium dodecyl sulfate. The solubilized cell extracts were
passed through a 21-gauge needle andlayeredonto15to30%
sucrose gradients and centrifuged at 20°Cin an SW27 rotor
at20,000 rpmfor 16 h, whichpelleted the 60SviralRNA,or
for12h,whichplacedthe 60SviralRNAone-third fromthe
bottom of the tube. Alternatively,the solubilized cellswere
deproteinized by extraction with 2 volumes of phenol fol-lowed by2volumes of chloroform.
Chromatographyonsepharose 2B and CF-11 cellulose. The
viralRNA wasseparatedaccordingtosize by
chromatogra-phy on Sepharose 2B columns (1.5 by 90 cm; Pharmacia,
Uppsala, Sweden) as previously described (19). Before
chromatography the extracted RNAwastreatedwith 10 ,ug
of DNase (RNase free; Bethesda Research Laboratories,
Inc., Gaithersburg, Md.) per ml in 50 mM sodium acetate
(pH 6.5)-10 mM
MgCl2-2
mM CaC12 for 30 min at 37°C, followed by the addition of EDTA to 50 mM and sodiumdodecyl sulfate toafinalconcentration of 1%. Viral replica-tive intermediate(RI) andreplicative form (RF) elutedin the
void volume and was collected by ethanol precipitation in
the presence ofunlabeled carrier tRNA.
Chromatography of viralRIs and RFsonCF-11 cellulose
columns (Whatman, Inc., Clifton, N.J.) has been described before (21). Briefly, columns of CF-11 cellulose in 3-ml
plastic syringes (2.5 mlofCF-11 cellulose) wereextensively
washed with STE buffer (0.1 M NaCl, 0.05 M Tris
hydro-chloride [pH 6.8], 10 mM EDTA) containing 1%
beta-mercaptoethanol followed by35%ethanol-STE buffer. After
application ofthe RNAsample, whichhadbeen adjustedto
35% ethanol in STE buffer, the columns were washed
sequentially with 35% ethanol-STE (15 ml) and with 15%
ethanol-STE(15ml)before elution oftheRF RNAwithSTE
bufferalone(9ml).The RF RNAwasethanolprecipitatedin
the presenceof25 ,ugof carriertRNA. Viral RIswhichhad
been obtainedby chromatography ofcellextracts on
Seph-arose2B,weretreated with 0.02or0.4 ,ugofRNase A per ml at25°C for15 min in 0.15 MNaCI-0.01MTris
hydrochloride
(pH 7.4) before being chromatographed onCF-11 cellulose.Determination of theamountofalkali-resistantradioactivity
in RNApreparations employed incubation ofthesamplesat
37°C for 16 hin 0.3 M KOH and collection onfilters of the
acid-insoluble radioactivity.
Determination ofMHV minus-strand RNA synthesis. The
procedures used for measuring MHV minus-strand RNA
were similar to thosedescribed for detection ofalphavirus
minus-strand synthesis (20, 21). Purified, unlabeled, virion
plus-strandRNAwasdissolvedafter ethanolprecipitationin 1 mM EDTA at a concentration of 1 mg/ml (25 units of
absorbancy at 260nmpermg) and storedat -20°Cin small
samples. Viral RF RNA was obtained either as described
aboveorfrom infected cell RNA which had been extracted
with phenol and chloroform, treated with RNase A, and
chromatographedtwiceonCF-11 cellulose.Hybridization of
theRFRNA,whichwasdenatured byheatingat100°C for 3
to5minfollowed by quick coolingat0°C,was at68 to 70°C
for 30min,followedbyanincubation of 30 minat25°C. The
annealing reaction employed 10 ,ug of unlabeled purified virion plus-strand RNA and was performed in a final volume of1ml of0.4 M
NaCl-1
mMEDTA.RESULTS
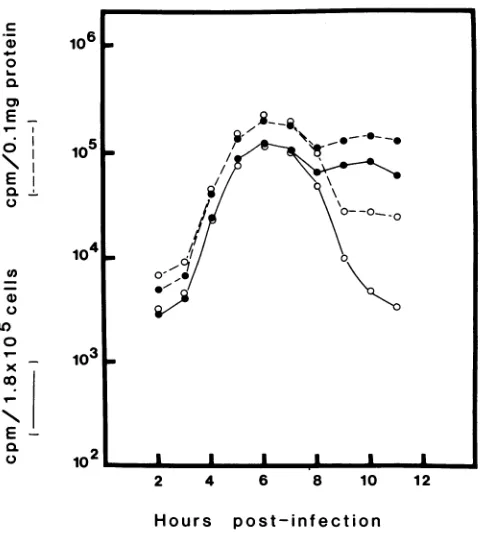
The time course of viral RNA synthesis of 17CL1 cells
infected with 100 PFU of the A59 strain of MHV per cell is
shownin Fig. 1. Before 3 hp.i.,viral RNA synthesiscould
not be detected above the level of incorporation of
[3H]uridine
observed in mock-infected 17CL1 cells.Begin-ning at about 3 h
p.i.,
the rate of viral RNAsynthesis
increased until about 5 h p.i. Decreasingthe multiplicity of
infection to S PFU/cell
delayed by
about 1 h the time atwhich viral RNA
synthesis
wasfirstobserved. Atabout 6 hp.i.,cellfusionbegantooccur,and
by
8to9 hp.i.
thegiant,
multinuclear cells weresloughed
from themonolayer.
