Electron Microscopy: the Structure of Lactococcal Phage TP901-1
Cecilia Bebeacua,a,cLivia Lai,aChristina Skovgaard Vegge,bLone Brøndsted,bMarin van Heel,aDavid Veesler,c* Christian Cambillauc
Division of Biological Sciences, Imperial College London, South Kensington Campus, London, United Kingdoma; Department of Veterinary Disease Biology, University of Copenhagen, Frederiksberg, Denmarkb; Architecture et Fonction des Macromolécules Biologiques, UMR 6098 CNRS and Universités Aix-Marseille I and II, Campus de Luminy, Francec
Tailed phages are genome delivery machines exhibiting unequaled efficiency acquired over more than 3 billion years of
evolu-tion. Siphophages from the P335 and 936 families infect the Gram-positive bacterium
Lactococcus lactis
using receptor-binding
proteins anchored to the host adsorption apparatus (baseplate). Crystallographic and electron microscopy (EM) studies have
shed light on the distinct adsorption strategies used by phages of these two families, suggesting that they might also rely on
dif-ferent infection mechanisms. Here, we report electron microscopy reconstructions of the whole phage TP901-1 (P335 species)
and propose a composite EM model of this gigantic molecular machine. Our results suggest conservation of structural proteins
among tailed phages and add to the growing body of evidence pointing to a common evolutionary origin for these virions.
Fi-nally, we propose that host adsorption apparatus architectures have evolved in correlation with the nature of the receptors used
during infection.
B
acteriophages of the order
Caudovirales
are exquisitely
evolved nanomachines possessing a tail appendage used to
recognize the host and ensure genome delivery with high
specific-ity. They are the most abundant biological entity on earth, with an
estimated 10
31tailed phages in the biosphere (
1
). Tail morphology
serves as a basis to classify
Caudovirales
phages into three distinct
families:
Myoviridae, having a complex contractile tail (e.g., T4
[
2
])
Podoviridae, bearing a short noncontractile tail (e.g., P22 [
3
,
4
]); and
Siphoviridae, characterized by their long noncontractile
tails (e.g., SPP1 [
5
]).
The first steps of phage infection require interactions between
the phage receptor-binding proteins (RBPs) and the receptors
at the host cell surface. Despite the diverse infection mechanisms
displayed by
Siphoviridae, using surface proteins and/or cell wall
saccharides as receptors, their tail architecture is rather conserved.
It is characterized by a long noncontractile tube, assembled by
stacking several dozen homohexameric major tail protein (MTP)
rings, and a central core formed by a few copies of the tape
mea-sure protein (TMP) extending between both tail extremities and
determining its length. The proximal tail end harbors the
homo-hexameric terminator that stops tube elongation during assembly,
whereas the distal tail end is characterized by the presence of the
tail adsorption apparatus. In phages of Gram-positive bacteria,
this structure is composed of the distal tail protein (Dit), as well as
the tail fiber, and is termed the baseplate or tip, depending on the
presence or absence of peripheral proteins, respectively.
During the last few years, we have characterized the
mecha-nisms underlying the initial steps of infection by bacteriophages
targeting Gram-positive bacteria. Structural studies of the host
adsorption apparatus of the
Lactococcus lactis
phages p2 and
TP901-1 revealed distinct baseplate architectures and diverse
strategies used by the two virions to initiate infection (
6
–
11
). The
phage p2 baseplate undergoes large conformational changes in the
presence of Ca
2⫹ions to appropriately orient its RBPs and
estab-lish multiple interactions with host saccharides at the onset of
infection (
8
). In contrast, the TP901-1 baseplate harbors RBPs
already pointing in the direction of the host, suggesting that the
organelle is in a conformation ready for host adhesion (
11
).
In vivo
infection experiments confirmed and extended these observations
by demonstrating that Ca
2⫹ions are required for host adhesion
among p2-like phages (936 species) but have no influence on
TP901-1-like phages (P335 species). Upon host recognition, a
fir-ing signal is generated and propagated along the tail up to the
connector to inject the double-stranded DNA (dsDNA) genome
into the host cell, which leads to the production of progeny virions
(
5
,
12
).
The highly flexible nature of
Siphoviridae
tails makes structural
characterization of such phage particles difficult and explains the
paucity of data reported for the organelle (
5
). We report here the
electron microscopy (EM) reconstructions of the entire TP901-1
virion using single-particle protocols and a methodology specially
implemented to characterize its tail. Mature TP901-1 virions have
thin angular capsid shells filled with dsDNA and long tails when
imaged by transmission electron microscopy. Based on our EM
reconstructions and bioinformatics analyses, we propose
pseudo-atomic models for most parts of this
Siphoviridae
virion. The
con-servation of canonical phage structural protein modules supports
the evolutionary connection proposed between all tailed phages
and provides insights into the putative TP901-1 assembly and
maturation pathway. We also put forward the idea that a striking
correlation exists between host adsorption device architectures
and the strategies employed to recognize and adsorb onto the host.
Received10 October 2012Accepted31 October 2012
Published ahead of print7 November 2012
Address correspondence to Christian Cambillau, [email protected], or David Veesler, [email protected].
* Present address: David Veesler, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA 92037, USA.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02836-12
on November 7, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Native phage production and purification.TP901-1 phage were purified as previously described (13). Briefly, phage were induced with 3g/ml mitomycin C from lysogenicL. lactis901-1 grown at 30°C in GM17 broth. Following cell lysis, the phage particles were precipitated and purified by isopycnic centrifugation using a CsCl gradient.
