Thomas Hoenen,a,bReed S. Shabman,cAllison Groseth,a,bAstrid Herwig,bMichaela Weber,bGordian Schudt,bOlga Dolnik,b Christopher F. Basler,cStephan Becker,band Heinz Feldmanna
Laboratory for Virology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rocky Mountain Laboratories, Hamilton, Montana, USAa; Institut für Virologie, Philipps Universität Marburg, Marburg, Germanyb; and Department of Microbiology, Mount Sinai School of Medicine, New York, New York, USAc
Inclusion bodies are a characteristic feature of ebolavirus infections in cells. They contain large numbers of preformed nucleo-capsids, but their biological significance has been debated, and they have been suggested to be aggregates of viral proteins with-out any further biological function. However, recent data for other viruses that produce similar structures have suggested that inclusion bodies might be involved in genome replication and transcription. In order to study filovirus inclusion bodies, we fused mCherry to the ebolavirus polymerase L, which is found in inclusion bodies. The resulting L-mCherry fusion protein was functional in minigenome assays and incorporated into virus-like particles. Importantly, L-mCherry fluorescence in transfected cells was readily detectable and distributed in a punctate pattern characteristic for inclusion bodies. A recombinant ebolavirus encoding L-mCherry instead of L was rescued and showed virtually identical growth kinetics and endpoint titers to those for wild-type virus. Using this virus, we showed that the onset of inclusion body formation corresponds to the onset of viral genome replication, but that viral transcription occurs prior to inclusion body formation. Live-cell imaging further showed that inclu-sion bodies are highly dynamic structures and that they can undergo dramatic reorganization during cell diviinclu-sion. Finally, by labeling nascent RNAs using click technology we showed that inclusion bodies are indeed the site of viral RNA synthesis. Based on these data we conclude that, rather than being inert aggregates of nucleocapsids, ebolavirus inclusion bodies are in fact com-plex and dynamic structures and an important site at which viral RNA replication takes place.
E
bolaviruses are negative-sense RNA viruses (NSVs) that cause severe hemorrhagic fever in humans and nonhuman primates, with case fatalities for Zaire ebolavirus (ZEBOV) reaching up to 90% (11). They are classified as biosafety level 4 (BSL-4) agents, as well as category A potential bioterrorism agents (7). Unfortu-nately, there are only experimental therapies and vaccines avail-able for ebolaviruses and, therefore, a further understanding of their life cycle will be instrumental in the development of novel therapeutic approaches (23).Ebolaviruses have a nonsegmented single-stranded RNA ge-nome that encodes seven structural and two nonstructural pro-teins (44). During all phases of the virus life cycle, the genome is encapsidated by the nucleoprotein (NP) and, together with the viral polymerase L and the polymerase cofactor VP35, as well as the VP24 protein, forms ribonucleoprotein complexes (RNPs) (3, 4). Within virus particles, RNPs are found in the form of con-densed nucleocapsids, which are surrounded by the matrix pro-tein VP40, which orchestrates morphogenesis and budding of vi-rus particles (17, 45). The sole viral glycoprotein GP, which is embedded in the viral envelope, is responsible for attachment to target cells as well as entry and fusion with cellular membranes during entry (34). The mRNAs for these proteins are sequentially transcribed from the 3=end of the RNA genome, and transcription terminates at conserved transcription stop signals and is reiniti-ated at nearby conserved transcription start signals (29). Since reinitiation does not occur in all cases, it is believed that, as with other NSVs, a gradient of mRNAs is produced, with the NP mRNA, which is encoded by the first gene at the 3=end of the genome, being the most abundant mRNA, whereas the L mRNA, which is encoded by the last gene, is the least abundant mRNA species (29).
One prominent feature of ebolavirus infections is the forma-tion of inclusion bodies, which can be detected by light
micro-scopy (2,32,43), fluorescence microscopy (28), and electron mi-croscopy (2,12,13,32,33,43). These inclusion bodies contain NP, VP35, VP30, and L (5,14), and electron microscopic studies indicate that inclusions contain nucleocapsids or nucleocapsid-like structures (12,33,35,43,46). On the one hand this has led to the proposal that inclusions constitute viral factories and fulfill important tasks in the virus life cycle (40), but on the other hand, inclusions are often regarded as a cellular mechanism to dispose of large quantities of misfolded proteins (25). Until now, no func-tional studies on ebolavirus inclusion bodies have been per-formed, and their function in the virus life cycle, if any, remains unknown.
For viruses other than NSVs, viral factories have been well studied. For double-stranded DNA viruses, such as poxviruses and iridoviruses, complex viral factories have been described. These form in the cytoplasm, are surrounded by a vimentin cage, and pass through different stages during virus infection (39). For single-stranded positive-sense RNA viruses, such as togaviruses and flaviviruses, viral factories are typically associated with intra-cellular membranes, such as the endoplasmic reticulum or Golgi apparatus (reviewed in reference39).
Unfortunately, with NSVs very little is known about the exis-tence of viral factories. However, recently it was shown for rabies
Received18 June 2012 Accepted12 August 2012
Published ahead of print22 August 2012
Address correspondence to Heinz Feldmann, feldmannh@niaid.nih.gov.
S.B. and H.F. contributed equally to this work.
Supplemental material for this article may be found athttp://jvi.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01525-12
on November 7, 2019 by guest
http://jvi.asm.org/
virus infection that Negri bodies, which are morphologically sim-ilar to the inclusion bodies seen during ebolavirus infection, are the site of viral RNA synthesis (26). Similar findings were also reported for another rhabdovirus, vesicular stomatitis virus (VSV) (18). We, therefore, sought to investigate the role of inclu-sion bodies in the filovirus life cycle, taking advantage of recent technological advances, such as live-cell microscopy, which is a novel technology with respect to the study of BSL4-pathogens, and the availability of a reverse genetics system for ZEBOV that allows for the generation of recombinant viruses. To this end, we generated a recombinant ZEBOV that expresses a fluorescently tagged polymerase, providing us with the opportunity to analyze the details of inclusion body formation, and we used this recom-binant virus to show that inclusion bodies are highly dynamic structures that first appear about 10 h postinfection and are in-deed a site of virus genome replication and, therefore, an impor-tant aspect of the virus life cycle.
MATERIALS AND METHODS
Cells and viruses.Vero E6 (African green monkey kidney), 293T (human embryonic kidney), and Huh-7 (human hepatoma) cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Life Technologies) sup-plemented with 10% fetal bovine serum (FBS; PAN Biotech), 2 mML -glu-tamine (Q; Life Technologies), and 100 U/ml penicillin and 100g/ml streptomycin (PS; Life Technologies) and grown at 37°C with 5% CO2.