Wefound that we could prevent MHV-induced cell fusion
by
maintainingthepH ofthe culture medium below
neutrality.
This wasaccomplished by using
one-fifth of theregular
concentration of sodium bicarbonate in Dulbecco modifiedminimum essential medium and
keeping
theCO2
level at10%.The datapresentedin
Fig.
1demonstrate thatlowering
thepHdidnotaffect viral RNA
synthesis
in17CL1cells,
butdid extend thetime
during
whichviral RNAsynthesis
couldbe
conveniently assayed.
Underconditions of loweredpH,
the rate of viral RNA
synthesis during
the lateperiod
remained approximately constant
(Fig.
1). Therate of viralRNA
synthesis
in infected cells maintained inpH
7.2me-diumwasthesame asin
pH
6.8 mediumuntil about8 hp.i.,
when therate of viral RNA
synthesis
declined(Fig.
1). Theproduction
of infectious virus did notappear to be affectedby
preventing
MHV-induced cell fusionandoccurredatthesame time in
pH
6.8 medium as it did inpH
7.2 medium.However, infected cells maintained at
pH
6.8 continued toc
0)
L.
0)
E.
-6'1
E i
0
(a
0
LO
0
x
00
",N
0
106
105
102
2 4 6 8 10 12
Hours post-infection
FIG. 1. Timecourseof MHV RNA synthesisat pH 7.2or6.8. Cultures of 17CL1 cells in 35-mm petri disheswere infected with MHV (multiplicity of infection of100) and maintained at 37°C in mediumatpH7.2(0)oratpH 6.8 (0)asdescribedin Materials and
Methods. The cultures were labeled with [3H]uridine for 60-min periods between 1 and 11 hp.i., and the cellswere solubilized in NET buffer with sodiumdodecyl sulfate. The acid-insoluble radio-activity in 1/10 of the sample was determined ( ). The protein
content in 1/10 of each sample was determinedby the method of Lowryetal.(15),and theacid insolubleradioactivity expressedper 0.1mgofprotein (---).
I I I I I I
m
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.316.556.364.631.2]releaseMHV for alongertime andproducedhigher titers of
MHV than did cells maintained at pH 7.2. Thus, MHV
resembles avian infectious bronchitis (1) and transmissible
gastroenteritis (18) viruses in their ability to grow to higher
titers at low pH. The results which follow were obtained
with low-pH conditions.
Wedetermined the relative proportionofsynthesis ofthe
viral genome RNA and the subgenomic mRNA species during differentperiods of the MHVreplication cycle.
Sim-ilar to the results published by Leibowitz et al. (14), we
found sevenspecies of MHVRNAbyacid-urea-agarose gel
or by
formaldehyde-agarose
electrophoresis of oligo (dT) cellulose-selectable RNA ortotal RNAfrom MHV-infected cells that hadbeen pulse-labeledwith32p,
orwith[3HJuridine(data not shown). No incorporation of[3H]uridine into60S
genomic or subgenomic RNA was detected before 2to 3 h
p.i. In ourlaboratory the A59 strain ofMHV synthesized approximately as much 60S RNA on a weight basis as
subgenomic RNA.
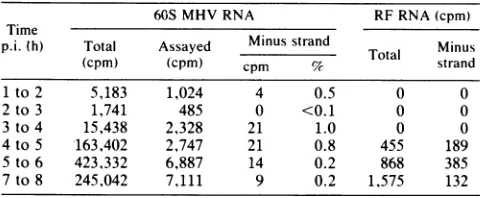
We next determined whenminus-strandRNAwas
synthe-sized in the MHV
replication
cycle. Becauseofthereportbytrayton
et al. (5), we were especially careful to look for minus-strand RNA synthesis early in the infection.MHV-infected cells were pulse-labeled with
[3H]uridine
for 1-hperiods between 1 and 8 h p.i. The total cell extract was
analyzed by rate zonal centrifugation onsucrose gradients.