Specimen preparation. (i) Negative staining.Approximately 3l of sample was applied onto glow-discharged carbon-coated grids and incu-bated for 1 min. The grids were blotted, and 10l of a 2% uranyl-acetate solution was added and incubated for 30 s. The excess stain was then blotted, and the grids were transferred to the microscope or stored.
(ii) Capsid cryo-EM.The sample (3l) was applied and incubated on glow-discharged Quantifoil grids for 1 min and subsequently blotted for 3 s before being plunged into liquid ethane for vitrification using an FEI Vitrobot.
Data collection. (i) Negative-stain data. Approximately 1,000 charge-coupled device (CCD) images were collected using a Phillips CM200 microscope with a field emission gun operated at 200 kV (Im-perial College London) and a 4,000 by 4,000 TVIPS CCD camera. We used a magnification of⫻38,000, resulting in a pixel size of 2.32 Å (4.64 Å coarsened by 2) over a range of nominal defocus values be-tween 0.5 and 1.5m.
(ii) Capsid cryo-EM.We collected 200 CCD images using the same setup as for stain data but with a magnification of⫻50,000 (resulting in a pixel size of 1.765 Å) and nominal defocus values ranging from⫺1.5m to⫺3m.
Image processing.All processing was carried out using IMAGIC soft-ware (14). Defocus estimation and correction for the microscope contrast transfer function (CTF) was carried out using the IMAGIC CTF2D_FIND and CTF2D_FLIP programs. Particles were selected using the program PICK_M_ALL and filtered, normalized, and masked before further pro-cessing. The number of particles used in each reconstruction is presented inTable 1.
(i) Full phage.In order to evaluate the overall dimensions of the tail and the number of MTP rings, 1,000 particles were manually selected from the images where the phage were observed in isolation and with a relatively straight tail. The full-phage particles were extracted into boxes of 1,200 by 1,200 pixels, coarsened by 2 to speed up processing, and submit-ted to single-particle analysis imposing 6-fold (c6) symmetry. Particles were then pretreated as described above, submitted to five rounds of alignment by classification (15), and subsequently multivariate statistical analysis (MSA) classified with 10 images per class (16). An initial model was generated by back-projecting a selected class average with c6 symme-try. The initial model was reprojected, and the reprojections were used for the initial angular assignment of the aligned particles by projection matching (16,17). As previously described (8), particles were positioned in a side view orientation with the symmetry axis perpendicular to the projection direction. Therefore, maps were reprojected along the equator (IMAGIC Euler angleequal to 90°) with a difference of 2°. Subsequent cycles of refinement over the entire data set, including alignment, projec-tion matching, and model calculaprojec-tions, were iterated until stabilized.
(ii) Fragment processing.The EM map generated for the full phage was cut into 7 continuous segments of 72 by 72 by 72 pixels corresponding to the connector (segment 1), the tail (segments 2 to 6), and the baseplate
(segment 7). The aligned particles resulting from the full-phage refine-ment described above were cut in the same way and remasked to generate 7 subsets. The 1st (connector) and 7th (baseplate) subsets were further refined by projection matching using the corresponding segment of the map obtained by cropping the full-phage initial map. Refinement was carried out for several rounds over 2°, imposing c12 symmetry for the connector and c6 symmetry for the baseplate. For further analysis and interpretation, however, we used the baseplate that we previously ob-tained (6). Fragments 2 to 6 (corresponding to the tail) were combined (⬃4,000 particles) and submitted to helical processing.
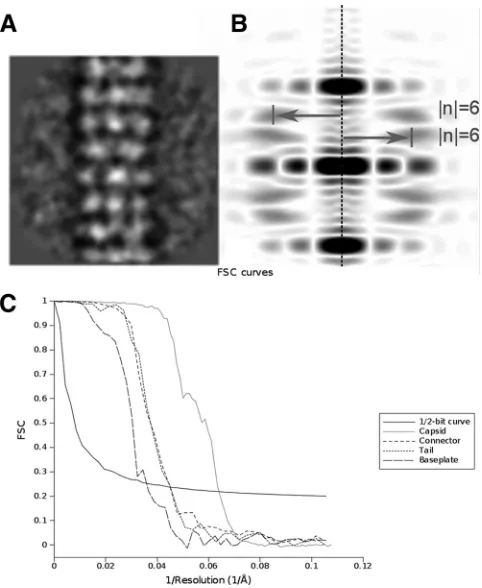
(iii) Tail helical processing.The 4,000 particles combined as de-scribed above were submitted to helical processing (Table 1). The helical map was produced using the package IHRSR⫹⫹(18). The rotational symmetry used was c6, and as the particles were already aligned, the max-imum allowed in-plane rotational angle was set to 10°. The initial helical parameters were determined using the Brandeis Helical Package (19) to calculate the Bessel orders of the basic layer lines (6 and⫺6) (Fig. 1Aand B) and the Ruby-Helix package to estimate a repeat distance of 110 Å (20). These were later refined by IHRSR to a helical rise of 38 Å and a rotation between subunits of 22.4°.
