0022-538X/11/$12.00 doi:10.1128/JVI.05501-11
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
A Sequence within the Varicella-Zoster Virus (VZV) OriS Is a
Negative Regulator of DNA Replication and Is Bound by a
Protein Complex Containing the VZV ORF29 Protein
䌤
Mohamed I. Khalil,
1,4* Ann Arvin,
2Jeremy Jones,
3and William T. Ruyechan
1Department of Microbiology and Immunology and the Witebsky Center for Microbial Pathogenesis and Immunology, University at Buffalo, Buffalo, New York 142141; Departments of Pediatrics and Microbiology and Immunology, Stanford University School of
Medicine, G311, Stanford, California 943052; Department of Molecular Pharmacology, Beckman Research Institute,
City of Hope, 1500 East Duarte Road, Duarte, California 910103; and Department of Molecular Biology,
National Research Center, Dokki, Cairo, Egypt4
Received 23 June 2011/Accepted 7 September 2011
The architecture of the varicella-zoster virus (VZV) origin of DNA replication (OriS) differs significantly from that of the herpes simplex virus (HSV) DNA replication origin. Novel aspects of the VZV OriS include a GA-rich region, three binding sites for the VZV origin-binding protein (OBP) all on the same strand and oriented in the same direction, and a partial OBP binding site of unknown function. We have designated this partial binding site Box D and have investigated the role it plays in DNA replication and flanking gene expression. This has been done with a model system using a replication-competent plasmid containing OriS and a replication- and transcription-competent dual-luciferase reporter plasmid containing both the OriS and the intergenic region between VZV open reading frames (ORFs) 62 and 63. We have found that (i) Box D is a negative regulator of DNA replication independent of flanking gene expression, (ii) the mutation of Box D results in a decrease in flanking gene expression, thus a sequence within the VZV OriS affects transcription, which is in contrast to results reported for HSV-1, (iii) there is a specific Box D complex formed with infected cell extracts in electrophoretic mobility shift assay experi-ments, (iv) supershift assays show that this complex contains the VZV ORF29 single-strand DNA-binding protein, and (v) the formation of this complex is dependent on the presence of CGC motifs in Box D and its downstream flanking region. These findings show that the VZV ORF29 protein, while required for DNA replication, also plays a novel role in the suppression of that process.
Varicella-zoster virus (VZV), human herpesvirus 3, is a member of theAlphaherpesvirinaeand is the etiologic agent of two distinct clinical syndromes in humans: chicken pox (vari-cella), occurring during primary infection, and shingles (zos-ter), occurring after reactivation from latency (12). The VZV genome is a 125-kb linear double-stranded DNA molecule and is predicted to code for at least 71 proteins. The viral DNA is made up of long and short unique segments designated ULand US, respectively, both of which are bounded by inverted and
terminal repeat sequences IRs/TRs (14). The VZV genome contains two copies of an origin of DNA replication (OriS), flanked by ORF62 and ORF63 genes, within the IRs/TRs re-peats bounding the USsegment (13, 14, 57, 58). VZV has been
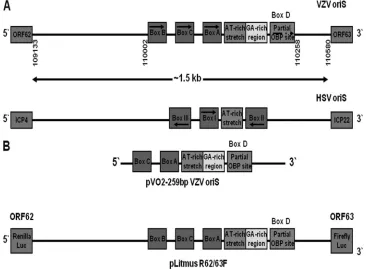
shown to encode homologues of all seven viral gene products required for herpes simplex virus type 1 (HSV-1) DNA repli-cation. These include the viral DNA polymerase and its asso-ciated processivity factor, a heterotrimeric helicase/primase complex, a single-strand DNA-binding protein (SSB), and a site-specific origin-binding protein (OPB) (20). However, while the two viruses have similar genomic organizations and encode similar DNA replication factors, the architecture of the VZV and HSV-1 OriS regions differs significantly (Fig. 1A).
The two VZV OriS contain an AT-rich palindrome and three 10-bp consensus binding sites [5⬘ -C(G/A)TTCGCACT-3⬘] for the VZV OBP encoded by VZV ORF51 (57, 59). These binding sites, designated Boxes A, B, and C, all are located upstream of the AT-rich palindrome. They are identical (Boxes A and B) or nearly identical (Box C) to the consensus binding site for the HSV-1 UL9 OBP, with which the VZV
ORF51 OBP shares 54.8% similarity and 46.5% identity (7, 30, 59). In addition to theseciselements, there is a characteristic GA-rich sequence immediately downstream of the AT-rich region in the VZV origin which is not present in the HSV-1 origin (14, 19).
All three OBP binding sites in the VZV origin are oriented in the same direction and are present on the same strand of the viral DNA. In contrast, in the HSV-1 OriS, binding sites for the UL9 OBP (Boxes I, II, and III) occur both upstream and downstream of the AT-rich region. Boxes I and II are located upstream and downstream on opposite strands of the DNA and are oriented in opposite directions. Box III is located upstream of Box I but is oriented in the same direction and occurs on the same DNA strand as Box II. Mutational analysis has shown that efficient HSV-1 origin-dependent DNA repli-cation requires the presence of all three UL9 binding sites as well as the central AT-rich region (3, 15, 22, 38, 45, 58, 61).
Origin-dependent DNA replication in VZV requires only the AT-rich region and Box A. Box C is not essential, but its presence increases replication efficiency, while Box B appears to be completely dispensable (57, 59). A putative partial down-* Corresponding author. Mailing address: Department of
Microbiol-ogy, 138 Farber Hall, University at Buffalo, SUNY, Buffalo, NY 14214. Phone: (716) 829-2376. Fax: (716) 829-2158. E-mail: mikhalil@buffalo .edu.