The radiolabeled RNA sedimenting at60S wascollected by ethanol precipitation, and the 60S RNA was allowed to
anneal to an excess ofunlabeled virion plus strands. Less
than 1% of the radiolabeled RNA sedimenting at 60S was
protected from
RNa$e
A digestion (Table 1). However,when purified viral RF RNA obtained from MHV-infected
cellslabeled with
[3H]uridine
from2.5 to 6hp.i. (seebelow)wasdenaturedandannealedunderidenticalconditions, 45% of the radiolabel was protected from RNase A digestion (data not shown). We conclude that at no time in the
infectious cycle did
MHV-infected
cells contain significantamounts of genome-length, single-stranded, minus-strand
RNA. Therefore, purification and analysis oftheviral
dou-ble-strahded RIswasundertaken to
determine
thekinetics of viralminus-strand synthesis.We found that cells infected with our stocks of MHV
synthesized large amounts ofgenome-sized 60S RNA, and thatthisRNA
coeluted
with theviralRIsin thevoidvolumefractions of Sepharose2B,Sepharose2B-CL,orUltrogelA2
TABLE 1. Absence ofsingle-stranded 60S minus-strand RNA"
60SMHV RNA RFRNA(cpm)
Time
p.i.(h) Total Assayed Minus strand Minus
Total
Mirnus
(cpm) (cpm) cpm % strand
1 to 2 5,183 1,024 4 0.5 0 0
2to3 1,741 485 0 <0.1 0 0
3 to4 15,438 2,328 21 1.0 0 0
4to5 163,402 2,747 21 0.8 455 189
5to 6 423,332 6,887 14 0.2 868 385
7 to 8 245,042 7,111 9 0.2 1,575 132
Cultures of 17CL1 cells in 60-mm petri dishes were infected at a
multiplicityofinfection of100with MHV strain A59 and were treated with
actinomycin D(2 tg/ml)beginningat 1h p.i. Pulses of1hweregive.awith [3Hjuridine (200 ,uCi/ml) as described in Materials and Methods. The 60S
RNA was recoveredafterratezonalcentrifugationonsucrosegradients;the
RF RNA was obtained after RNase treatment of 40 to 10S RNA and
chromatography on CF-11 cellulose. Hybridization was as described in
Materials and Methods.
Fx:
25
10 15
20
25
30
4
94S
-,9S
F1 "'
-_- 2
--*3
26S
*_
~5
-_-~6
_bZ-~7
FIG. 2. FormaldehydeagarosegelelectrophoresisofMHVRNA
species. MHV-infected cellswerelabeledwith 1mCiof32pperml
inphosphate-freemedium for 1 hat6 hp.i.inthe presence of 10,ug
ofactinomycinD perml. The cellsweresolubilizedinNET buffer
with5%osodiumdodecylsulfateandlayeredonto15to30%osucrose
gradientsin NETbuffercontaining0.2%o sodiumdodecylsulfateand
centfifuged inan' SW27rotor at 20,000 rpmfor 16 h at20°C. The
gradient was fractionated into 1-ml fractions. Small samples of
fractions 2, 5, 10, 15, 20, 25, and 30were diluted and made 50% formamideand 2.2 Mformaldehyde,heatedto55°C for15min,and
subjectedtoelectrophoresison l1o agarosegelsin 2.2 M
formalde-hyde in 20 mM morpholinopropanesulfo.-ic acid-5 mM sodium acetate-2 mM EDTA(pH7.3).Thefirstlane showssizemarkersof SemlikiForestvirus 49Sand26S RNA.
columns
(data
not shown).Therefore,,
to separate the 60Ssingle-strand
RNA from thepartially
double-stranded viralRIs and to obtain an enrichment of the viral Rls, we first
fractionated the intracellular viral RNA
species by
sucrosegradient
centrifugation
andpooled
the.region
of thegradient
containing
the Rls.Figure
2showsaformaldehyde-agarose
gel
of various fractions ofasucrosegradient
of32P_labeled,
MHV-infected cell extracts. Fraction 37 was the top, and
fraction 1 wasthebottom,of the
gradient.
TheviralRls andRFs were identifiedas RNase A-resistant
radioactivity
andwerefoundtosediment between 40 and 10S
(fractions
15to30 in
Fig.
2).Wethenroutinely
usedconditions ofsedimen-tation that resulted in the
pelleting
of the 60S RNA aild thatclearly separated
the RIregion
from the 60S RNA. Wheninfected cultureswere
pulsed
for S min with [3H]uridine
at6h
p.i.,
38%o of the labeled RNA that sedimented between40and 10Swasfoundtoelute in thevoidvolum'e of
Sepharose
2Bcolumns(datanotshown);52% of this labeled RNAwas
resistantto
digestion
with RNase A,but it wascompletely
digestable
with alkali. Whenrechromatographed.,
all of thelabeled RNA was
again
recovered in the void volume.Trhe
5-min
pulse-labeled
viralRls sedimentedheterogeneously
onsucrose
gradients,
with apeak
at about 26S, asreported
previously by
Baric et al. (2). Their behaviorduring
chro-matography
onCF-11 cellulosewasalsoconsistent with thatexpected
ofanRImolecule:40% of the labeled RNA elutedin the15% ethanol-STE
fraction,
and60%oeluted in the STEfraction.However, pretreatmentwith lowlevels of RNase A
(0.4
jig/ml)
resulted in all of the labeled RNAeluting
in theon November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.63.303.562.661.2]106
E
0. 0
z
:2
I
C')
105
104
MHV, A59
strain6 totalRNA
'\
RF RNA 0minus-strand RNA VU
\A~
I 0
0
1n, -
-I \
0 '
I I 4i I I I
1 2 3 4 5 6 7 8
HOURS, post-infection
FIG. 3. Kinetics of MHV minus-strand and plus-strand RNA synthesis. Cultures of 17CL1 cells in 100-mm petri dishes were
infected with MHV(multiplicity of infection of 100). The cultures were treated with actinomycin D (2 ,ug/ml) for 1 h before pulse-labeling with[3H]uridinefor30minstartingat3.5 hp.i. The viralRI RNA was obtained by sedimentation onlinear 15 to 30% sucrose gradients and digested withRNase A. The RF RNA wasobtainedby chromatographyonCF-11cellulose, heat denatured,and annealed to an excessof unlabeledvirionRNA asdescribed in Materials and Methods. The[3H]uridine-labeledRNAthatwasresistantto diges-tion withRNase Aafterhybridizationwasconsideredtobe minus-strandRNA.