[image:2.585.39.287.87.156.2](iv) Reconstruction of the capsid.A total of 1,500 particles were man-ually selected, extracted into boxes of 256 by 256 pixels, and submitted to MSA classification (Table 1). An initial model was created by back-pro-jecting a single class average with icosahedral symmetry. The initial model
TABLE 1Summary of the data-processing strategies employed for the various TP901-1 reconstructions
Phage
component Method Symmetry
Resolution (Å)
No. of particles
Capsid Cryo-EM Icosahedral 15 1,500
Connector Negative-staining EM c12 21 1,000
Tail Negative-staining EM Helical 20 4,000
Baseplate Negative-staining EM c6 25 10,000
FIG 1EM parameters of TP901-1 structure. (A and B) Determination of the helical parameters of the TP901 tail. An average of aligned tail tube segments (A) was used to generate the Fourier transform (B). The meridional line is marked by a dotted line. The layer lines are marked by arrows that also indicate their Bessel orders (6 and⫺6). This indexation showed the 6-fold rotational symmetry of the TP901 helical tail. (C) Fourier shell correlation (FSC) curves of the final three-dimensional (3D) reconstructions obtained by correlation of two different 3D reconstructions created by splitting the particle set into two subsets. The resolution was estimated by the 1/2-bit cutoff threshold criterion as 15 Å for the capsid, 21 Å for the connector, 21 Å for the helical tail, and 25 Å for the baseplate.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.301.542.328.622.2]was refined by projection matching over the entire data set with an angu-lar sampling rate of 1° for several rounds, imposing icosahedral symmetry until stabilized.
(v) Resolution.The resolution of the reconstructions was estimated by Fourier shell correlation and the 1/2-bit threshold correlation criterion (21) (Fig. 1C).
Fitting and analyses were carried out using the UCSF Chimera package (22) (Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health).
Sequence alignments were performed using the profile-profile align-ment and fold recognition algorithm FFAS03 (Fold and Function Assign-ment System), as well as HHpred. Typically, predictions with FFAS03 scores lower than⫺9.5 contain⬍3% false positives.
Accession numbers.The capsid, connector, and tail reconstructions have been deposited at the Electron Microscopy Data Bank (EMDB) with accession codes EMD-2133, EMD-2227, and EMD-2228, respectively.
RESULTS
The capsid.
Bacteriophage capsids are robust containers designed
to carry and protect the viral genome that is packaged at liquid
crystalline density within its interior (
23
). We computed a
recon-struction of the TP901-1 capsid at 15-Å resolution using
⬃
1,500
particle images and applying icosahedral symmetry (
Fig. 2A
). The
mature capsid is approximately 660 Å wide along its 5-fold axes
and is made of 60 hexamers and 11 pentamers of the ORF36 major
capsid protein (MCP), organized with a T
⫽
7 symmetry, as well as
a dodecamer of the portal protein occupying a unique vertex. Due
to the 60-fold averaging applied during the reconstruction
pro-cess, the portal density was averaged out, and its structure was
independently investigated by reconstructing the connector
re-gion only. The capsid interior is filled with the dsDNA genome
organized as concentric layers regularly spaced at
⬃
25 Å (
Fig. 2B
),
as typically observed in other
Caudovirales
phages (
23
).
The plethora of MCP structures reported to date demonstrate
the conservation of the so-called HK97 “Johnson fold” among
tailed phages, herpesviruses, and some archaeal counterparts (
23
–
25
). Hence, we expect the TP901-1 MCP to exhibit a similar fold,
and this is further supported by the detection of weak sequence
similarity (but with high confidence) with the T7 and HK97 MCP
sequences using the FFAS03 server and HHpred (
Table 2
) (
26
). A
pseudoatomic model of the TP901-1 MCP shell was thus
pro-duced by rigid-body fitting of the icosahedral asymmetric unit of
the mature HK97 capsid (Protein Data Bank [PDB] 1OHG)
within the EM reconstruction (
Fig. 2C
). The seven subunits of the
icosahedral asymmetric unit are well accommodated in the capsid
density and form a 32-Å-thick shell surrounding the viral genome.
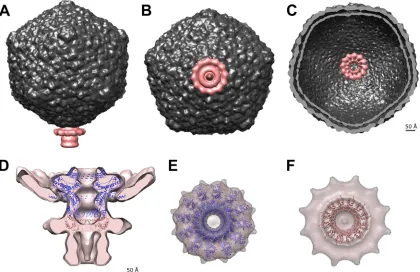
The head-to-tail connecting region.
The connector ensures
the cohesion of the phage capsid with its tail and is often made of
three different components organized as successive rings: the
por-tal protein and two head completion proteins. It is located at a
unique capsid vertex, where it replaces a penton motif (
Fig. 3A
to
C
). We achieved a reconstruction of the connector using
⬃
1,000
particles and applying 12-fold symmetry along the connector
channel axis.
The portal is a dodecameric protein, disclosing a conserved
fold in tailed phages and herpesviruses (
23
,
24
), that is involved in
DNA packaging during assembly and allowing release at the onset
[image:3.585.75.514.63.210.2]FIG 2The 15-Å-resolution cryo-EM reconstruction of the TP901-1 mature capsid. (A) Surface rendering of the icosahedral reconstruction low-pass filtered at 15 Å viewed along an icosahedral 2-fold axis. The capsid measures 660 Å along its 5-fold axis. (B) Cross section of the capsid reconstruction showing the layers of the dsDNA genome organized as concentric shells. (C) Pseudoatomic model of the TP901-1 mature capsid fitted in the reconstruction.