䌤Published ahead of print on 21 September 2011.
12188
on November 7, 2019 by guest
http://jvi.asm.org/
stream OBP site (5⬘-TGTTCGCGCG-3⬘) is present proximal to the AT-rich stretch within the VZV OriS sequence which has 70% identity with the authentic OBP sites upstream of the AT-rich stretch. It is oriented in the same direction and is on the same DNA strand as the three upstream sites. However, previous studies have shown that both recombinant full-length VZV OBP and the C-terminal half of the OBP, which contains the OBP DNA-binding domain, failed to bind to this sequence (7, 59). Thus, the role(s) that this sequence plays in the VZV life cycle is unknown.
In this work, we have designated this site Box D and inves-tigated its role in VZV origin-dependent DNA replication and in the expression of the OriS-flanking genes, ORF62 and ORF63. ORF62 encodes the VZV major transcriptional acti-vator which is essential for viral growth and the expression of the VZV genome (9, 42, 48, 49, 55). ORF63 encodes the VZV IE63 protein, influences viral and cellular transcription, and is required for the establishment of latency in the cotton rat model (2, 8, 10, 16, 25, 33, 67). The results demonstrate that the Box D sequence (i) acts to suppress DNA replication and (ii) aids in driving expression from the ORF62 and ORF63 promoters.
Electrophoretic mobility shift assay (EMSA) and supershift assays showed that there is a major characteristic infected cell complex, formed with Box D-containing probes, containing the VZV single-stranded DNA-binding protein (ORF29), suggest-ing a regulatory role in the replication of the viral genome. The VZV ORF29 protein is the homologue of HSV-1 ICP8, with
which it shares 50% identity. The ORF29 protein is believed to be essential for the initiation of viral DNA replication by anal-ogy with ICP8 (20, 30). ORF29 transcripts and protein have been reported to be present in latently infected human dorsal root ganglion (DRG) neurons (36), and ORF29 is involved in the establishment of latency in the cotton rat model (11).
These results also raise the possibility that the interaction of the protein complex containing the ORF29 protein with Box D plays a role in the establishment of VZV latency based on data from existing models of VZV pathogenesis.
MATERIALS AND METHODS
Cells and viruses.MeWo cells, a human melanoma cell line that supports the replication of VZV, were grown in Eagle’s minimal essential medium supple-mented with 10% fetal bovine serum as previously described (56). VZV strain MSP was propagated in MeWo cell monolayers as described by Lynch et al. (37) and Peng et al. (47).
Nuclear extracts.Nuclear extracts were prepared from infected and unin-fected MeWo cells as previously described (37). Briefly, pelleted cells were washed once with 30 volumes of phosphate-buffered saline (PBS). Packed cells were resuspended in one packed-cell volume of buffer A (10 mM HEPES, pH
7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol) at 4°C and allowed to
swell on ice for 15 min. Cells then were lysed by 10 rapid passages through a 25-gauge hypodermic syringe, and the homogenate was sedimented briefly at
12,000⫻g. The crude nuclear pellet was resuspended in two-thirds of one
packed-cell volume (determined at the time of cell harvest) of buffer C (20 mM
HEPES, pH 7.9, 25% [vol/vol] glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM
EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol) followed by incubation on ice with stirring for 30 min. The nuclear debris was pelleted by
centrifugation for 5 min at 12,000⫻g, and the supernatant (nuclear extract) was
dialyzed against buffer D (20 mM HEPES, pH 7.9, 20% [vol/vol] glycerol, 0.1 M
FIG. 1. (A) Comparison of the architecture of the VZV and HSV-1 OriS origins of DNA replication. The positions of the OBP binding site boxes on the two strands and their orientations are indicated. The numbers under the VZV OriS structure represent the positions of the beginning and the end of the intergenic region between ORF62 and ORF63 genes and the beginning and the end of the OriS structure of the Dumas IRs copy of the origin of replication; these locations are duplicated in TRs. (B) Schematic diagram showing the structures of the VZV OriS sequences within the pVO2 and pLitmus R62/63F plasmids.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.108.474.71.341.2]KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothre-itol) for 2 h. The dialyzed extract then was quick-frozen in liquid nitrogen and
stored at⫺70°C. Nuclear extracts from infected MeWo cells maintained in
medium containing 400g/ml phosphonoacetic acid (PAA) were prepared using
the same procedure.
Plasmids.The pVO2 plasmid containing a 259-bp fragment encompassing
nucleotides 5502 to 5760 of the TRs/IRssequence of the Dumas strain (13) was
provided by Nigel Stow (MRC Virology Unit, Institute of Virology, University of Glasgow). This fragment contains the VZV OriS region, including OBP Boxes A and C, the AT-rich palindrome, and 125 bp of downstream sequence. The pVO2 parental plasmid, pAT153, was purchased from MoBiTec (Goettingen, Ger-many). The pLitmus R62/63F plasmid (26) contains the complete 1.5-kb inter-genic region of DNA between the ORF62 and ORF63 genes of the pOka strain, including the VZV oriS structure inserted between genes encoding the Renilla and firefly luciferases (Promega), respectively, so that the luciferase genes act as reporters of ORF62 and ORF63 transcription. Plasmids containing Box D and Box A site-specific mutations within the OriS region were generated by mutating the wild-type pVO2 and/or pLitmus R62/63F plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using the following
primer sets: Box D mutation, 5⬘-AGAGAGAGAGAGTTTCTTGTTAAAGCG
TGTTCCCGCGATGTCGCG-3⬘and 5⬘-CGCGACATCGCGGGAACACGCT
TTAACAAGAAACTCTCTCTCTCT-3⬘; Box A mutation, 5⬘-GGCATGTGTC
CAACCACCGTTAAAACTTTCTTTCTATATATATAT-3⬘ and 5⬘-ATATAT
ATATAGAAAGAAAGTTTTAACGGTGGTTGGACACATGCC-3⬘.