STEfraction,consistentwiththe formationofacompletely double-stranded RF molecule. Under these conditions,
DNAelutes in the 15% ethanol-STE fraction. Bycombining
DNase treatmentbefore Sepharose 2Bchromatographyand
theuse ofCF-11 chromatography tofurtherpurifythe viral
double-stranded RNAfromDNAandsingle-strandedRNA,
we wereable to isolate labeled minus strands as part of the
viral RIs or RFs. MHV-infected cells were labeled with
[3H]uridine
from2.5to6hp.i.,and the 40to 10S RNAwascollected after sedimentation of infected cell extracts on
sucrose gradients and subjected to chromatography on CF-11 cellulose. The labeled RFs represented about 1% of
thelabeled RNA in theoriginalinfected extracts. Samples of
the purified RF RNA corresponding in amount to that
present inabout 3 x
106
cells were analyzed in hybridizationassays with increasing amounts (1 to 50 ,uglml) of purified
virionplus strands. Maximum amounts of hybridization (45 to 65% of the labeled RF RNA) were found when 5 ,ug or
more of plus-strand RNA per ml was used in each assay.
Since the labeling period encompassed essentiallytheentire
period of minus-strand synthesis, approximately 50% of the total labeled RF RNA would be expected to be in minus-strand RNA. Having successfully isolated MHV RIs and
RFs and detected viral minus-strand RNA in the amounts
expected, we nextdetermined thekinetics of MHV
minus-strand synthesis.
Figure 3 presents the results of our investigation on the
kinetics ofMHVminus-strandandplus-strandRNA
synthe-sis. Infected cultures were pulsed with [3H]uridine for
30-minperiods between3 and 8 hp.i.Theviral RF RNA(1.4
to 3% of the total labeled viral RNA) was isolated by
chromatography onCF-11 cellulose. Beginning atabout 3 h
p.i., the rate of incorporation of radiolabel into RF RNA
begantoincrease,and itparalleledthe increase in therateof
overall viral RNA synthesis. Minus-strand RNA synthesis
wasdetectable at 3hp.i. and increased in proportion to the
labeling of RF RNA. The overall rates of viral RNA
synthe-sis (the vast majority of which was plus-strand RNA), of
accumulation of RF RNA, and of minus-strand synthesis
reached a maximum at about 6 h p.i. The rate of
minus-strand synthesis declined after6 h p.i. to about 20% ofthe
maximumrate.
We next asked whether coronavirus RNA synthesis
re-quired continued protein synthesis. The effect of adding cycloheximideatdifferent times after infectionontherateof synthesis ofMHV RNA is shown in Fig. 4. Inhibition of
protein synthesis before 3 h p.i. prevented MHV RNA
synthesis, whereas inhibition ofprotein synthesis after 3 h
p.i.prevented therateofMHV RNAsynthesis from
increas-5 105
E
0.
C)
z
cc0
:2
1._
I Co,
10
OCH
6h'CH
5hOCH
4hCH 3h
1 3 5 7 9
HOURS, post-infection
FIG. 4. Effectofcycloheximide (CH)onMHV RNAsynthesis. 17CL1 cells in 35-mmpetri dishes (2.5 x 101cells perdish) were infected with MHV(multiplicity ofinfection of100). The cultures werepulse labeled with [3H]uridine(100 ,uCi/ml)for 60 min(0)in thepresence ofactinomycin D(20,ug/ml). At3, 4, 5, and 6 h p.i. (arrows), cycloheximide at 100 ,ug/ml was added to sets of three cultures. Eachsetofinfected cultures waslabeledfor60min with
[3H]uridineinthe presence ofactinomycinDandcycloheximide at
thetimes indicated (-).