TABLE 2Sequence analyses of TP901-1 structural proteins
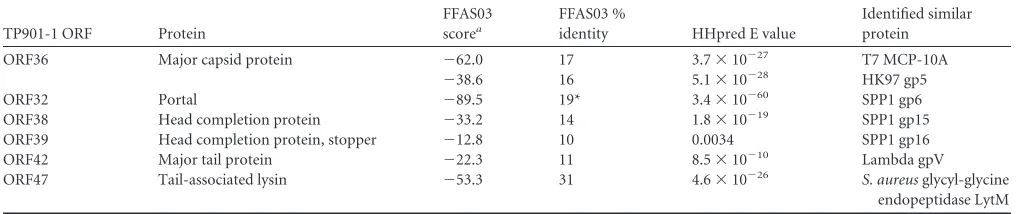
TP901-1 ORF Protein
FFAS03 scorea
FFAS03 %
identity HHpred E value
Identified similar protein
ORF36 Major capsid protein ⫺62.0 17 3.7⫻10⫺27 T7 MCP-10A
⫺38.6 16 5.1⫻10⫺28 HK97 gp5
ORF32 Portal ⫺89.5 19* 3.4⫻10⫺60 SPP1 gp6
ORF38 Head completion protein ⫺33.2 14 1.8⫻10⫺19 SPP1 gp15
ORF39 Head completion protein, stopper ⫺12.8 10 0.0034 SPP1 gp16
ORF42 Major tail protein ⫺22.3 11 8.5⫻10⫺10 Lambda gpV
ORF47 Tail-associated lysin ⫺53.3 31 4.6⫻10⫺26 S. aureusglycyl-glycine
endopeptidase LytM
aFFAS03 scores lower than⫺9.5 are considered significant.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.39.551.608.715.2]of infection. The TP901-1 portal (ORF32) has an overall length
comparable to that of the equivalent protein in phage SPP1 (452
versus 503 residues), and the two sequences share 26% identity
and 45% similarity. We used an SPP1 dodecameric portal model
(
27
) to fit into the proximal region of the connector
recon-struction, revealing good agreement between the atomic model
and the EM map at this resolution (
Fig. 3D
and
E
) and
con-firming the structural similarity between TP901-1 and SPP1
portal proteins suggested by FFAS03 and HHpred (Table 2).
Sequence analysis of the TP901-1 portal indicates that no
P22-like coiled-coil structure is present in its C-terminal region,
suggesting that the phage relies only on the turbine region of
the dodecamer to trigger packaging termination when the
ge-nome reaches the headful density (
4
,
28
,
29
).
The remaining region of the connector reconstruction
re-ported here was assumed to account for the two rings of head
completion proteins and the tail terminator. Sequence analyses
using the FFAS03 and HHpred servers allowed us to link TP901-1
ORF38 and ORF39 to the SPP1 head completion proteins gp15 and
gp16, respectively (Table 2). These two proteins form dodecameric
rings with known structures
in vivo
(
12
). We docked the SPP1
gp15 (PDB 2KBZ) and gp16 (PDB 2KCA) rings directly
under-neath the portal dodecamer in the connector EM map. The SPP1
gp15 head completion protein ring matches the dimensions of the
corresponding TP901-1 connector moiety reasonably well (
Fig.
3D
and
F
), while the SPP1 gp16 dodecamer is only partially
accounted for by the density (data not shown). Due to the
sym-metry mismatch between the connector and the tail, we have not
attempted to fit any tail terminator model in the connector
recon-struction, and this region is analyzed below.
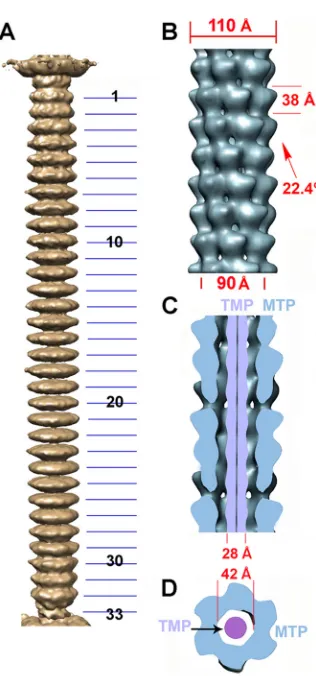
The tail.
Bacteriophage tails ensure genome delivery to the
target cell with an efficacy unequaled in the viral world. To
inves-tigate the tail structure, we first produced a low-resolution,
6-fold-averaged reconstruction of the whole TP901-1 phage from
se-lected virions exhibiting a straight (unbent) tail. We used this
reconstruction to assess that the tail tube is made of 34 stacks
comprising a tail terminator hexamer, located at the interface with
the connector, and 33 MTP hexamers forming the rest of the tube.
We then boxed small tail segments (each including seven
com-plete MTP stacks) from the tails and combined them in one data
set subsequently processed with the appropriate helical symmetry.
The TP901-1 tail extends over 1,180 Å (
Fig. 4A
) between the
connector and the baseplate, and its diameter varies between 110
Å (at the level of the MTP rings) and 90 Å (at the intersections
between rings) (
Fig. 4B
). The MTP hexameric stacks are rotated
by 22.4° between each other from the distal to the proximal tail
extremities, and the interhexamer distance is 38 Å (
Fig. 4B
). The
tail tube delineates a 42-Å-wide central channel that is continuous
with the connector and baseplate channels to form the genome
ejection pathway (
5
,
11
,
12
,
27
,
30
,
31
) (
Fig. 4C
and
D
). We
attrib-uted the 28-Å-wide elongated density filling the tail interior to the
TMP, the molecular ruler controlling the tail length during
assem-bly (
32
) (
Fig. 4C
and
D
). The TMP is believed to be oligomeric and
to form a long helical region extending through the tail tube and
FIG 3The 20-Å-resolution reconstruction of the TP901-1 connector. (A to C) The connector occupies a unique capsid vertex. (A) Side view. (B) View from the distal extremity along the tail tube. (C) Cross section of the capsid showing the portal protruding into it. (D) The SPP1 portal and the first head completion protein dodecamer (SPP1 gp15) are fitted into the connector reconstruction. Note the additional density surrounding the gp15 ring that likely corresponds to the collar/whiskers. The stopper (equivalent to SPP1 gp16) and the tail terminator are postulated to account for the remaining density. (E) View along the tail axis showing the fitting of the SPP1 portal dodecamer into the corresponding TP901-1 EM density (the region corresponding to the capsid density has been computationally removed for clarity). (F) View along the tail axis showing the fitting of the SPP1 first head completion protein into the corresponding TP901-1
EM density.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.83.503.68.340.2]anchored by one globular domain at each extremity. Consistent
with what has been observed in phage SPP1, no contacts were
observed between the TMP and the surrounding MTP rings,
ei-ther due to their nonexistence or because of a symmetry
discrep-ancy between the two structures. Weak interactions between the
TMP and the MTP hexamers probably facilitate ejection of the
former before DNA ejection through the tail channel. Although of
lower resolution, the overall dimensions of the TP901-1 tail
com-ponents are in good agreement with their equivalents in the SPP1
tail, supporting the validity of our reconstruction.