All primers were synthesized by IDT (Coralville, IA). The positions and sequences of the mutations in the VZV OriS sequence contained within the pVO2 and pLitmus R62/63F plasmids used in transfections were verified by sequencing at the Roswell Park sequencing facility.
DpnI replication assays.MeWo cells were transfected with Lipofectamine reagent (Invitrogen, Carlsbad, CA) by following the manufacturer’s instructions. Four microliters of Lipofectamine reagent were used per microgram of trans-fected DNA in each transfection. In transfections performed in
100-mm-diam-eter petri dishes, 2.1⫻106
MeWo cells per dish were seeded in 12 ml of complete growth medium. The cells were 80% confluent at the time of transfec-tion. Three hours before transfection, the medium was replaced with fresh medium. Origin-dependent replication experiments were performed as described by Stow and McMonagle (58), Stow and Davison (57), and Khalil et al. (28). The
cells were transfected with 5 g of wild-type or mutant pVO2 or pLitmus
R62/63F plasmid. At 6 h posttransfection, cells were superinfected with VZV strain MSP. VZV superinfections were performed by adding 0.4 infected cells per 1 uninfected cell to each monolayer. Total cellular DNA was prepared 48 h after superinfection by the addition of 5 ml of lysis buffer (10 mM Tris-HCl, pH
7.5, 1 mM EDTA, 0.5% SDS, 20g/ml RNase I) per dish. Lysates were
trans-ferred to 15-ml conical test tubes and incubated for 1 h at 37°C. Proteinase K in TE buffer (10 mM Tris HCl and 1 mM EDTA) was added to a final
concentra-tion of 100g per ml, and the NaCl concentration was adjusted to 0.15 M. The
mixture then was incubated at 50°C for 3 h.
The DNA was isolated by phenol-chloroform extraction followed by ethanol precipitation. The DNA was digested with DpnI and EcoRI as described by Stow and Davison (57) and analyzed by Southern blot hybridization. Transfers were done using TurboBlotter kits obtained from Whatman, Inc. (Sanford, ME). The blots for the experiments done with the pVO2 plasmids were probed with a
400-bp PCR product derived from pVO2 using the primers 5⬘-GTGCTCCTGT
CGTTGAGGACCCGG-3⬘ and 5⬘-CCTCTGACTTGAGCGTCGATTTTT-3⬘
and end labeled with [␣-32P]ATP using T4 kinase (Invitrogen, Carlsbad, CA).
The pVO2 fragment includes a portion (nucleotide positions 1465 to 1864) of the pVO2 plasmid containing the Col E1 origin of replication and was designed to detect both intact DpnI-resistant linearized pVO2 (3.9 kb) resulting from the replication of the input plasmid and an 872-bp fragment resulting from the DpnI-sensitive unreplicated input plasmid. The blots for the experiments done with the pLitmus R62/63F plasmids were probed with a 476-bp PCR product
prepared from pLitmus R62/63F using the primers 5⬘-TAGGCCACCACTTCA
AGAACTCTGT-3⬘and 5⬘-AGCAAAAGGCCAGCAAAAGGCCAGG-3⬘and
end labeled with [␣-32P]ATP using T4 kinase (Invitrogen, Carlsbad, CA). The
pLitmus R62/63F fragment includes a portion (nucleotide positions 1832 to 2307) of the pLitmus R62/63F plasmid containing the pUC19 origin of replica-tion and was designed to detect both intact DpnI-resistant linearized pLitmus R62/63F (6.9 kb) resulting from replication of the input plasmid and a 651-bp fragment resulting from the DpnI-sensitive unreplicated input plasmid.
The resulting bands were quantified by PhosphorImager (Molecular Dynam-ics, Sunnyvale, CA) analysis. The ratio of replicated plasmid to input plasmid represents the replication efficiency of the test plasmid. The data from repre-sentative experiments are presented as the means from triplicate DpnI
replica-tion assays. Error bars indicate standard errors. Statistical significance was
de-termined by one-way analysis of variance followed by Tukey’spost hoctest.
Transfection, superinfection, and reporter gene assays. Transfection/superin-fection experiments followed by luciferase reporter gene assays were performed as previously described (64). Transfections were performed using 12-well plates.
Briefly, 2⫻105
cells were seeded in each well 24 h before transfection. The
pCMV-SPORT-Gal vector (Gibco, Carlsbad, CA) was used as an internal
control reporter for transfections. One microgram of each reporter vector
(pLitmus R62/63F) and 0.4g of-galactosidase (-Gal)-expressing plasmid
was transfected in each assay with Lipofectamine reagent (Invitrogen, Carlsbad, CA) by following the manufacturer’s instructions to act as a control for trans-fection efficiency. The cells were superinfected 24 h posttranstrans-fection with VZV strain MSP. VZV superinfections were performed by adding 0.4 infected cells per 1 uninfected cell to each monolayer.