-m
on November 10, 2019 by guest
http://jvi.asm.org/
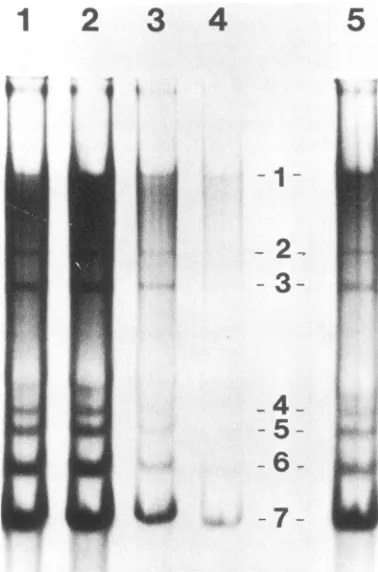
[image:4.612.60.299.73.450.2] [image:4.612.329.545.429.646.2]ing. To determine whether the synthesis of the MHV
genomic 60S RNA wasdifferentially sensitivetoinhibitionof
protein synthesis compared with the viral subgenomic
mRNA, infected cultures were treated with cycloheximide
beginningat5 hp.i. and pulsed with [3H]uridinefor60-min
periods inthe presence ofcycloheximide between5and 7 h
p.i. Extracts of theinfected cells were analyzed by
formal-dehyde-agarose gel electrophoresis (Fig. 5). Both the 60S
RNA and the subgenomic mRNAspecieswere produced in
untreated cultures. With increased time of cycloheximide
treatment, diminished incorporation of radiolabel into all
viral plus-strand RNA species was found. There was an
approximately 50% reduction in the overall rate of RNA
synthesisfor every 30 minofcycloheximidetreatment. This
reductionwas notdue tothelossof cells from the monolay-ers,sinceequal amountsofrRNA were presentin extractsof
cellstreated or nottreated with cycloheximide. The
synthe-sis of all species of MHV mRNA was found to decrease
proportionally with increasing time of cycloheximide treat-ment (Fig. 6). Thus, all species of MHV plus strands
appeared to be equallysensitive to inhibition by the protein
synthesis inhibitor.
Thecycloheximide-treated cultureswerealso analyzed for
1
2
3
4
tl
i
i
I-1--
2--
3-5
I
i
.i
-
4
4. j=, .~~~~~~~~~~~~~~~~~~~~~~~~i
-
6
- 4-
7
-FIG. 5. Cycloheximide sensitivity of MHV plus-strand RNA synthesis. MHV-infected cells weretreated with 100 ,ugof cyclo-heximidepermlat5hp.i.and labeled with[3H]uridine (100,uCi/ml) For 1 h at 5 (lanes 1 and 3) and 6(lanes 2, 4 and 5) h p.i. in the
presence(lanes 3, 4,and5)orabsence (lanes1and2)of cyclohex-imide. The cells were solubilized in NET buffer containing 5%
sodium dodecylsulfate and extracted twice with phenolandtwice with chloroform. The RNA was ethanol precipitated, denatured with 50% formamide-2.2 Mformaldehyde at55°C for 15min, and electrophoresedon1%agarosegelscontaining2.2 Mformaldehyde. Lane 5 isthe same aslane 4,except theexposure wasfour times longer.
501
0
x
E
0
z
._
0
._
I
Ch
201
10o
5
2
5 6 7 5 6 7
Hours, post-infection
FIG. 6. Comparison of the cycloheximide sensitivity of MHV minus-strand and plus-strand RNA synthesis. Cultures of 17CL1 cellswereinfected with MHVas described in Materials and Meth-ods. Cycloheximide (100 p.g/ml)wasaddedtothree cultures begin-ningat 5 h p.i. The cultures were labeled with ['H]uridine (200 ,uCi/ml) for 30 min between 5 and 6.5 h p.i. The viral RNA was
resolvedon15to30%sucrosegradients.A, Incorporation obtained in the60S (A, A) and the 16S(0, *) regions of the gradients in untreated( ) andcycloheximide-treated (---) cultures. B, Incor-poration of [3H]uridine into minus strands was determined as
described in thelegendtoFig. 3 and in Materials and Methods for untreated(0) andcycloheximide-treated (0) cultures.
their ability to continue minus-strand synthesis in the
ab-senceof continued protein synthesis.Cultureswereinfected
with MHV andwere incubated at37°C until 5 h p.i., when
cycloheximidewasaddedtoone-halfof the cultures.Pulses
of[3H]uridineweregivenfor30-min periodsbetween 4.5and
6 hp.i.tothe untreated cultures and between 5 and6.5 hp.i.
to the cycloheximide-treated cultures (Fig. 6). The rate of
synthesis ofMHV60Sand 16S (RNA 4, 5, 6, 7) plus-strand
RNA declined at a linear rate with increasing time of
cycloheximidetreatment,but continuedtobesynthesizedat
a relatively constant rate in the absence of the protein
synthesisinhibitor(Fig. 6A). Moreover(Fig. 6B),
coronavi-rus minus-strand synthesis was inhibited by cycloheximide treatment, and its synthesis appeared to be at least three times more sensitive than was plus-strand RNA synthesis. We conclude that both coronavirus plus- and minus-strand
synthesis required continued proteinsynthesis.