The host adsorption device.
The baseplate is the control
cen-ter for infectivity and is in charge of host recognition, attachment,
and initiation of infection. Combining our recently reported
TP901-1 baseplate crystal structure with the EM reconstruction
(
6
) shows the detailed organization of this 280-Å-wide and
150-Å-high organelle exhibiting an overall 6-fold symmetry (
Fig. 5A
and
B
). From the proximal to the distal ends, it is formed by 18
copies of BppU (ORF48) arranged around a central Dit hexamer
(ORF46) and holding 54 RBPs (ORF49) organized as 18 trimers
(
11
) (
Fig. 5A
to
C
). The RBPs orient their 54 receptor-binding
sites toward the distal extremity in a way suitable for establishing
interactions with the pellicle layer of the host without requiring
conformational changes (
11
,
33
). The tail-associated lysine (Tal;
ORF47) forms a 150-Å-long trimeric tail fiber appended to the Dit
ring and extending beyond the baseplate core at its distal
extrem-ity. We modeled the tail fiber N-terminal domains using the
closed p2 ORF16 trimer, which is expected to share a virtually
identical fold and to undergo a similar conformational change to
open the DNA ejection conduit during infection (
8
,
11
,
24
,
30
,
31
).
While no structure is available to model the tail fiber central
re-gion, the last
⬃
150 residues of each monomer form a domain
belonging to the peptidase M23 family that is probably involved in
peptidoglycan digestion at the onset of infection to allow the
vi-rion to access the cytoplasmic membrane (Table 2) (
13
,
34
).
DISCUSSION
Overall structure of the TP901-1 phage.
The structure
determi-nation of the TP901-1 phage capsid, tail, connector, and baseplate
FIG 4The 20-Å-resolution reconstruction of the TP901-1 tail. (A) Sixfold-averaged reconstruction of the TP901-1 phage tail from a few selected virions exhibiting an almost straight tail, making it possible to obtain the number of MTPs. (B) Detailed view of the reconstruction of a segment of the tail (7 MTP rings) using helical symmetry. The helical parameters of the tail are shown. (C) Cross section of the tail segment (blue) along its long axis revealing the internal TMP (violet). (D) Cross section of the tail segment orthogonal to its long axis (the color scheme is the same as in panel C).
FIG 5The 20-Å-resolution reconstruction of the TP901-1 distal tail region (the baseplate). The TP901-1 baseplate crystal structure was rigid-body fitted in the reconstruction. The color code is as follows: Dit (green), BppU (red), and BppL (light blue). The MTP hexamers were modeled using the Hcp type VI secretion system protein (dark blue). The N-terminal region of the tail fiber was modeled using the phage p2 ORF16 in closed conformation (pink). (A) Side view. (B) View along the tail axis from the distal extremity toward the capsid. (C) Cross section of the baseplate EM map. The assignment of the EM density to the different ORFs was performed using the X-ray structure.
on November 7, 2019 by guest
http://jvi.asm.org/
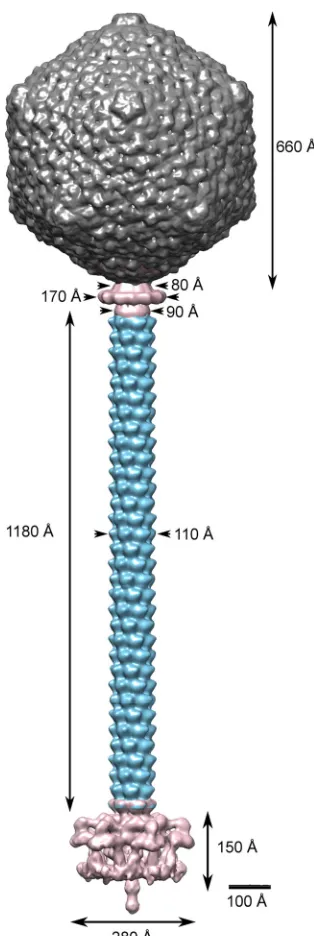
[image:5.585.84.242.59.397.2] [image:5.585.58.529.497.674.2]makes it possible to have an intermediate-resolution view of this
large viral molecular machine (
Fig. 6
). The TP901-1 capsid is 660
Å wide along its 5-fold axes and is virtually identical to the phage
HK97 capsid (
25
). Indeed, the TP901-1 MCP seems to harbor the
so-called HK97 Johnson fold that is conserved among
Caudovirales
phages and in some viruses infecting eukaryotes and archaea (
23
–
25
). Analysis of the TP901-1 MCP sequence revealed that the
pro-tein does not harbor a scaffolding domain fused at its N terminus,
in contrast to the HK97 situation. Instead, the virion genome
ex-hibits an upstream open reading frame (ORF) encoding a protein
product of
⬃
200 residues predicted to possess a high helical
con-tent and likely acting as a scaffolding protein. Based on these
observations, we propose that the TP901-1 capsid assembly and
maturation pathway are reminiscent of those of phage P22, which
expels the intact scaffolding proteins upon initiation of dsDNA
packaging rather than via proteolysis (
35
).