The cells were collected 48 h after superinfection and lysed in 250l of the
lysis buffer (50 mM HEPES, pH 7.4, 250 mM NaCl, 1% NP-40, 1 mM EDTA). Control experiments without infection were done for each plasmid to determine basal expression levels. Dual-luciferase assays were performed using the
dual-luciferase reporter assay system (Promega) with 20l of cell extract, 50l of
luciferase assay reagent II, and 50l of Stop & Glo reagent in each assay. The
-Gal assays were performed using the-Gal assay kit (Invitrogen, Carlsbad,
CA) by following the microtiter plate protocol recommended by the manufac-turer. Dual-luciferase activities were normalized to the beta-galactosidase activ-ities. Transfection experiments were repeated at least three times, and each set of transfection conditions in a given experiment was performed in triplicate. The data from representative experiments are presented as the means from triplicate gene reporter assays. Error bars indicate standard errors. Statistical significance
was determined by one-way analysis of variance followed by Tukey’spost hoctest.
EMSA, supershift, and competition analyses. Fifty-one-base-pair oligonu-cleotide probes containing wild-type and mutant Box D elements (IDT,
Cor-alville, IA) were used in EMSAs. Probes were end labeled with [␣-32
P]ATP using T4 kinase (Invitrogen, Carlsbad, CA). One hundred femtomoles of the labeled
probes containing wild-type and mutant Box D (⬃1⫻105
dpm) were incubated
with 15g of uninfected or infected MeWo cell nuclear extract or PAA-treated
infected cell nuclear extract in a 10-l reaction mixture in binding buffer [40 mM
HEPES, pH 7.9, 100 mM NaCl, 10 mM MgCl2, 200g/ml bovine serum albumin
(BSA), 12% glycerol, 0.05% NP-40, 1 mM dithiothreitol, and 3g poly(dI-dC)].
In competition assays using the labeled Box D probe, the ratio of cold probe used to labeled probe was 200:1. The samples were analyzed by electrophoresis on a 5% polyacrylamide (37.5:1 acrylamide/bisacrylamide) gel followed by autora-diography. All competitions were performed in triplicate, and the relative amounts of specific complexes were quantified by PhosphorImager analysis.
Antibodies and antisera used in the supershift assays included anti-ORF29 rabbit polyclonal antisera directed against the C terminus of the ORF29 protein (30) and antisera directed against the full-length IE62 protein (56). Rabbit polyclonal anti-ORF51 antibodies prepared against the peptides of the first 9 amino acids corresponding to the N-terminal sequence and 20-amino-acid pep-tides corresponding to the C-terminal sequence of the ORF51 protein, as well as anti-IE62 antisera prepared against the DNA-binding domain (DBD) of IE62, were prepared by Bethyl Laboratories Inc. (Montgomery TX). Rabbit polyclonal antibodies against E2F1, E2F2, E2F3, E2F4, and E2F5 and were purchased from both Santa Cruz Biologicals (Santa Cruz, CA) and Abcam (Cambridge, MA). Antibody against the cellular transcription factor Zfp64 was purchased from
Abcam (Cambridge, MA). Antisera were added in 2.5-l aliquots to 10-l
reaction mixtures containing infected cell nuclear extracts and the 51-bp probe
containing the wild-type Box D sequence. For purified antibodies, either 2.5g
(Abcam) or 5g (Santa Cruz) antibody was added to the reaction mixtures. The
samples then were analyzed by electrophoresis on a 5% polyacrylamide (37.5:1 acrylamide/bisacrylamide) gel followed by autoradiography.
RESULTS
Mutation of the Box D site results in an increase in origin-dependent DNA replication in the absence and presence of flanking gene transcription. Previous work (28, 59) showed that the deletion or mutation of the upstream Box A binding site for the VZV OBP inhibits VZV origin-dependent DNA replication in DpnI resistance replication assays. Here, we in-vestigated the role of the partial downstream OBP site, which we have designated Box D, in that process. We also assessed
on November 7, 2019 by guest
http://jvi.asm.org/
the effect of the presence of transcribable flanking genes on Box D function, since Summers and Leib (60) and Nguyen-Huynh and Schaffer (43) showed that flanking gene expression had no effect on HSV origin-dependent DNA replication. The first set of experiments employed the pVO2 plasmid, which contains a 259-bp portion of the intergenic region between the ORF62 and ORF63 genes (Fig. 1B). The pVO2 OriS insert includes the upstream Box A and Box C sites, the AT-rich palindrome, and 125 bp downstream of the AT-rich sequence, including Box D. The plasmid lacks the TATA boxes and the proposed promoter elements of the OriS-flanking genes ORF62 and ORF63 and their coding sequences (31, 32, 41). The assays were performed as previously described via the initial transfection of MeWo cells with the origin-containing plasmid followed by VZV superinfection (28).
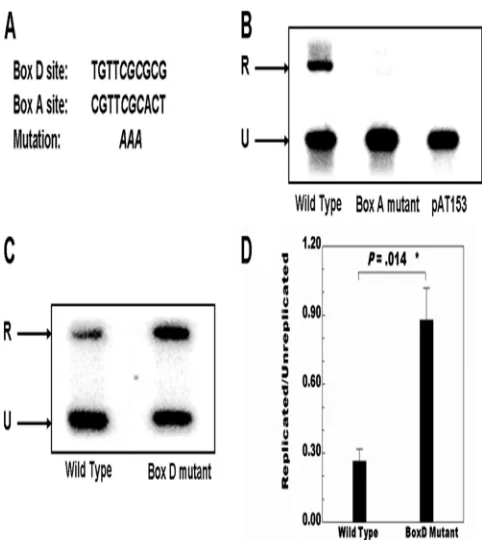
DpnI replication assays using pVO2 containing the wild-type VZV OriS showed a readily detectable signal at the position predicted for the replicated DNA (Fig. 2). In contrast, assays performed under the same conditions using the parental pAT153 plasmid lacking the VZV OriS resulted in no signal at
the position of the replicated DNA. As a second negative control, experiments with pVO2 containing a 3-base replace-ment of the core CGC sequence with AAA in the Box A OBP binding site were performed. The CGC triplet was shown by Chen and Olivo (7) to be essential for interaction with the VZV OBP. As previously reported (28), this also resulted in a loss of the replicated DNA band correlating with the results of Stow et al. (59), who showed via deletion that Box A is re-quired for OriS-dependent DNA replication.