DISCUSSION
The results of our studies on MHV indicated that the
synthesis of both plus- and minus-strand RNA was first
detectedbeginningatabout 3 hp.i.at37°C.Thesynthesisof
bothplus-and minus-strand RNAwasobserved toincrease
between 3 and 5 hp.i. The increase in the number ofminus strands ledtotheaccumulationof increased numbers of viral RIswhichweremostly engagedinplus-strandRNA synthe-sis. At all times investigated, the synthesis of 60S genome
RNA was proportional in total incorporation to thatofthe
most prominent subgenomic mRNA, the 16S mRNA or
A B
+CH +CH
-¢ k sa5 ' V
04
-&\ 0
E\9
\\
a
clE
\\
C.) 100A^
<s
\
z I
cc 5.
c)
\C
X 0
D
\
I 2 .
coa
I * . I U
*or
*6
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.321.558.73.311.2] [image:5.612.87.276.328.614.2]mRNA 7.Sincethereisanapproximate 10-folddifferencein
themolecular weight of the 60S genome RNA and the 16S
mRNA(14), cells infected with our strain ofMHV
synthe-sized10timesmore16SmRNA thangenome RNA and thus
synthesized 3 to 4 times more genome RNA than that
reported by Leibowitz et al. (14). The pattern of overall
MHV RNA synthesis was identical to that reported by
others, who found that the synthesis of viralmRNAspecies first became detectable at 4 h p.i. with agarosegel electro-phoresis(14). Our resultsalsoagreewith thoseof Stern and Kennedy (27), Wege et al. (30), Spaan et al. (25), and
Leibowitzetal. (14) in findinga nonequimolar synthesis of
the seven viral mRNA species at all times during the
replication cycle.
ViralRIscontainedessentiallyallofthe viralminus-strand
RNA. No single-stranded 60S minus-strand RNA was
de-tectedin infected cell extracts atimmediate early, early, or
lateperiods in the viral replication cycle. Our failuretofind single-stranded 60S minus-strand RNA early in infection is
notinagreementwiththesuggestion ofBrayton et al. (5) that
there is an early time when only minus-strand RNA is
synthesized. In the absence of viral plus strands, there
shouldbefree, single-stranded minus-strandRNAmolecules iftheyweresynthesized inamountssufficienttobe detected
and ifthey were 60S in size. If, on the other hand, MHV
minus-strand polymerase wasformed but notactivein vivo atimmediate early times, its activity mightbe detectable in
vitro. Our studies on the temporal appearance of viral
plus-strandandminus-strandRNA areinagreementwiththe
results ofDennis and Brian (7, 8) on the temporal
appear-ance of viral RNA synthesis in porcine transmissible gastroenteritis virus-infected cells.
The synthesis of MHV minus-strand RNA was readily detectableby4hp.i.,ataboutthe sametimethatplus-strand synthesis was detectable. The proportion of
[3H]uridine
incorporated into minus strands relative to plus strands inthe RF RNAremainedat40 to50% duringtheearly period
of the viral replication cycle, when the rate ofplus-strand
RNAsynthesiswas increasing exponentially (four-to
eight-fold every 30 min). Because the rate of minus-strand RNA
synthesiswas also increasing exponentially, pulse labels of
30 min or longer would result in the majority of minus
strands becoming labeled during the pulse period. Hence,
eventhough minus strands, unlikeplus strands,remained in
the RIs and were not released as single-stranded species
after their synthesis, the relative numbers of minus strands that were synthesized during the pulse period must have
exceeded the number ofminus-strand templates that were
synthesized
before thepulse period.
Thesynthesis
ofminusstrandswasfollowedby accumulation of increased incorpo-ration of radiolabel intoreplicative intermediates. However,
the synthesis of minus strands declined after 6 h p.i., the
sametime that the overallrate ofplus-strandsynthesisand
of
incorporation
of[3H]uridine
into viral RIs became con-stant. This pattern is similar to that ofalphaviruses (20).Becauseplus-strand synthesis continued afterthe synthesis
of minus-strand RNA had significantly decreased and
be-cause the incorporation of
[3H]uridine
into viral RIsre-mained relativelyconstant, we conclude that the viral minus
strands are stable and functionastemplates for a long time
after their synthesis. Although there was a significant
de-crease in the rate of viral minus-strand synthesis in
MHV-infected cells, it did not decrease to the very low levels
observed during the late period in alphavirus-infected cells
(20).
The synthesisof both plus-strand and minus-strand RNA
inMHV-infected cells required continued proteinsynthesis.
This was clearly different from the requirementsfor newly
synthesized proteins for continued synthesis of minus
strands but not ofplus strands, during alphavirus replication
(20,21).The decrease of MHV minus-strandsynthesisafter
theaddition of cycloheximidewasthree tofourtimesgreater than the decrease of plus-strand synthesis. Also, the
de-creaseinthe rate ofplus-strand synthesisafter the addition
ofcycloheximideoccurredonlyafter 20 to30min, whereas
minus-strandsynthesis was moreimmediately affected.