During live-cell imaging, cells were cultivated in Leibovitz’s medium without phenol red (Life Technologies) with 100 U/ml penicillin and 100
g/ml streptomycin, 20% FBS, and 400M 6-hydroxy-2,5,7,8-tetra-methylchromane-2-carboxylic acid (Trolox; Sigma).
Recombinant viruses were based on ZEBOV (strain Mayinga; acces-sion numberAF272001). All work with viruses was performed in the BSL-4 laboratories at the Philipps Universität Marburg, Germany, and the Rocky Mountain Laboratories (RML), Division of Intramural Research (DIR), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH).
Plasmids.Expression plasmids for the minigenome and viral proteins have been previously described (19, 21). For construction of the L-mCherry expression plasmid, the coding sequence for mCherry (acces-sion numberAY678264) was introduced into the L open reading frame between nucleotides 5115 and 5116 (amino acids P1705 and Q1706) by using standard cloning techniques. The wild-type full-length clone plas-mid (pAmp-rgZ-SXBS) encoding wild-type ZEBOV (rgZEBOV-WT) is described elsewhere (R. S. Shabman, T. Hoenen, O. Jabado, H. Feldmann, and C. F. Basler, submitted for publication), and the full-length clone plasmid encoding the cRNA for a recombinant ZEBOV (pAmp-rgZ-SXBS-L-mCherry) containing L-mCherry instead of L was generated by subcloning the relevant portion of the L-mCherry expression plasmid into pAmp-rgZ-SXBS via unique restriction sites within L (HpaI and PacI).
Expression, detection, and functional characterization of recombi-nant L-mCherry in mammalian cells.For expression, 293T cells in 6-well dishes (for Western blotting experiments) or Huh-7 cells in 35 mm u-dishes (Ibidi; for fluorescence microscopy experiments) at a confluence of about 50% were transfected with Transit LT1 (Mirus) according to the manufacturer’s instructions using 3l Transit LT1 perg of DNA and 0.6
g pCAGGS-NP, 0.6g pCAGGS-VP35, 0.35g pCAGGS-VP30, and 4
g pCAGGS-L or pCAGGS-L-mCherry for Western blotting experi-ments, or 0.125g pCAGGS-NP, 0.125g pCAGGS-VP35, 0.125g pCAGGS-VP30, and 1g pCAGGS-L or pCAGGS-L-mCherry for fluo-rescence microscopy experiments. In all cases, the total amount of DNA was kept constant by adding empty pCAGGS vector to the transfection mixtures. Western blotting was performed as previously described (19) 48 h after transfection, using a monoclonal antibody against mCherry (Bio-Vision) at a dilution of 1:100 and a secondary Alexa Fluor 680-coupled
goat-anti-mouse antibody (Molecular Probes) at a dilution of 1:5,000. Fluorescent signals were detected and quantified using the Odyssey infra-red imaging system (Li-Cor Biosciences). For fluorescence microscopy, cells were visualized on an inverted Axiovert 200 M microscope (Zeiss). Minigenome and transcription- and replication-competent virus-like particle (trVLP) assays with naive target cells were performed as previ-ously described (19,21). Briefly, for a minigenome assay 293T cells were transfected with expression plasmids carrying NP, VP35, VP30, and L as well as T7 polymerase and a T7-driven minigenome encoding firefly lu-ciferase. At 48 h posttransfection, luciferase activity, reflecting viral mini-genome replication and transcription, was measured. For the trVLP assay, producer cells were transfected as for a minigenome assay, but in addition, expression plasmids for VP40, VP24, and GP were cotransfected. This led to the formation of trVLPs, which contained minigenomes in the form of RNP complexes. At 72 h posttransfection, trVLPs were used to infect naive Vero E6 target cells, and at 48 h postinfection luciferase activity in target cells, reflecting minigenome replication in producer cells, packaging of RNP complexes into trVLPs, budding of trVLPs, and their entry into target cells as well as primary transcription in target cells, was measured.
Rescue of recombinant viruses.Rescue of recombinant viruses was performed as described elsewhere (15; Shabman et al., submitted). Briefly, 50% confluent Vero E6 cells were transfected using Transit LT1 according to the manufacturer’s instructions, using 6l Transit LT1 perg of DNA and the following plasmids: 125 ng pCAGGS-NP, 125 ng pCAGGS-VP35, 75 ng pCAGGS-VP30, 1,000 ng pCAGGS-L, 250 ng pCAGGS-T7, and 250 ng full-length plasmid. At 24 h posttransfection, the medium was ex-changed, and after 7 days the supernatant was transferred onto fresh Vero E6 cells. Upon development of cytopathic effect (CPE; after 7 to 14 days), supernatant from these cells was clarified and stocks were frozen in liquid nitrogen. RNA from these stocks was isolated, and the entire genome was sequenced to ensure there were no unwanted mutations.
Infection of cells.Cells were seeded in 6-well plates or 4-well Labtek-II chamber slides (Thermo Scientific) 1 day before infection. Cells were in-fected at a multiplicity of infection (MOI) of 20 (based on the 50% tissue culture infective dose [TCID50]) in volumes of 1 ml (6-well plates) or 250 l (chamber slides) of DMEM/PS/Q without FBS for 1 h at 37°C, or on ice (for time course experiments). After infection, the inoculum was removed and the cells were washed twice with phosphate-buffered saline (PBS), before DMEM/PS/Q with 2% FBS was added to the cells, and cells were incubated at 37°C with 5% CO2.
Confocal microscopy, immunofluorescence analysis, and live-cell imaging of infected cells.For confocal microscopy, Vero E6 cells were seeded in chamber slides and infected as described above. At the indicated times, cells were fixed in 10% formalin for 24 h, the fixative was ex-changed, and cells were removed from the BSL-4 laboratory following approved standard operating procedures. Outside the BSL-4 laboratory, chamber slides were washed twice with PBS and either directly mounted or stained for immunofluorescence analysis as previously described (19). For detection of VP35, a mouse monoclonal VP35 antibody (27) at a dilution of 1:100 and a secondary goat anti-mouse–Alexa Fluor 488 (Life Technologies) conjugate at a dilution of 1:1,000 were used, while for stain-ing of the endoplasmic reticulum the 1D3 anti-protein disulfide isomer-ase antibody (Enzo Life Sciences) coupled to DyLight 488 at a dilution of 1:500 was used. Staining for the Golgi apparatus was performed as previ-ously described (41) using Alexa Fluor 488-coupled wheat germ aggluti-nin (Life Technologies) at a concentration of 100g/ml. Coverslips were mounted on the slides with ProLong Gold antifade reagent (Life Technol-ogies) with 4=,6-diamidino-2-phenylindole (DAPI), and slides were then visualized on a Zeiss LSM 710 confocal microscope using a 40⫻oil im-mersion objective, 408-, 488-, and 561-nm laser lines and sequential re-cording mode, and the ZEN software package. At least 6 random micro-scopic fields were analyzed per time point, with an average of 5.2 cells per field and an optical slice thickness of 1m. DAPI staining was used to ensure that the analyzed cells were not undergoing mitosis.