In the next series of experiments, triplicate DpnI assays were performed using wild-type pVO2 and pVO2 plasmids contain-ing the core CGC-to-AAA mutation within the Box D se-quence. As shown in Fig. 2C and D, the mutation of Box D increased the level of origin-dependent replication approxi-mately 3-fold, and this difference was statistically significant (P⫽0.014). The influence of the Box D sequence on origin-dependent DNA replication then was examined in the pres-ence of flanking gene expression. We used the pLitmus R62/ 63F plasmid containing the entire intergenic region between the ORF62 and ORF63 genes, which includes the VZV OriS and the promoters of the two genes (26). The positions of ORF62 and ORF63 coding sequences were replaced by the Renilla (R) and firefly (F) luciferase genes, respectively. As shown in Fig. 3A, the mutation of the triplet CGC to AAA within Box A in the context of the pLitmus reporter plasmid resulted in the loss of DNA replication. This result shows, for the first time, that Box A is required for VZV origin-depen-dent DNA replication in the presence as well as the absence of flanking gene expression, and that the presence of Boxes B and C does not compensate for its functional absence.
The CGC-to-AAA mutation within Box D in the pLitmus R62/63F plasmid, in contrast, resulted in an increase in the level of origin-dependent replication to an extent equivalent to that observed with the pVO2 plasmid (Fig. 3A and B). The increase seen in the DNA replication efficiency with the Box D mutation using both the pVO2 and pLitmus R62/63F plasmids indicates the involvement of Box D in the negative regulation of VZV origin-dependent DNA replication. The expression of reporter genes was confirmed by luciferase assays and showed that both reporters were expressed to very high levels (1,600 and 1,800-fold increases for ORF62 and ORF63 reporters, respectively) in the presence of VZV superinfection compared to basal levels in the absence of superinfection (data not shown).
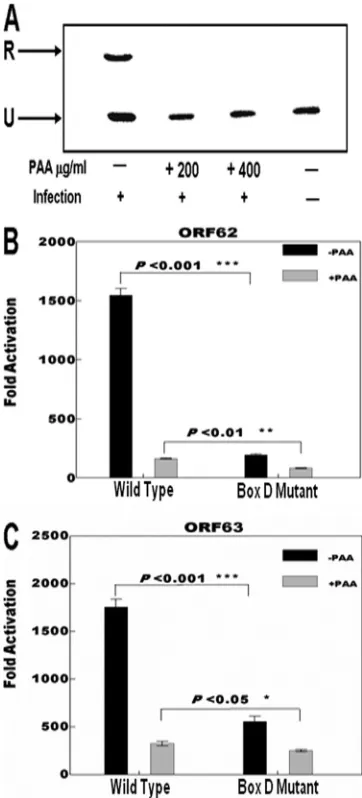
The Box D sequence is involved in expression of the ORF62 and ORF63 genes.Experiments next were performed to deter-mine if Box D and origin-dependent DNA replication influ-ence the expression of the flanking ORF62 and ORF63 genes. We studied the effects of the Box D mutation in the context of superinfection following the transfection of pLitmus R62/63F reporter plasmid in the presence and absence of phosphono-acetic acid (PAA). DpnI replication assays using the wild-type plasmid confirmed that PAA at concentrations of 200 and 400
g/ml completely inhibited DNA replication under our exper-imental conditions (Fig. 4A). Luciferase reporter assays exam-ining the effect of the Box D mutation on the transcription of the OriS-flanking genes with VZV superinfection using pLitmus R62/63F wild-type and mutant plasmids were per-formed in the absence and presence of 400g/ml of PAA. The results are shown in Fig. 4B and C. The activities of the pro-FIG. 2. Results of DpnI replication assays performed with the
pVO2 plasmid. (A) The sequences of Box D and Box A and the core CGC motif mutation. (B) Southern blot analysis of control assays with wild-type pVO2, pVO2 containing the core Box A CGC triplet, and the parental pAT153 plasmid which lacks the VZV OriS sequences. The upper band (R) indicates the position of DpnI-resistant DNA resulting from replication. The lower band (U) indicates the position of unreplicated input plasmid. (C) Typical results showing the South-ern blot analysis of the effects of the mutation of the core CGC motif within Box D. (D) Histogram summarizing the data from three inde-pendent DpnI replication assays comparing the effects of the mutation of Box D to the level of replication observed with the wild-type se-quence. Cells were harvested 48 h after superinfection. Error bars indicate standard errors. Statistical significance was determined by a one-way analysis of variance followed by Tukey’spost hoctest.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.41.282.66.336.2]moter in the presence of VZV infection are reported as the induction (n-fold) of the luciferase activities in reference to the basal activity observed without infection.