The TP901-1 connector structure is globally similar to that of
SPP1: the SPP1 portal dodecamer, as well as the most proximal
dodecamer of the head completion protein (SPP1gp15), is
reason-ably well accommodated in the corresponding regions of the
TP901-1 reconstruction. The most distal ring of head completion
protein should logically correspond to the SPP1 gp16 dodecamer
(the “stopper”) according to sequence comparisons and to the
observed density occluding the DNA exit channel in the
recon-struction. However, the EM density appears to only partially
ac-count for it, probably due to the limited quality and resolution of
the map in this region. Interestingly, an additional ring-like
struc-ture surrounds the connector at the level of the gp15 dodecamer,
and we propose that this additional region of density might result
from binding of additional proteins forming the collar/whiskers
observed in micrographs of TP901-1, which have been averaged
out during the reconstruction (
13
,
36
). This additional ring might
also be due to large conformational changes occurring upon tail
attachment.
Considerable efforts have been made to understand how the
dsDNA genome is driven from the phage capsid up to the host
cytoplasm during infection. In the case of phage SPP1, it has been
demonstrated that the high pressure with which the genome is
packaged into the head is not enough to power its complete entry
into the target cell (
37
). Other proteinaceous factors have been
shown to participate in this phenomenon in bacteriophages T5
and T7 by pulling DNA into the host cell (
38
,
39
). All the proteins
building the central channel that allows DNA transit from the
capsid to the host cell form an
⬃
40-Å-wide central channel with
conserved negative electrostatic properties. No structure of the
hexameric (biologically relevant) form of the MTP has been
re-ported so far. However, considering the low pIs observed for most
MTPs (e.g.,
⬃
4.8 in TP901-1 or
⬃
4.7 in SPP1), it is likely that this
protein contributes to the overall negative potential of the central
ejection tunnel. The fact that the genome transit pathway is
neg-atively charged has an obvious functional implication: as the
dsDNA backbone is also negatively charged due to the presence of
phosphate groups, a strong repulsion occurs, with the
corre-sponding phage regions to which it is exposed favoring genome
transit.
Besides ejection, this property might have an impact on phage
assembly. A survey of the pIs of various TMP proteins revealed
that these tail components are strongly basic (e.g., pI
⬃
8.8 in
TP901-1 or pI
⬃
10 in SPP1). Therefore, association of the central
TMP oligomer with the surrounding tail tube (formed of stacked
MTP hexamers) is likely to rely, at least partially, on strong
com-plementary electrostatic interactions.
The distal tail architecture is governed by the host
adsorp-tion strategy.
All bacteriophages belonging to the family
Sipho-viridae
share a canonical tail organization but differ mainly in
their distal tail part. Dairy phages belonging to the P335 and 936
species harbor complex baseplates in comparison to other phages
of the same family, and this seems to correlate with the different
host adsorption strategies. Indeed, they interact only with host
cell wall saccharides, putatively the phosphosaccharides present
in the pellicle of Gram-positive bacteria, for specific recognition
and attachment (
8
,
10
,
33
,
40
–
42
). Furthermore, many other
FIG 6Assembled complete structure of the TP901-1 phage. The complete phage was assembled by fitting the individually refined reconstructions into the map obtained for the full phage.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.83.240.63.531.2]siphophages, such as SPP1 and the lactococcal phages belonging
to the c2 species, bind reversibly to saccharidic receptors in a first
step before interacting irreversibly with a membrane protein that
initiates infection (
5
,
43
–
49
). As the affinity between phage
anti-receptors and their saccharidic partners is generally moderate (in
the low micromolar range), several RBPs are involved in binding
to ensure strong interactions based on avidity (
50
,
51
). In contrast,
the interaction between antireceptors and their proteinaceous
re-ceptors is strong, as illustrated by the tight binding reported
be-tween the T5 pb5 protein and the outer membrane transporter
FhuA (
52
) or
gpJ and LamB (
53
).
Siphoviridae
members can
therefore be dichotomized in two categories based on the
obser-vation of their distal tail architecture. On one hand, some phages
harbor a large baseplate accommodating up to several dozen RBPs
to interact only with the saccharidic part of the host cell wall. On
the other hand, bacteriophages devoid of any baseplate and
pos-sessing a simplified tail tip rely on irreversible binding with a
transmembrane protein present in the target cell to ensure their
commitment.
ACKNOWLEDGMENTS
This work was supported in part by grants from the Marseille-Nice Génopole, the CNRS, and the Agence Nationale de la Recherche (grants ANR-07-BLAN-0095, Siphophages, and ANR-11-BSV8-004-01, Lactophages). A Ph.D. grant from the Ministère Français de l’Enseignement Supérieur et de la Recherche (no. 22976-2006) was awarded to D.V. M.V.H. acknowledges financial support from the EU/NOE (NOE-PE0748), from the Dutch Ministry of Economic Af-fairs (Cyttron Project; BIBCR_PX0948), and from the BBSRC (grant BB/G015236/1).
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Infor-matics at the University of California, San Francisco (supported by NIH P41 RR-01081).
REFERENCES
1.Brussow H, Hendrix RW.2002. Phage genomics: small is beautiful. Cell
108:13–16.
2.Kostyuchenko VA, Leiman PG, Chipman PR, Kanamaru S, van Raaij MJ, Arisaka F, Mesyanzhinov VV, Rossmann MG. 2003. Three-dimensional structure of bacteriophage T4 baseplate. Nat. Struct. Biol.