For live-cell imaging, Huh-7 cells were seeded into 35-mm u-dishes
Hoenen et al.
on November 7, 2019 by guest
http://jvi.asm.org/
(Ibidi) 24 h prior to infection. Cells were infected in 400l Opti-MEM without phenol red (Life Technologies) for 1 h, after which the inoculum was removed and 500l Leibovitz’s medium was added. Cells were then visualized on a DMI6000B Leica inverted fluorescence microscope in a 37°C temperature-controlled environment using a 63⫻oil immersion objective.
Size measurement of inclusion bodies.For determining the cross-sectional areas of inclusion bodies, confocal images were imported into ImageJ 1.44p (1) at a resolution of 2,048 by 2,048 pixels, corresponding to 67.5 by 67.5m. Pictures were enhanced using a mean-type fast filter and standard settings and with contrast enhancement at a setting of 0.1% saturated pixels. A threshold was then applied to the pictures to isolate the inclusion bodies, which were selected using the wand tool, and their areas were measured.
RNA extraction and quantitative RT-PCR.Vero E6 cells were seeded in 6-well plates and infected as described above. At the indicated times postinfection, the supernatant was removed, cells were washed with PBS, scraped into 600l RLT buffer (Qiagen), and frozen at⫺80°C. For re-moval from the BSL-4 laboratory, samples were thawed, 600l of 70% ethanol was added, and samples were removed from the BSL-4 laboratory following approved standard operating procedures. RNA was then ex-tracted using the Qiagen RNeasy kit following the manufacturer’s instruc-tions. Quantitative two-step reverse transcription-PCR (RT-PCR) was performed as described elsewhere (Shabman et al., submitted). For re-verse transcription, either an oligo(dT) primer (for detection of mRNA) or a primer complementary to the negative-sense viral RNA (vRNA) was used, followed by quantitative real-time PCR using primers directed against NP (Shabman et al., submitted). In parallel, quantitative RT-PCR directed against-actin mRNA was performed, to control for differences in the amount of total input RNA. The relative copy number of target RNA was calculated from the change in threshold cycle (⌬CT) value be-tween-actin mRNA and target RNA, with the log of relative vRNA copies at 0 h postinfection set to 1.
Treatment of cells with drugs.Actinomycin D (Sigma) was used at a final concentration of 0.02 mg/ml to stop cellular RNA synthesis (24). Cytochalasin D was used at 5M to inhibit actin polymerization, and nocodazole was used at 33M to inhibit microtubule polymerization (6). All drugs were prepared in dimethyl sulfoxide (DMSO), and DMSO alone was used as a control in all experiments involving drug treatment.
Ethynyl uridine staining.Nascent RNA staining was performed using the Click-iT nascent RNA detection kit (Life Technologies), following the manufacturer’s instructions. Vero E6 cells were infected as described above in 4-well chamber slides. At 17.5 h postinfection, the medium was replaced with 400l medium containing actinomycin D. At 18 h postin-fection, 400l of medium containing 8l of a 100 mM ethynyl uridine stock solution in water (Life Technologies) was added, and cells were incubated for 1 h at 37°C and 5% CO2. Subsequently, cells were fixed in
10% formalin for 15 min, washed once with PBS, permeabilized with 0.5% Triton X-100, washed again with PBS, and then incubated with Alexa Fluor 488-azide in 400l Click-iT buffer for 30 min. Afterwards, cells were washed once with Click-iT rinse buffer and 4 times with PBS, before they were fixed for 24 h in 10% formalin. After that, the fixative was replaced with fresh 10% formalin, and the cells were removed from the BSL-4 laboratory following standard operating procedures and imaged by confocal microscopy as described above.
RESULTS
Cloning and expression of a fusion protein of the ebolavirus
polymerase L and mCherry.Inclusion bodies have long been
de-scribed for filovirus infection; however, their functional signifi-cance is unknown. To facilitate the study of ebolavirus inclusion bodies, we decided to fuse a fluorescent protein to the viral poly-merase L, in order to allow detailed analysis of the kinetics and dynamics of inclusion body formation in both live and fixed cells. Previously, it was shown that the Reston ebolavirus (REBOV) L contains a flexible linker region that allows insertion of molecular tags without loss of polymerase activity (14). Based on the data for REBOV, as well as multiple sequence alignments with other NSV polymerases (data not shown), a putative flexible linker in ZEBOV L was identified between amino acids S1678 and K1712 (Fig. 1A). The coding sequence for mCherry was inserted into this region, and the resulting L-mCherry fusion protein was expressed in cells in the presence or absence of other RNP proteins. Expression of L-mCherry could be readily detected by fluorescence analysis 18 h posttransfection, with L being distributed throughout the cells in a punctuate pattern and signal intensity significantly above the level
FIG 1Cloning and expression of L-mCherry. (A) Schematic representation of L-mCherry. Shown are both the wild-type ZEBOV L protein and the L-mCherry fusion protein. The putative linker region is shown in green, and the arrow marks the site where mCherry (shown in purple) is inserted. Numbers indicate amino acid positions. (B) Fluorescence analysis of Huh-7 cells expressing L-mCherry. Cells were transfected with expression plasmids encoding L-mCherry or wild-type L and NP, VP35, and VP30, as indicated. Cells were visualized 18 h posttransfection. Shown are phase-contrast, mCherry signal, and a merged image. (C) Western blot analysis of 293T cells expressing L-mCherry. Cells were transfected with expression plasmids encoding L-mCherry or wild-type L and NP, VP35, and VP30, as indicated. At 48 h posttransfection, cell lysates were subjected to SDS-PAGE and Western blotting with an antibody against mCherry.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.113.474.65.246.2]of autofluorescence occasionally observed in cells expressing an untagged L (Fig. 1B). Interestingly, L-mCherry was only detect-able when other RNP proteins were cotransfected. In order to investigate the contributions of other RNP proteins, cells were transfected with different combinations of L-mCherry and RNP proteins, and L-mCherry expression was detected by Western blotting (Fig. 1C). L-mCherry could only be detected when VP35 was cotransfected, suggesting that this protein, which is a known cofactor of L (31), is required for efficient and stable expression of L-mCherry.