The mutation of Box D decreased the expression of the ORF62 and ORF63 luciferase reporters (8- and 3-fold, respec-tively) compared to the wild-type levels in the absence of PAA. In the presence of PAA, the activity of the ORF62 reporter was reduced 8-fold in the case of the wild-type sequence. The Box D mutation reduced the activity an additional 50% under these conditions. Wild-type levels of the ORF63 reporter were
[image:5.585.90.235.69.422.2]reduced 5- to 6-fold in the presence of PAA, and as was the case with the ORF62 reporter, the presence of the Box D mutation further reduced the ORF63 reporter activity by 30 to 40%. These data indicate that Box D influences the expression FIG. 3. Results of DpnI replication assays performed using the
pLitmus R62/63F plasmid. (A) Typical results showing the Southern blot analysis of the effects of the site-specific mutation of Box D. Wild type refers to the pLitmus R62/63F plasmid containing the full inter-genic region between the coding sequences of ORF62 and ORF63 including the full OriS. Box D mutant refers to pLitmus R62/63F containing a triple-point mutation of the core CGC triplet within Box D. Box A mutant refers to pLitmus R62/63F containing a triple-point mutation of the core CGC triplet within Box A. pLitmus Ren/FF indicates the parental pLitmus Ren/FF lacking the intergenic region between the ORF62 and ORF63 genes. The upper band (R) indicates the position of DpnI-resistant DNA resulting from replication within the MeWo cells. The lower band (U) indicates the position of unrep-licated input plasmid. (B) Histogram summarizing the data from three independent DpnI replication assays analyzed at 48 h after VZV su-perinfection. Error bars indicate standard errors. Statistical signifi-cance was determined by a one-way analysis of variance followed by Tukey’spost hoctest.
FIG. 4. Effect of DNA replication and Box D mutation on flanking gene expression. (A) Typical data from Southern blot analysis of DpnI replication assays with wild-type pLitmus R62/63F showing the effect of the presence of 200 and 400g/ml of phosphonoacetic acid (PAA) on VZV origdependent DNA replication. The upper band (R) in-dicates the position of DpnI-resistant DNA resulting from replication within the MeWo cells. The lower band (U) indicates the position of unreplicated input plasmid. (B) Results of triplicate assays comparing the effects of the presence of the Box D mutation on the expression levels of theRenillaluciferase reporter gene present at the position of the ORF62 gene. (C) Results of triplicate assays comparing the effects of the presence of the Box D mutation on the expression levels of the firefly luciferase reporter gene present at the position of the ORF63 gene. The promoter activities resulting from the presence of VZV superinfection are reported as the induction (n-fold) of the luciferase activity above the basal level in the absence of infection. The black bars represent experiments done in the absence of PAA, and the gray bars represent experiments done in the presence of 400g/ml PAA. Sta-tistical significance was determined by a one-way analysis of variance followed by Tukey’spost hoctest. Error bars indicate standard errors.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.329.510.70.469.2]of both the ORF62 and ORF63 genes during viral replication and confirm that origin-dependent DNA replication is impor-tant for high levels of flanking gene transcription. The 150- to 300-fold levels of activation of the ORF62 and ORF63 report-ers above basal levels observed with superinfection in the ab-sence of DNA replication presumably are due to the preab-sence of VZV immediate-early and, possibly, early proteins which are expressed prior to VZV DNA replication (52). The results also show that there is a small, but statistically significant, loss of expression of both reporter genes upon the mutation of Box D in the absence of DNA replication.
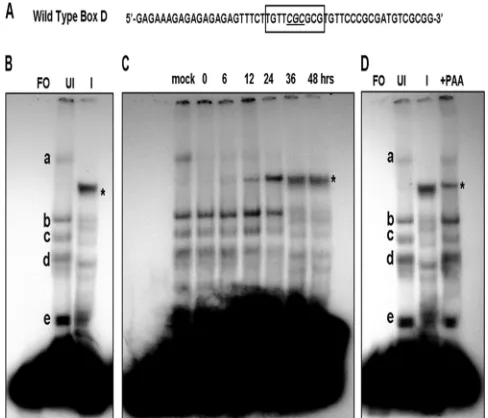
A specific and infected cell complex is formed with the VZV Box D sequence.Based on the results described above,
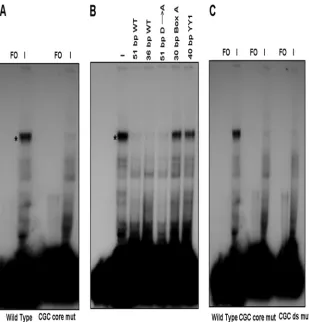
exper-iments were performed to determine if specific infected cell proteins or protein complexes could interact with Box D. In the first series of experiments, EMSAs were performed using a 51-bp duplex oligonucleotide probe containing the 10-bp Box D site and upstream and downstream flanking sequences (Fig. 5A and Table 1). Shift patterns were obtained using uninfected and VZV-infected MeWo cell nuclear extracts. There was a significant difference in the pattern of complexes obtained with the two extracts. Five complexes (a through e) were consis-tently observed with uninfected nuclear extracts (Fig. 5B). The majority of the signals for these complexes were relatively weak, suggesting that the cellular factors involved were present in low abundance or have weak affinity for the Box D-contain-ing oligonucleotide. In contrast, one major complex was formed in the presence of VZV-infected nuclear extracts, while the signal from the uninfected complexes was signifi-cantly diminished. Time-course analysis of VZV-infected cell extracts showed that the infected cell complex was observed at low levels in the extracts at 12 h postinfection. It was the major complex formed 24 h postinfection and beyond with the con-comitant loss of the uninfected cell complexes (Fig. 5C). The complex also was formed using cell extracts infected for 48 h that were derived from cells treated with PAA. However, the amount of complex observed was lower than that seen with untreated extracts, and several uninfected cell complexes also were observed (Fig. 5D). Thus, while DNA replication was not required for the formation of the infected cell Box D complex, it is necessary for the higher levels observed with infected cell extracts not treated with PAA.
Sequence requirements for infected cell complex formation.