10:688 – 693.
3.Lander GC, Khayat R, Li R, Prevelige PE, Potter CS, Carragher B, Johnson JE.2009. The P22 tail machine at subnanometer resolution re-veals the architecture of an infection conduit. Structure17:789 –799. 4.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A,
Potter CS, Carragher B, Johnson JE.2006. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science312: 1791–1795.
5.Plisson C, White HE, Auzat I, Zafarani A, Sao-Jose C, Lhuillier S, Tavares P, Orlova EV.2007. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J.26:3720 –3728.
6.Bebeacua C, Bron P, Lai L, Vegge CS, Brondsted L, Spinelli S, Cam-panacci V, Veesler D, van Heel M, Cambillau C.2010. Structure and molecular assignment of lactococcal phage TP901-1 baseplate. J. Biol. Chem.285:39079 –39086.
7.Campanacci V, Veesler D, Lichière J, Blangy S, Sciara G, Moineau S, van Sinderen D, Bron P, Cambillau C.2010. Solution and electron-microscopy characterization of lactococcal phage baseplates expressed in Escherichia coli. J. Struct. Biol.172:75– 84.
8.Sciara G, Bebeacua C, Bron P, Tremblay D, Ortiz-Lombardia M, Lichiere J, van Heel M, Campanacci V, Moineau S, Cambillau C.2010. Structure of lactococcal phage p2 baseplate and its mechanism of activa-tion. Proc. Natl. Acad. Sci. U. S. A.107:6852– 6857.
9.Shepherd DA, Veesler D, Lichiere J, Ashcroft AE, Cambillau C.2011. Unraveling lactococcal phage baseplate assembly by mass
spectrome-t r y . M o l . C e l l . P r o spectrome-t e o m i c s 1 0: M 1 1 1 . 0 0 9 7 8 7 . d o i : 1 0 . 1 0 7 4 / mcp.M111.009787.
10. Spinelli S, Desmyter A, Verrips CT, de Haard HJ, Moineau S, Cam-billau C.2006. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mamma-lian viruses. Nat. Struct. Mol. Biol.13:85– 89.
11. Veesler D, Spinelli S, Mahony J, Lichiere J, Blangy S, Bricogne G, Legrand P, Ortiz-Lombardia M, Campanacci V, van Sinderen D, Cam-billau C.2012. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. U. S. A.
109:8954 – 8958.
12. Lhuillier S, Gallopin M, Gilquin B, Brasiles S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S.
2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. U. S. A.106:8507– 8512.
13. Vegge CS, Brondsted L, Neve H, McGrath S, van Sinderen D, Vogensen FK.2005. Structural characterization and assembly of the distal tail struc-ture of the temperate lactococcal bacteriophage TP901-1. J. Bacteriol.187: 4187– 4197.
14. van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M.1996. A new generation of the IMAGIC image processing system. J. Struct. Biol.116: 17–24.
15. Dube P, Tavares P, Lurz R, van Heel M.1993. The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J.12: 1303–1309.
16. van Heel M.1984. Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy13: 165–183.
17. Harauz G, Ottensmeyer FP.1983. Interpolation in computing forward projections in direct three-dimensional reconstruction. Phys. Med. Biol.
28:1419 –1427.
18. Egelman EH. 2007. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J. Struct. Biol.157:83–94.
19. Owen CH, Morgan DG, DeRosier DJ.1996. Image analysis of helical objects: the Brandeis Helical Package. J. Struct. Biol.116:167–175. 20. Metlagel Z, Kikkawa YS, Kikkawa M.2007. Ruby-Helix: an
implemen-tation of helical image processing based on object-oriented scripting lan-guage. J. Struct. Biol.157:95–105.
21. van Heel M, Schatz M.2005. Fourier shell correlation threshold criteria. J. Struct. Biol.151:250 –262.
22. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE.2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem.25:1605–1612. 23. Veesler D, Johnson JE.2012. Virus maturation. Annu. Rev. Biophys.
41:473– 496.
24. Veesler D, Cambillau C.2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev.75:423– 433.
25. Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE.
2000. Topologically linked protein rings in the bacteriophage HK97 cap-sid. Science289:2129 –2133.
26. Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A.2011. FFAS server: novel features and applications. Nucleic Acids Res.39:W38 –W44. 27. Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J,
Dodson EJ, Tavares P, Antson AA.2007. Structural framework for DNA translocation via the viral portal protein. EMBO J.26:1984 –1994. 28. Olia AS, Prevelige PE, Johnson JE, Cingolani G. 2011.
Three-dimensional structure of a viral genome-delivery portal vertex. Nat. Struct. Mol. Biol.18:597– 603.
29. Tang J, Lander GC, Olia AS, Li R, Casjens S, Prevelige P, Jr, Cingolani G, Baker TS, Johnson JE.2011. Peering down the barrel of a bacterio-phage portal: the genome packaging and release valve in p22. Structure
19:496 –502.
30. Goulet A, Lai-Kee-Him J, Veesler D, Auzat I, Robin G, Shepherd DA, Ashcroft AE, Richard E, Lichiere J, Tavares P, Cambillau C, Bron P.2011. The opening of the SPP1 bacteriophage tail, a prevalent mechanism in Gram-positive-infecting siphophages. J. Biol. Chem.
286:25397–25405.
31. Veesler D, Robin G, Lichiere J, Auzat I, Tavares P, Bron P, Campanacci V, Cambillau C.2010. Crystal structure of bacteriophage SPP1 distal tail
on November 7, 2019 by guest
http://jvi.asm.org/
protein (gp19.1): a baseplate hub paradigm in Gram-positive infecting phages. J. Biol. Chem.285:36666 –36673.