Impact of mCherry on the function of L.In order to assess the
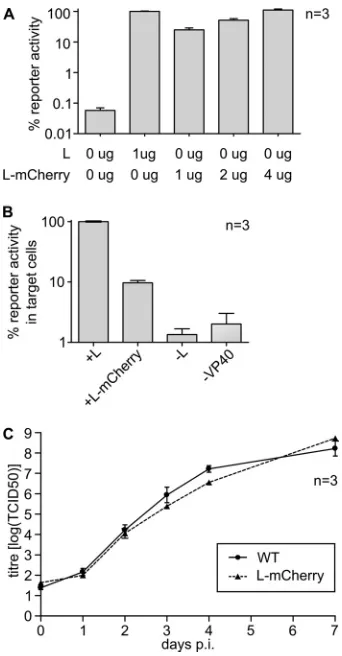
impact of mCherry insertion on L function, we compared the ability of L-mCherry to perform viral genome replication and transcription in a minigenome assay. To this end, cells were trans-fected with expression plasmids for all viral RNP proteins and a T7-driven expression vector for a ZEBOV minigenome encoding firefly luciferase, as well as an expression plasmid for T7 polymer-ase. Therefore, in this assay firefly luciferase activity served as a readout for viral genome replication and transcription. We ob-served that reporter activity in cells transfected with L-mCherry was somewhat reduced compared to cells transfected with wild-type L (Fig. 2A). However, reporter activity remained several or-ders of magnitude above the background signal, showing that L-mCherry was still able to efficiently promote viral genome replication and transcription. Further, reporter activity could be restored by increasing the amount of transfected L-mCherry (Fig. 2A).
In order to assess other parts of the virus life cycle, we used a trVLP assay (20). This assay is an extension of the minigenome assay and is based on coexpression of all viral structural proteins as well as a minigenome in mammalian cells (producer cells). This leads to the production of trVLPs, which contain both the viral glycoprotein GP as well as nucleocapsids containing a minigenome and are able to infect naive target cells. In these target cells, the minigenome under-goes primary transcription driven by the RNP proteins that are intro-duced into the target cells with the trVLPs. Thus, reporter activity in target cells reflects not only replication of the minigenomes in the producer cells but also formation of trVLPs, incorporation of all RNP components into these trVLPs, their entry into target cells, and fi-nally, primary transcription in target cells. When we compared L-mCherry with wild-type L in this assay, we observed a reduction in reporter activity in target cells similar to that observed in the classical minigenome assay (Fig. 2B); however, reporter activity was still sig-nificantly above that of the negative control, showing that L-mCherry is incorporated into trVLPs and supports primary transcription.
Based on these results, we next rescued a recombinant ZEBOV expressing L-mCherry (rgZEBOV-L-mCherry) instead of wild-type L (rgZEBOV-WT). Rescue was successful, and the recombi-nant virus showed similar growth kinetics and endpoint titers as a recombinant wild-type ZEBOV that was rescued in parallel (Fig. 2C). Also, progression of CPE showed no differences be-tween those two viruses (see Fig. S1 in the supplemental material), demonstrating that rgZEBOV-L-mCherry is not attenuated com-pared to rgZEBOV-WT in Vero E6 cells.
Characterization of inclusion bodies in infected cells.After
infection of cells with rgZEBOV-L-mCherry, we were able to de-tect L-mCherry in inclusion bodies throughout the cytoplasm. To study the kinetics of inclusion body formation, time course stud-ies were performed in which Vero E6 cells were infected with rgZEBOV-L-mCherry and fixed at 2-h intervals for 24 h after
in-fection. Fixed cells were analyzed by fluorescence microscopy, and the first L-mCherry-positive cells, although very rare, were ob-served after 10 h (Fig. 3A). These first inclusion bodies were rather small, with a mean cross-sectional area of 0.8m2, and all inclu-sion bodies had a cross-section of less than 3m2(Fig. 3B). Larger inclusions with a cross-sectional area of more than 10m2 ap-peared at about 12 to 14 h postinfection, and late in infection inclusion bodies filled the majority of the cytoplasm, with individ-ual inclusion bodies having cross-sectional areas of greater than 100m2. Interestingly, while the overall mean size of inclusion bodies increased over time, particularly at later time points two populations of inclusion bodies were apparent, i.e., very large
in-FIG 2Functional analysis of L-mCherry. (A) Minigenome assay. 293T cells were transfected with expression plasmids encoding all ZEBOV RNP proteins, including the indicated amounts of either wild-type L or L-mCherry, a T7-driven minigenome, and a T7 RNA polymerase. At 48 h posttransfection, cells were lysed, and reporter activity, reflecting viral genome replication and tran-scription, was measured. (B) trVLP assay results. 293T producer cells were transfected as described for panel A, but in addition, expression plasmids for VP40 and GP were cotransfected. At 72 h posttransfection, naive Vero E6 target cells were infected with trVLP-containing supernatants of these cells. At 48 h postinfection, reporter activity in target cells, reflecting viral genome replication and production of trVLPs containing RNP complexes including L-mCherry, entry of trVLPs into target cells, and primary transcription in these target cells was measured. As negative controls, either the expression plasmid for L or VP40 was omitted when transfecting producer cells. (C) Growth kinetics of a recombinant ZEBOV expressing L-mCherry. Vero E6 cells were infected with either a recombinant WT or a ZEBOV expressing L-mCherry instead of L. Supernatants were collected at the times indicated and titers were determined based on TCID50analysis. In all panels, means and standard errors from three independent experiments are shown.