Mi-nus strands were minor species in MHV-infected cells,
accounting for 2% or less of the total incorporation of
radiolabel intoviral RNA.Thus,because there may beonly
a few RIs active in minus-strand synthesis, a decrease in
their functionmightbe morereadilyapparent.Alternatively,
there may be a greater sensitivity of the minus-strand
polymerase activity than of the plus-strand polymerase activitytoproteinsynthesis inhibition.Thesynthesis ofboth
genome60SRNAandthesubgenomicmRNAsappearedto
beequally sensitivetoinhibition of
protein synthesis.
Thus,coronavirusplus-strand synthesisappears to utilizea
mech-anism that also differs from that used by the negative-stranded RNA rhabdoviruses. The replication of
genome-sizedplus-andminus-strandRNAby the rhabdovirus
vesic-ular stomatitis virus required continued synthesis of viral
nucleocapsid protein (3, 4, 16, 32), butthe transcription of
viralmRNAbythesecells did notrequire continued protein synthesis (17, 33).
Thesensitivity of coronavirusRNAsynthesisto cyclohex-imidemorecloselyresembled that seen forpoliovirus RNA
synthesis.Ithas beensuggestedthat thefailuretosynthesize
the polyprotein precursor to the VPg leads to the rapid inhibition ofpoliovirus RNA synthesis (29). Therefore, the
poliovirus replicasemayfunction foroneroundofsynthesis
andrequire newly synthesized viral polypeptidestoinitiate
each round of viral RNAsynthesis. Coronavirus
transcrip-tion mayinvolveaviralpolymerasethatis eitherturned over
or short lived or may
require
a newlysynthesized
viralproteintoinitiate viral RNA synthesis. Thepresenceofthe
5' cap structure on coronavirus plus-strand RNA species
(12) argues against afunction fora VPg-like polypeptidein
coronavirus plus-strand RNA synthesis. Since the
mecha-nisms hypothesized for coronavirus mRNA synthesis
in-volvethesynthesis ofaleadersequence anditsmovement to aregion adjacenttothestartofthe
respective
mRNAcoding
sequence in thetemplate RNA(2), continued
synthesis
ofaprotein thatfunctions in
specific
initiation or translocationmay be required for coronavirus transcription.
ACKNOWLEDGMENTS
Weespecially thankLawrenceS. Sturman for hisencouragement and generous provision of virus and cells and K. V. Holmes for advice. DeboraBrunerprovided excellent technical assistance, and
CherylZimmermantyped the manuscript.
This investigation was supported by the United States-Japan Medical Sciences Program through Public Health Service grant
AI-15123 from the National Institutes of Health and by Public
Health Service Research CareerDevelopment AwardAI-00510(to D.L.S.)from the NationalInstitutes ofHealth.
LITERATURECITED
1. Alexander, D.J., andM.S. Collins. 1975. Effect ofpHonthe growth and cytopathogenicity of avian infectious bronchitis virus inchickkidneycells. Arch. Virol.49:339-348.
2. Baric,R.S.,S. A. Stohlman,and M. M.C. Lai. 1983. Charac-terization ofreplicative intermediate RNAofmurine hepatitis virus: presence of leader RNA sequences on nascent chains. J.
on November 10, 2019 by guest
http://jvi.asm.org/
Virol. 48:633-640.
3. Blumberg, B. M.,C. Giorgi, and D.Kolakofsky.1983. NProtein of vesicular stomatitis virus selectively encapsidates leader RNAin vitro. Cell 32:559-567.
4. Blumberg, B.M., M.Leppert, and D. Kolakofsky. 1981. Inter-action of leader RNA and nucleocapsid protein may control VSVgenome replication. Cell 23:837-845.
5. Brayton, P.R.,M.M. C. Lai, C. D. Patton, andS.A.Stohlman. 1982. Characterization of two RNA polymerase activities in-duced bymousehepatitis virus. J. Virol. 42:847-853.
6. Cheley, S., R. Anderson,M. J. Cupples, E.C.M. Leechan, and V. L. Morris. 1981. Intracellularmurine hepatitis virus-specific RNAscontaincommon sequences. Virology 112:596-604.
7. Dennis, D. E., and D. A. Brian. 1981. Coronavirus cell-associatedRNA-dependent RNA polymerase. Adv. Exp. Biol. Med. 142:155-170.
8. Dennis, D. E., and D. A. Brian. 1982. RNA-dependent RNA polymerase activity in coronavirus-infected cells. J. Virol. 42:153-164.
9. Dubois-Dalcq, M., K. V. Holmes, and B. Rentier. 1984. Assem-bly of coronaviridae, p. 100-119. In Assembly of enveloped viruses. Springer-Verlag, New York.
10. Jacobs, L., W. J. M.Spaan, M. C. Horzinek, and B. A. M.van
der Zeijst. 1981. Synthesis of subgenomic mRNAs of mouse
hepatitis virus is initiated independently: evidence from UV transcription mapping. J. Virol. 39:401-406.