EMSA experiments were performed using the wild-type and mutant Box D 51-bp oligonucleotide probes (Table 1) to de-termine the effect of the CGC-to-AAA mutation on the for-mation of the infected cell complex. As shown in Fig. 6, the levels of the infected cell complex observed with the mutant probe were significantly lower than those observed with the wild-type probe. The quantification of the results from three independent experiments indicated that levels were reduced to approximately 25% of those observed with the wild-type se-quence (Table 2). Thus, the formation of the infected cell complex with the wild-type and mutant probes correlated with the effects seen on origin-dependent DNA replication and flanking gene expression. However, the residual levels of com-plex formed with the mutant probe suggested that sequences flanking Box D also are important.
[image:6.585.40.284.70.279.2]Competition EMSAs were performed to test the flanking FIG. 5. EMSAs using a 51-bp probe containing the wild-type Box
D sequence and flanking regions in the presence of uninfected and VZV-infected MeWo cell nuclear extracts. (A) Sequence of the probe showing the position of Box D. The core CGC triplet is underlined. (B) Detection of complexes formed in the presence of uninfected and infected protein extracts. Letters indicate the major complexes iden-tified with uninfected cell extracts. The characteristic major complex formed in the presence of infected cell extracts is indicated by the asterisk. FO, free oligonucleotide; UI, uninfected MeWo cell nuclear extract; I, VZV-infected nuclear extract. (C) Time course showing the development of the infected-cell complex and loss of the uninfected cell complexes during a 48-h period. (D) EMSA experiment done in the presence of 400g/ml PAA showing that low levels of the infected cell complex form in the absence of DNA replication.
TABLE 1. Sequences of oligonucleotide probes used in EMSA and competition assays
Oligonucleotideb Sequencea
51-bp wild type (1)...5⬘-GAGAAAGAGAGAGAGAGTTTCTTGTTCGCGCGTGTTCCCGCGATGTCGCGG-3⬘ 36-bp wild type (1, 2) ...5⬘-AGTTTCTTGTTCGCGCGTGTTCCCGCGATGTCGCGG-3⬘
51-bp D3A Box D (1) ...5⬘-GAGAAAGAGAGAGAGAGTTTCTCGTTCGCACTTGTTCCCGCGATGTCGCGG-3⬘ 30-bp Box A (1, 2)...5⬘-GTCCAACCACCGTTCGCACTTTCTTTCTAT-3⬘
40-bp YY1 site (1, 2) ...5⬘-ATATATATTCCAAATGGAGCGGCAGGCTTTTTAAAATCGA-3⬘
CGC core mut (1) ...5⬘-GAGAAAGAGAGAGAGAGTTTCTTGTTAAAGCGTGTTCCCGCGATGTCGCGG-3⬘ CGC ds mut (1) ...5⬘-GAGAAAGAGAGAGAGAGTTTCTTGTTCGCGCGTGTTCCAAAGATGTCGCGG-3⬘
a
The positions and limits of Box D and Box A are indicated by boldface. The core CGC motif within Box D and a second CGC motif in the downstream flanking sequence are underlined. The site-specific mutations are indicated in underlined italics. The position of the YY1 site is indicated in italics.
b
Numbers in parentheses refer to sequences of the Dumas strain (1) and pOka strain (2).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.45.541.615.700.2]sequence requirements for the formation of the infected cell Box D complex. Competitors (Table 1) included the wild-type 51-bp probe, a 51-bp probe with the Box A sequence substi-tuted for the Box D sequence, a 36-bp probe lacking the up-stream GA-rich sequence, and a 30-bp probe containing the box A sequence and its flanking regions. A 40-bp probe con-taining the YY1 site that is present downstream of Box D (28) was used as a nonspecific competitor. Typical EMSA results
[image:7.585.136.445.67.389.2]are shown in Fig. 6B. These experiments were performed in triplicate, and the relative amount of the infected cell complex in the presence of the various competing oligonucleotides was quantified relative to the amount of complex formed in the absence of competitor. The results are presented in Table 2 and show that under the experimental conditions used, the presence of the cold Box D wild-type probe reduced the level of the infected cell complex to 13% of that observed in the absence of competitor. The 36-bp Box D probe lacking the 5⬘ GA-rich stretch reduced complex formation to levels similar to those seen with the full-length wild-type competitor, indicating that the GA-rich sequence plays no role in the formation of the infected cell complex. Also, the use of the 51-bp competitor probe containing the Box D-to-Box A substitution with the Box D flanking sequence reduced levels to 36%. The wild-type Box A probe and the probe containing the YY1 site both showed essentially no ability to compete. Although both the 51-bp Box D-to-Box A substitution probe and the wild-type Box A probe contain the core CGC motif of the Box D site, only the 51-bp Box D-to-Box A substitution probe still has the ability to com-pete for the formation of the infected cell complex. These results suggest that the upstream and downstream flanking sequences of the Box D site play a role in the formation of this FIG. 6. EMSAs examining the sequence requirements for the formation of the infected-cell complex. Wild-type, mutant, and competitor probes are listed in Table 1. (A) Typical data from EMSA experiments using the Box D probe and the Box D CGC core mutant probe. The major infected-cell complex is indicated by the asterisk. FO, free oligonucleotide; I, VZV-infected nuclear extract. (B) Typical data from competition assays with a variety of mutated and truncated Box D-containing probes and controls. The sequences of the competing probes are listed in Table 1. All cold competing probes were present at a concentration 200-fold in excess of that of the labeled probe. (C) Typical data showing the effect of mutation of the downstream CGC motif compared to that of the Box D core CGC motif.