Hoenen et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.336.507.64.392.2]clusion bodies with cross-sectional areas of 20m2or more and a large number of small structures with cross-sectional areas around 1m2. In order to better analyze the formation of inclusion bod-ies, we then utilized live-cell microscopy. To this end, cells were infected as before, visualized using a live-cell microscope, and again at 10 h after infection weak L-mCherry signals were visible (Fig. 3C). Infected cells initially had small inclusions identical to the ones observed by confocal microscopy, while at later time points (about 14 to 18 h postinfection) larger inclusions were observed in the cytoplasm. In order to further study the details of inclusion body formation, live-cell analysis was performed using a finer temporal resolution of 3 min per picture. We observed that the small inclusions observed early during infection were mobile and appeared to fuse together to form the larger inclusions seen later in infection (Fig. 4; see also Movie S1 in the supplemental material). Beginning at 20 to 24 h postinfection, L-mCherry sig-nals were observed throughout most of the cytoplasm of the cells, either in larger inclusion or in small dot-like structures. These dot-like structures appeared to be shed from larger inclusions (see Movie S2 and Fig. S2 in the supplemental material) and moved rapidly throughout the cells (see Movie S3 in the supplemental material). This rapid movement was sensitive to treatment with nocodazole but not cytochalasin D (see Movie S3), indicating that it is dependent on microtubules but not actin filaments.
In a number of instances, we observed infected cells undergo-ing mitosis (see Movie S4 and Fig. S3 in the supplemental mate-rial). Surprisingly, inclusion bodies underwent major reorganiza-tion during mitosis. While they showed a widely dispersed pattern
early during mitosis, about 120 min before cytokinesis (presum-ably at the beginning of the metaphase), they condensed into large inclusions. During cytokinesis, these inclusions again dispersed, and viral material was equably distributed into the daughter cells. Together with our inhibitor data, this observation suggests that inclusion body morphology is directly influenced by the cytoskel-etal rearrangements taking place during cell division.
To confirm the presence of viral proteins other than L in inclusion bodies, we investigated the distribution of VP35, which has been described to be a component of viral inclusion bodies and to interact with L (5,14), in infected cells. Colocal-ization between L-mCherry and VP35 was readily detectable, with VP35 being more abundant at the edges of inclusion bod-ies (Fig. 5). Interestingly, while all inclusion bodies that con-tained L-mCherry also concon-tained VP35, a subset of VP35-pos-itive inclusions did not contain L-mCherry, suggesting that there are different classes of inclusion bodies composed of dif-ferent subsets of viral proteins.
Formation of larger inclusion bodies coincides with viral
ge-nome replication.Based on the fact that they contain all the
pro-teins minimally required for viral RNA synthesis, inclusion bodies have sometimes been suggested to serve as viral factories and to play an important role in the viral replication cycle (40), although direct evidence for such a function has so far been lacking. There-fore, we assessed a possible role for inclusion bodies in viral ge-nome transcription and replication and investigated whether in-clusion body formation correlated with these processes, which are the major aspects of the virus replication cycle. To this end, cells
FIG 3Time course analysis of inclusion body formation. (A) Onset of inclusion bodies. Cells were infected with rg-ZEBOV-L-mCherry. At 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, and 24 h postinfection, cells were fixed, removed from the BSL-4 laboratory, counterstained with DAPI, and analyzed by confocal microscopy. Shown are the percentages of cells showing inclusion bodies from at least 5 random fields per time point. (B) Sizes of inclusion bodies. Images obtained as described for panel A were analyzed with ImageJ, and the cross-sectional areas of inclusion bodies at different time points were measured. Bars indicate the mean sizes of inclusion bodies at a given time. (C) Live-cell imaging of inclusion body formation. Cells were infected with rgZEBOV-L-mCherry and continuously monitored by live-cell fluorescence microscopy. Pictures show the same cell at the indicated time points after infection.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.137.449.63.338.2]were infected with rgZEBOV-L-mCherry and RNA was harvested from these cells every 1 to 4 h for 24 h (samples were taken at 0, 1, 2, 4, 6, 8, 10, 12, 16, 20, and 24 h postinfection). In order to quantify transcription, viral mRNA was quantified using a two-step RT-PCR, first reverse transcribing RNA using an oligo(dT) primer and then amplifying NP-specific sequences by quantitative real-time PCR (qPCR) (Shabman et al., submitted). For quantifi-cation of viral genome repliquantifi-cation, a two-step RT-PCR was per-formed using a genome-specific primer for the RT step, followed by qPCR, and thus we only detected negative-sense genomes and not positive-sense mRNAs or antigenomes. Onset of viral tran-scription was detected between 2 and 4 h postinfection (Fig. 6A), long before the first inclusion bodies were observed. In contrast,
viral genome replication only started to be detectable, based on the RT-PCR assay we used, between 12 and 16 h postinfection (Fig. 6B), which coincided with the appearance of larger inclusion bodies, suggesting that these structures are involved in viral ge-nome replication.
Viral inclusion bodies are the site of viral RNA synthesis.In
order to more directly show that viral inclusions are involved in viral genome replication, we performed nascent RNA staining. To this end, Vero E6 cells were infected with rgZEBOV-L-mCherry, and 17.5 h postinfection cellular transcription was shut down by adding actinomycin D to the medium. At 18 h postinfection, cells were then fed ethynyl-uridine, which is incorporated into nascent RNA. After 1 h, cells were fixed, and ethynyl-uridine was stained
FIG 4Fusion of smaller inclusion bodies into larger inclusions. Cells were infected with rgZEBOV-L-mCherry. After formation of early inclusion bodies, these were monitored by live-cell imaging. Fusion events are marked with arrows.
FIG 5Colocalization of inclusion bodies with VP35. Vero E6 cells were infected with rgZEBOV-L-mCherry. At 18 h postinfection, cells were fixed and stained with a mouse anti-VP35 antibody and an anti-mouse Alexa Fluor 488-coupled secondary antibody. Nuclei were counterstained with DAPI.
Hoenen et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.136.451.66.381.2] [image:6.585.114.474.601.702.2]using a highly specific alkyl-azide reaction. Subsequently, the cells were removed from the BSL-4 laboratory, and nascent RNA was visualized by confocal microscopy. In the absence of actinomycin D, newly synthesized RNA was mainly detected in the nucleus, particularly the nucleoli (Fig. 7). Weak signals for nascent RNA were also detected in inclusion bodies. In cells in which cellular transcription had been eliminated by using actinomycin D, signals were clearly visible in inclusion bodies, showing that they are an important site of viral RNA synthesis (Fig. 7). Interestingly, newly synthesized RNA was only detectable in larger inclusions, with cross-sectional areas between 10 and 60m2, which appeared
between 12 and 16 h postinfection. In contrast, smaller inclusion bodies appeared to be void of newly synthesized RNA, again sug-gesting that there are functionally distinct classes of inclusion bod-ies, although we cannot absolutely exclude the possibility that fur-ther RNA synthesis below the detection limit of our assay occurs in these smaller inclusion bodies.