32. Pedersen M, Ostergaard S, Bresciani J, Vogensen FK.2000. Mutational analysis of two structural genes of the temperate lactococcal bacteriophage TP901-1 involved in tail length determination and baseplate assembly. Virology276:315–328.
33. Chapot-Chartier MP, Vinogradov E, Sadovskaya I, Andre G, Mistou MY, Trieu-Cuot P, Furlan S, Bidnenko E, Courtin P, Pechoux C, Hols P, Dufrene YF, Kulakauskas S.2010. Cell surface of Lactococcus lactis is covered by a protective polysaccharide pellicle. J. Biol. Chem.285:10464 – 10471.
34. Kenny JG, McGrath S, Fitzgerald GF, van Sinderen D.2004. Bacterio-phage Tuc2009 encodes a tail-associated cell wall-degrading activity. J. Bacteriol.186:3480 –3491.
35. Greene B, King J.1994. Binding of scaffolding subunits within the P22 procapsid lattice. Virology205:188 –197.
36. Johnsen MG, Neve H, Vogensen FK, Hammer K.1995. Virion positions and relationships of lactococcal temperate bacteriophage TP901-1 pro-teins. Virology212:595– 606.
37. Sao-Jose C, de Frutos M, Raspaud E, Santos MA, Tavares P. 2007. Pressure built by DNA packing inside virions: enough to drive DNA ejec-tion in vitro, largely insufficient for delivery into the bacterial cytoplasm. J. Mol. Biol.374:346 –355.
38. Letellier L, Boulanger P, Plancon L, Jacquot P, Santamaria M.2004. Main features on tailed phage, host recognition and DNA uptake. Front. Biosci.9:1228 –1339.
39. Molineux IJ.2001. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol.40:1– 8.
40. Ricagno S, Campanacci V, Blangy S, Spinelli S, Tremblay D, Moineau S, Tegoni M, Cambillau C. 2006. Crystal structure of the receptor-binding protein head domain from Lactococcus lactis phage bIL170. J. Virol.80:9331–9335.
41. Spinelli S, Campanacci V, Blangy S, Moineau S, Tegoni M, Cambillau C.2006. Modular structure of the receptor binding proteins of Lactococ-cus lactis phages. The RBP structure of the temperate phage TP901-1. J. Biol. Chem.281:14256 –14262.
42. Tremblay DM, Tegoni M, Spinelli S, Campanacci V, Blangy S, Huyghe C, Desmyter A, Labrie S, Moineau S, Cambillau C.2006. Receptor-binding protein of Lactococcus lactis phages: identification and character-ization of the saccharide receptor-binding site. J. Bacteriol.188:2400 – 2410.
43. Baptista C, Santos MA, Sao-Jose C.2008. Phage SPP1 reversible adsorp-tion to Bacillus subtilis cell wall teichoic acids accelerates virus recogniadsorp-tion of membrane receptor YueB. J. Bacteriol.190:4989 – 4996.
44. Geller BL, Ivey RG, Trempy JE, Hettinger-Smith B.1993. Cloning of a chromosomal gene required for phage infection of Lactococcus lactis subsp. lactis C2. J. Bacteriol.175:5510 –5519.
45. Monteville MR, Ardestani B, Geller BL. 1994. Lactococcal bacterio-phages require a host cell wall carbohydrate and a plasma membrane pro-tein for adsorption and ejection of DNA. Appl. Environ. Microbiol.60: 3204 –3211.
46. Sao-Jose C, Baptista C, Santos MA.2004. Bacillus subtilis operon encod-ing a membrane receptor for bacteriophage SPP1. J. Bacteriol.186:8337– 8346.
47. Sao-Jose C, Lhuillier S, Lurz R, Melki R, Lepault J, Santos MA, Tavares P.2006. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J. Biol. Chem.281:11464 – 11470.
48. Valyasevi R, Sandine WE, Geller BL.1991. A membrane protein is required for bacteriophage c2 infection of Lactococcus lactis subsp. lactis C2. J. Bacteriol.173:6095– 6100.
49. Vinga I, Baptista C, Auzat I, Petipas I, Lurz R, Tavares P, Santos MA, Sao-Jose C.2012. Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Mol. Micro-biol.83:289 –303.
50. Lortat-Jacob H, Chouin E, Cusack S, van Raaij MJ. 2001. Kinetic analysis of adenovirus fiber binding to its receptor reveals an avidity mech-anism for trimeric receptor-ligand interactions. J. Biol. Chem.276:9009 – 9015.
51. Veesler D, Dreier B, Blangy S, Lichiere J, Tremblay D, Moineau S, Spinelli S, Tegoni M, Pluckthun A, Campanacci V, Cambillau C.2009. Crystal structure and function of a DARPin neutralizing inhibitor of lac-tococcal phage TP901-1: comparison of DARPin and camelid VHH bind-ing mode. J. Biol. Chem.284:30718 –30726.
52. Plancon L, Janmot C, le Maire M, Desmadril M, Bonhivers M, Letellier L, Boulanger P.2002. Characterization of a high-affinity complex be-tween the bacterial outer membrane protein FhuA and the phage T5 pro-tein pb5. J. Mol. Biol.318:557–569.
53. Berkane E, Orlik F, Stegmeier JF, Charbit A, Winterhalter M, Benz R.
2006. Interaction of bacteriophage lambda with its cell surface receptor: an in vitro study of binding of the viral tail protein gpJ to LamB (Maltoporin). Biochemistry45:2708 –2720.
on November 7, 2019 by guest
http://jvi.asm.org/