11. Lai, M. M. C., C. D.Patton, R. S. Baric, and S.A. Stohlman. 1983. Presence of leader sequences in the mRNA of mouse
hepatitis virus. J. Virol. 46:1027--1033.
12. Lai, M. M. C., D. C. Patton, and S. A. Stohlman. 1982. Replication of mouse hepatitis virus: negative-stranded RNA
andreplicative formareofgenomelength. J. Virol. 44:487-492.
13. Lai, M. M. C., and S. A. Stohlman. 1978. RNA of mouse
hepatitis virus. J. Virol. 26:236-242.
14. Leibowitz, J. L., and K. C. Wilhemsen, and C. W. Bond. 1981. The virus specific intracellular species oftwo murine
corona-viruses: MHV-A59 and MHV-JHM. Virology 114:39-51. 15. Lowry,0.H., N.J. Rosebrough, A.L.Farr, and R. J. Randall.
1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275.
16. Peluso,R.W., andS.A.Moyer.1984. Vesicularstomatitis virus proteins required for the in vitro replication of defective inter-fering particlegenomeRNA,p.153-160. In D. H. L.Bishopand R. W. Compans (ed), Nonsegmented negative strand viruses. AcademicPress, Inc., New York.
17. Perlman, S. M., and A. S. Huang. 1973. RNA synthesis of vesicular stomatitis virus. V. Interactions between transcription andreplication. J. Virol. 12:1395-1400.
18. Pocock, D. H., and D. J. Grawes. 1975. The influence ofpHon
thegrowth and stability of transmissiblegastroenteritis virus in vitro. Arch. Virol. 49:239-247.
19. Sawicki, D. L., and P. J.Gomatos. 1976. Replication of Semliki Forestvirus: polyadenylateinplus-strand RNA and polyuridyl-atein minus-strand RNA. J. Virol. 20:446-464.
20. Sawicki, D. L., and S. G. Sawicki. 1980. Short-lived minus-strand polymerase for Semliki Forest virus. J. Virol. 34:108-118.
21. Sawicki, S. G., D. L. Sawicki, L. Kaariainen, and S. Keranen. 1981. A Sindbis virus mutant temperature sensitive in the regulation of minus-strand RNA synthesis. Virology 115: 161-172.
22. Siddell, S., H. Wege, and V. terMeulen.1982. Thestructure and replication of coronaviruses. Curr. Top. Microbiol. Immunol. 99:131-163.
23. Siddell, S., H. Wege, and V. terMeulen. 1983.The biologyof coronaviruses.J. Gen. Virol.64:761-776.
24. Spaan, W., H. Delius, M. A. Skinner, J. Armstrong, P. Rottier, S. Smeekens, S. G. Siddell, and B.van der Zeijst. 1983. Tran-scription strategy of coronaviruses: fusion of non-contiguous sequences during mRNAsynthesis. Adv. Exp. Biol. 173:173-186.
25. Spaan, W. J. M., P. J. M. Rottier, M. C. Horzinek, and B. A. M. van derZeijst. 1981.Isolation andidentification ofvirus-specific mRNAs in cells infected with mouse hepatitis (MHV-A59). Virology 108:424-434.
26. Spaan, W. J. M., P. J. M. Rottier, M. C. Horzinek, and B. A. M. van der Zeijst. 1982. Sequence relationships between the genome and the intracellular RNA species 1, 3, 6, and 7 of mouse hepatitis virus strainA59. J.Virol. 42:432-439. 27. Stern, D. F., and S. I. T. Kennedy. 1980. Coronavirus
multipli-cationstrategy. II.Mappingtheavian infectious bronchitis virus intracellular RNA speciestothe genome. J. Virol. 36:440-449. 28. Sturman, L.S.,and K.Takemoto. 1972. Enhancedgrowth ofa murine coronavirusintransformedmousecells.Infect. Immun. 6:501-507.
29. Takegami, T., R. J. Kuhn, C. W. Anderson, and E. Wimmer. 1983. Membrane-dependenturidylylation of the genome-linked proteinVPgofpoliovirus.Proc.Natl. Acad.Sci.USA 80:7447-7451.
30. Wege, H.,S. G.Siddell,L.S.Sturman, and V. ter Meulen.1981. CoronavirusJHM:Characterization ofintracellular viral RNA. J. Gen. Virol.54:213-217.
31. Weiss, S. R., and J. L. Leibowitz. 1983. Characterization of murine coronavirus RNA by hybridization with virus-specific cDNAprobes.J. Gen. Virol. 64:127-133.
32. Wertz, G. W. 1983. Replication of vesicular stomatitis virus defective interfering particle RNA in vitro: transition from synthesis of defective interfering leader RNA to synthesis of full-length defectiveinterferingRNA.J. Virol. 46:513-522. 33. Wertz, G. W., and M. Levine.1973. RNAsynthesis by vesicular
stomatitis virusandasmallplaquemutant: effects of cyclohex-imide. J. Virol. 12:253-264.