DISCUSSION
While inclusion bodies have been observed and described since the discovery of ebolaviruses, until now no studies directly exam-ining their function have been performed. In order to address this problem, we developed a recombinant ZEBOV expressing a fluo-rescently tagged polymerase that allowed detailed studies of inclu-sion body formation and function. We chose to tag the polymer-ase L with mCherry, because for the closely related REBOV it had been shown that a hemagglutinin (HA) tag could be inserted into a putative flexible linker region in the protein without loss of ac-tivity (14). Also, given the tight packaging of RNP proteins in nucleocapsids, it is not clear whether other RNP proteins would tolerate an insertion of mCherry, which would increase their size (with respect to the number of amino acids) by 32% (NP), 69% (VP35), or even 82% (VP30), whereas for L this would constitute only a modest increase (11%). Further, a recombinant measles virus was successfully rescued that expressed a fusion of its poly-merase and the green fluorescent protein (GFP), which was in-serted into the same putative flexible linker region (9). Indeed, it was possible to express L-mCherry and detect it both by fluores-cence microscopy and Western blotting. In contrast to the HA-tagged REBOV polymerase (14), we observed some loss of reporter activity when using L-mCherry for minigenome replication and transcription; however, this loss was by no means complete, and a similar loss of polymerase activity has been reported for measles L-GFP (9). This difference in the activity of the various tagged L
FIG 6Analysis of mRNA and vRNA production in infected cells. Vero E6 cells were infected with ZEBOV-L-mCherry. At 0, 1, 2, 4, 6, 8, 10, 12, 16, 20, and 24 h postinfection, cells were lysed and total RNA was purified. mRNA was re-verse transcribed using an oligo(dT) primer, and vRNA was rere-verse tran-scribed using a vRNA specific primer, followed by quantitative real-time PCR using NP-specific primers. Shown are the means and standard errors from three independent experiments.
FIG 7Colocalization of nascent RNA and inclusion bodies. Vero E6 cells were infected with rg-ZEBOV-L-mCherry. At 17.5 h postinfection, actinomycin (ActD) was added to the cells to inhibit cellular RNA synthesis. At 18 h postinfection, ethynyl-uridine (EU) was added to the medium for 1 h before cells were fixed, permeabilized, and the EU incorporated into nascent RNAs was detected using Alexa Fluor 488-azide. Nuclei were stained with DAPI, and cells were visualized by confocal microscopy.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.134.450.448.684.2]constructs may be related to the size of the insertion (i.e., the HA tag versus mCherry or GFP), but it may also be related to the highly sensitive nature of the luciferase reporter in comparison to older reporter systems. Further, the fact that this loss in reporter activity could be overcome by increasing the amount of trans-fected L-mCherry suggests that the loss of activity might be due to protein expression or stability rather than an actual loss of poly-merase activity, since the amounts of the other RNP components did not have to be increased and the stoichiometries of RNP com-ponents have been shown to be essential for RNP function (30,38; Shabman et al., submitted). The availability of a fluorescently tagged L that can be detected both by fluorescence analysis and Western blotting is of particular significance, because until now it has not been possible to raise antibodies against L (5,14). Further, while we were previously able to detect an HA-tagged REBOV L protein by immunofluorescence (14), this protein could not be detected by Western blotting, so that the mCherry-tagged L pro-tein will be an important tool for future studies of the ebolavirus polymerase.
Interestingly, when we assessed the behavior of the recombi-nant ZEBOV expressing L-mCherry, no attenuation in tissue cul-ture was observed, suggesting that the recombinant virus might upregulate L-mCherry expression in comparison to wild-type L. Alternatively, in a minigenome assay, the overall protein expres-sion levels may be higher, which might make this system more prone to degradation due to improper processing and exacerbate differences between wild-type L and L-mCherry. Further, we cur-rently do not know whether there is a difference in the localization of viral protein synthesis in a minigenome assay, compared to viral infection, and this might also contribute to the observed differ-ences in phenotypes. The fact that the recombinant L-mCherry virus did not show attenuation in Vero E6 cells was in stark con-trast to findings with the recombinant measles virus expressing L-GFP, which was severely attenuated in tissue culture, showing a reduction in titer of about 2 to 4 logs at all times (9). Whether this was due to the use of different reporter proteins, slight differences in the exact position of insertion, or to intrinsic differences be-tween the polymerases of paramyxoviruses and filoviruses and their tolerance toward insertions in this region remains to be in-vestigated. Nevertheless, inserting reporter proteins in this linker region in the viral polymerase seems to be a strategy that is gener-ally tolerated by NSVs, and since this region is relatively easy to predict due to the high conservation of functional domains in NSV polymerases, this strategy appears to be a very promising approach for tagging the polymerases of other NSVs.
Fluorescent-tagged ebolaviruses have been available for a number of years (10); however, these viruses are based on the expression of GFP from additional open reading frames and thus have the potential to distort the transcriptional mRNA gradient. Indeed, while these viruses showed no attenuation in Vero E6 cells, they were somewhat attenuated in a STAT1 knockout mouse model and caused only mild disease in an otherwise-lethal non-human primate model (10). Therefore, it will be of great interest to assess rgZEBOV-L-mCherry in these animal models, since a fluorescent virus that is not attenuated in animals, or is attenuated to a lesser degree than already existing ones, would be an impor-tant tool for pathogenesis studies.
When we analyzed inclusion body formation in cells infected with ZEBOV-L-mCherry, we observed an onset of detectable in-clusion bodies at about 10 h. This is consistent with previous
re-ports using electron microscopy, which showed the first appear-ance of inclusion bodies in ZEBOV-infected cells after 9 h (40), but is much later than inclusion body formation for VSV and rabies virus, for which inclusion bodies are clearly visible after 2 and 4 h postinfection, respectively (18,26). Initially, inclusion bodies were small, but they fused together to form larger inclu-sions later in infection. While for other NSVs no live-cell studies of inclusion body formation have been performed, time course anal-ysis of VSV infection showed a large number of small inclusion bodies at early time points postinfection, but fewer and larger inclusion bodies later, suggesting that a similar phenomenon oc-curs with this virus (18). In contrast, for rabies virus most cells show only two or three distinct inclusion bodies early in infection, which grow in size, and only later in infection do many smaller inclusions appear (26). However, it remains unclear whether these are shed from larger inclusions, as we observed for the small dot-like structures apparent late in infection with ZEBOV-L-mCherry. For rabies virus, it has been shown that the appearance of these smaller structures is dependent on microtubule forma-tion, which is an interesting parallel to our observation that mi-crotubules are important for the motility of the small dot-like structures seen late in ebolavirus infection. Also, our observation of a dramatic reorganization of inclusions during mitosis further suggests that inclusions interact with the cytoskeleton. Interest-ingly, these data show that ebolavirus-infected cells, in contrast to cells infected with other NSVs, such as VSV (8), retain the ability to undergo mitosis. This capability may represent a novel means of virus dissemination. While further studies are clearly required to dissect these interactions of inclusion bodies with the cytoskel-eton, as well as the role of the reorganization of inclusion bodies during mitosis, it is interesting that the matrix protein VP40, which has been shown to be recruited to inclusion bodies (19) and to be required for the transport of nucleocapsids to the cell surface (36), interacts with microtubules (42). Together with the live-cell imaging data, this makes it tempting to speculate that the small dot-like structures apparent late in infection represent nucleocap-sids en route to budding sites.
Evidence for a functional involvement of inclusion bodies in the ebolavirus life cycle has so far been lacking. In this study, we have examined the possible role of ebolavirus inclusions in viral genome replication and transcription by detecting ethynyl-uri-dine incorporation into nascent RNA followed by detection with the highly specific alkyne-azide “click” chemistry, which is a novel technique that has so far only very rarely been used to study vi-ruses (16). Our data clearly showed that inclusion bodies are an important site of viral RNA synthesis, although only larger inclu-sion bodies seem to be active in RNA synthesis. However, at the first appearance of inclusion bodies, transcription had already been going on for several hours, showing that inclusions are most likely not required for transcription. This is consistent with results with VSV, for which it was shown that viral genome transcription initially took place throughout the cytoplasm, although at later time points it was redirected to inclusions (18). Another argument that speaks against an involvement of inclusions in viral genome transcription is the observation that phosphorylation of VP30 is required for recruitment of VP30 into inclusion bodies but that this inhibits the function of VP30 in transcription (28). This sug-gests that inclusion bodies actually play a functional role in the switch between viral genome transcription and replication and physically separate these two aspects of the virus life cycle. In con-Hoenen et al.
on November 7, 2019 by guest
http://jvi.asm.org/
trast to transcription, the appearance of larger inclusions with a cross-sectional area of more than 10m2, which are the ones that were positive for RNA synthesis, coincided with the onset of viral genome replication, and so it is very likely that inclusions are sites of viral genome replication, although a redirection of viral tran-scription toward inclusions, as seen with VSV, cannot be abso-lutely excluded at this point.
Interestingly, not all inclusion bodies were engaged in RNA synthesis, and during costaining for other viral proteins we ob-served VP35-positive inclusions that were devoid of L-mCherry, although all L-mCherry-positive inclusions contained VP35. For the closely related Marburg virus, ultrastructural studies have de-scribed different types of inclusion bodies, including sponge-like material connected to the surface of the endoplasmic reticulum, sheets and chains of nucleocapsids, amorphous electron-dense matrices surrounded by 45- to 60-nm spheres, and tightly packed tubules (12,22,33,43). In contrast, for ebolaviruses, inclusions have mostly been described as very regular matrices of tightly packed nucleocapsids (12,37,43), and at least until now different phenotypes, as suggested by our study, have not been observed ultrastructurally. Therefore, further studies linking observations from live-cell imaging and fluorescence microscopy, as well as nascent RNA staining on the one hand and ultrastructural analysis by electron microscopy on the other hand, will be required to further our understanding of the mechanisms by which inclusion bodies are involved in the virus life cycle.
In summary, we have generated a fluorescent ZEBOV L-mCherry fusion protein that retains its function and we rescued a recombinant virus expressing this fusion protein, which showed no attenuation in tissue culture. This virus was then used to show that L-mCherry-containing inclusion bodies are highly dynamic structures that fuse together to form larger inclusions as infection progresses, that these larger inclusions are sites of viral genome replication but likely not transcription, and that late in infection the inclusions shed highly motile small dot-like structures, which rapidly move through the cell in a microtubule-dependent manner and might represent nucleocap-sids en route to budding sites. Overall, our work demonstrates that inclusion bodies are important structures for the virus life cycle and, therefore, represent a promising target for antiviral interventions.
ACKNOWLEDGMENTS
We are very grateful to Hideki Ebihara (Laboratory of Virology, DIR, NIAID, NIH) for helpful discussions and advice. We also thank Friederike Feldmann (Office of Operations and Management, DIR, NIAID, NIH) and Les Shupert (Laboratory of Virology, DIR, NIAID, NIH), as well as Markus Eickmann, Gotthard Ludwig, and Michael Schmidt (Philipps Universität Marburg) for their technical assistance with the respective BSL-4 laboratories. Further, we are very grateful to Sonja Best and Travis Taylor (Laboratory of Virology, DIR, NIAID, NIH) for their help with confocal microscopy, to Anita Mora (Office of Operations and Manage-ment, DIR, NIAID, NIH) for help with preparing the supplemental movie files, and to Danielle Offerdahl and Marshall Bloom (Laboratory of Virol-ogy, DIR, NIAID, NIH) for providing reagents.
This research was supported in part by the Intramural Research Pro-gram of the NIH, NIAID. Further, this work was supported in part by NIH grants AI059536 and AI093786 to C.F.B., as well as by the Deutsche For-schungsgemeinschaft through the SFB 593, TP B12, and the Initiative for Excellence in Science and Technology of the state of Hessen (LOEWE).
REFERENCES
1.Abramoff MD, Magalhaes PJ.2004. Image processing with ImageJ. Bio-photonics Int.11:36 – 42.
2.Baskerville A, Fisher-Hoch SP, Neild GH, Dowsett AB.1985. Ultra-structural pathology of experimental Ebola haemorrhagic fever virus in-fection. J. Pathol.147:199 –209.
3.Beniac DR, et al.2012. The organisation of Ebola virus reveals a capacity for extensive, modular polyploidy. PLoS One7:e29608. doi:10.1371/ journal.pone.0029608.
4.Bharat TA, et al.2012. Structural dissection of Ebola virus and its assem-bly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. U. S. A.109:4275– 4280.
5.Boehmann Y, Enterlein S, Randolf A, Muhlberger E.2005. A reconsti-tuted replication and transcription system for Ebola virus Reston and comparison with Ebola virus Zaire. Virology332:406 – 417.
6.Carter GC, et al.2003. Vaccinia virus cores are transported on microtu-bules. J. Gen. Virol.84:2443–2458.
7.CDC. 14 June2012, posting date. Bioterrorism agents/diseases. Centers for Disease Control and Prevention, Atlanta, GA.http://www.bt.cdc.gov /agent/agentlist.asp.
8.Chakraborty P, et al.2009. Vesicular stomatitis virus inhibits mitotic progression and triggers cell death. EMBO Rep.10:1154 –1160. 9.Duprex WP, Collins FM, Rima BK.2002. Modulating the function of the
measles virus RNA-dependent RNA polymerase by insertion of green fluo-rescent protein into the open reading frame. J. Virol.76:7322–7328. 10. Ebihara H, et al.2007. In vitro and in vivo characterization of
recombi-nant Ebola viruses expressing enhanced green fluorescent protein. J. In-fect. Dis.196(Suppl. 2):S313–S322.
11. Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet
377:849 – 862.
12. Geisbert TW, Jahrling PB.1995. Differentiation of filoviruses by electron microscopy. Virus Res.39:129 –150.
13. Geisbert TW, Jahrling PB, Hanes MA, Zack PM.1992. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol.106:137–152. 14. Groseth A, et al.2009. The Ebola virus ribonucleoprotein complex: a
novel VP30-L interaction identified. Virus Res.140:8 –14.
15. Groseth A, et al. 2 August2012, posting date. The Ebola virus glycopro-tein contributes to but is not sufficient for virulence in vivo. PLoS Pathog.
8:e1002847.
16. Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ, de Haan CA.
2012. Visualizing coronavirus RNA synthesis in time by using click chem-istry. J. Virol.86:5808 –5816.
17. Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP.2000. A PPxY motif within the VP40 protein of Ebola virus interacts physically and func-tionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U. S. A.97:13871–13876.
18. Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplas-mic inclusions. PLoS Pathog.6:e1000958. doi:10.1371/journal.p-pat.1000958.
19. Hoenen T, et al.2010. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol.84: 7053–7063.
20. Hoenen T, Groseth A, de Kok-Mercado F, Kuhn JH, Wahl-Jensen V.
2011. Minigenomes, transcription and replication competent virus-like particles and beyond: reverse genetics systems for filoviruses and other negative stranded hemorrhagic fever viruses. Antiviral Res.91:195–208. 21. Hoenen T, et al.2006. Infection of naive target cells with virus-like
particles: implications for the function of Ebola virus VP24. J. Virol.80: 7260 –7264.
22. Kolesnikova L, Muhlberger E, Ryabchikova E, Becker S.2000. Ultra-structural organization of recombinant Marburg virus nucleoprotein: comparison with Marburg virus inclusions. J. Virol.74:3899 –3904. 23. Kondratowicz AS, Maury WJ.2012. Ebola virus: a brief review of novel
therapeutic targets. Future Microbiol.7:1– 4.
24. Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. doi:10.1371/ journal.pbio.0050220.
25. Kopito RR.2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol.10:524 –530.
26. Lahaye X, et al.2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcrip-tion and replicatranscrip-tion. J. Virol.83:7948 –7958.
27. Leung LW, et al.2011. Ebola virus VP35 suppresses IFN production from
on November 7, 2019 by guest
http://jvi.asm.org/
conventional but not plasmacytoid dendritic cells. Immunol. Cell Biol.
89:792– 802.
28. Modrof J, Becker S, Muhlberger E.2003. Ebola virus transcription acti-vator VP30 is a zinc-binding protein. J. Virol.77:3334 –3338.
29. Muhlberger E.2007. Filovirus replication and transcription. Future Virol.
2:205–215.
30. Muhlberger E, Lotfering B, Klenk HD, Becker S.1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocis-tronic minigenomes. J. Virol.72:8756 – 8764.
31. Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol.73: 2333–2342.
32. Murphy FA.1978. Pathology of Ebola infection, p 40 –52.InPattyn SR (ed), Ebola virus haemorrhagic fever. Elsevier, Amsterdam, Netherlands. 33. Murphy FA, Van der Groen G, Whitfield SG, Lange JV.1978. Ebola and Marburg virus morphology and taxonomy, p 53–71.InPattyn SR (ed), Ebola virus haemorrhagic fever. Elsevier, Amsterdam, Netherlands. 34. Nanbo A, et al.2010. Ebolavirus is internalized into host cells via
mac-ropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog.
6:e1001121. doi:10.1371/journal.ppat.1001121.
35. Noda T, Aoyama K, Sagara H, Kida H, Kawaoka Y.2005. Nucleocapsid-like structures of Ebola virus reconstructed using electron tomography. J. Vet. Med. Sci.67:325–328.
36. Noda T, et al.2006. Assembly and budding of Ebola virus. PLoS Pathog.
2:e99. doi:10.1371/journal.ppat.0020099.
37. Noda T, Halfmann P, Sagara H, Kawaoka Y.2007. Regions in Ebola virus VP24 that are important for nucleocapsid formation. J. Infect. Dis.
196(Suppl. 2):S247–S250.
38. Noda T, Kolesnikova L, Becker S, Kawaoka Y.2011. The importance of the NP: VP35 ratio in Ebola virus nucleocapsid formation. J. Infect. Dis.
204(Suppl. 3):S878 –S883.
39. Novoa RR, et al.2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell97:147–172.
40. Olejnik J, Ryabchikova E, Corley RB, Muhlberger E.2011. Intracellular events and cell fate in filovirus infection. Viruses3:1501–1531. 41. Parkkinen JJ, et al.1997. Polyamine-dependent alterations in the
struc-ture of microfilaments, Golgi apparatus, endoplasmic reticulum, and pro-teoglycan synthesis in BHK cells. J. Cell. Biochem.66:165–174. 42. Ruthel G, et al.2005. Association of Ebola virus matrix protein VP40 with
microtubules. J. Virol.79:4709 – 4719.
43. Ryabchikova E, Price B.2004. Ebola and Marburg viruses: a view of infection using electron microscopy. Batelle Press, Columbus, OH. 44. Sanchez A, Geisbert TW, Feldmann H.2007. Filoviridae: Marburg and
Ebola viruses, p 1410 –1448.InFields BN, Knipe DM, Howley PM (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. 45. Timmins J, Scianimanico S, Schoehn G, Weissenhorn W.2001.
Vesic-ular release of Ebola virus matrix protein VP40. Virology283:1– 6. 46. Watanabe S, Noda T, Kawaoka Y. 2006. Functional mapping of the
nucleoprotein of Ebola virus. J. Virol.80:3743–3751.
Hoenen et al.