TABLE 2. Quantification of residual infected cell complex remaining in the presence of competing cold duplex
oligonucleotide probes or upon mutation of the core and downstream CGC motifs
Competitor/mutationa
% Residual complex
51-bp wild type (b)...100.0⫾0.0 51-bp wild type (c) ... 13.2⫾5.9 51-bp D3A Box D (c) ... 36.5⫾10.5 36-bp wild type (c) ... 18.2⫾7.7 30-bp Box A (c)... 95.8⫾7.3 40-bp YY1 site (c) ... 97.6⫾10.5 CGC core mut (b)... 25.6⫾7.8 CGC ds mut (b) ... 26.7⫾8.4
a
(b), Binding probe; (c), competition probe.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.43.283.624.716.2]complex in addition to the core CGC motif. Since the compe-tition assay done with the 36-bp short Box D probe showed that the upstream flanking sequence (GA-rich region) has no role in the formation of this complex, the CGC motifs in the downstream flanking sequence may influence complex forma-tion.
To test this hypothesis, a 51-bp probe with the first down-stream CGC motif mutated to AAA was used in EMSAs. As shown in Fig. 6C, the mutation of the first downstream CGC motif resulted in a loss of complex formation similar to that observed upon the mutation of the core CGC motif within Box D. In contrast, when a probe lacking the second downstream CGC motif was used in competition assays, no loss of complex formation was observed (data not shown).
The VZV ORF29 protein is present in the infected cell Box D complex.The kinetics of the appearance of the infected cell complex indicated that the protein or proteins present therein
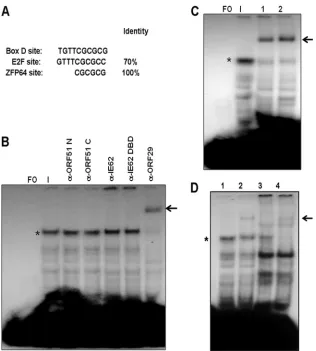
[image:8.585.134.450.66.417.2]were VZV-encoded proteins, cellular proteins that interact with viral proteins, cellular proteins modified by infection, or a mixture of these possibilities. The examination of the sequence of Box D and the flanking regions relevant to the formation of the infected cell complex revealed that the Box D sequence showed 70% homology to the consensus binding site for the E2F family of cellular transcription factors (Fig. 7A). However, in EMSAs using antibodies against E2F1, E2F2, E2F3, E2F4, and E2F5 obtained from two different commercial sources, we were unable to demonstrate either the supershifting or disrup-tion of the major complex observed with infected cell extracts (data not shown). We also were unable to show competition with a 25-bp probe containing the E2F consensus binding site (63). Thus, despite the fact that immunoblot analysis con-firmed the presence of each of these E2F family members in infected cell extracts (data not shown), we have no evidence indicating that they are present in the major infected cell FIG. 7. Identification of the VZV ORF29 single-strand binding protein as a component of the major infected cell complex. (A) Sequence of Box D compared to the consensus sequences of the cellular E2F transcription factor family and the ZFP64 cellular transcription factor which binds to CGCG runs. (B) EMSA supershift experiments using antibodies directed against the N and C termini of the VZV ORF51 origin-binding protein, full-length IE62 and the IE62 DNA-binding domain (DBD), and the C terminus of the VZV ORF29 protein. FO, free oligonucleotide. The major infected cell extract is indicated by an asterisk. The shifted complex is indicated by an arrow. (C) Supershift assays with the anti-ORF29 antiserum performed with different orders of addition. Lane 1, (protein extract plus antibody) plus probe; lane 2, (probe plus protein extract) plus antibody. (D) Typical results from supershift experiments using 0.5l of the ORF29 antiserum and nuclear protein extracts from infected cells incubated with and without 400g/ml PAA. Lane 1, EMSA with infected cell extract; lane 2, supershift with anti-ORF29 antisera; lane 3, EMSA with PAA-treated infected cell extract; lane 4, supershift with anti-ORF29 antisera. The infected cell complex is indicated by an asterisk, and the supershifted complex is indicated by an arrow.
on November 7, 2019 by guest
http://jvi.asm.org/
complex. Since CGC motifs within Box D and its downstream flanking sequence both were important for complex formation, antibodies against the cellular transcription factor Zfp64 also were used in EMSAs. This protein is a member of the Kruppel-associated box (KRAB) family and is a zinc finger protein with a consensus binding motif of CGCG (46). As with the E2F transcription factor family, no supershifts or disruption of com-plexes was observed (data not shown).
A second series of EMSA experiments was performed using a panel of antibodies specific for VZV gene products synthe-sized during the immediate-early and early phases of VZV replication (52). These included antibodies raised during the course of this work against 9-amino-acid peptides correspond-ing to the N terminus (amino acids 1 to 9) and 20-amino-acid peptides corresponding to the C terminus (amino acids 728 to 747) of the VZV ORF51 OBP, polyclonal antisera against full-length IE62 and the IE62 DBD (62), polyclonal antisera raised against a peptide corresponding to the C terminus of the VZV ORF29 single-stranded binding protein (30), polyclonal antisera raised against full-length IE63 (67), and antisera against the C terminus of the HSV-1 UL9 OBP (5, 6). No supershifts or disruption of complexes were observed with the anti-UL9 and anti-IE63 antisera (data not shown). Similarly, no shifts or disruption of either the major infected cell complex or several faster migrating complexes was observed with the anti-ORF51 and anti-IE62 antibodies (Fig. 7B). In contrast, the major complex was shifted by the anti-ORF29 antisera. This effect was independent of the order of the addition of the antisera to the other components of the reaction mixture (Fig. 7B and C). These results concerning the presence of the ORF29 protein within the major complex are consistent with the formation of the major complex in the presence of PAA (Fig. 5D), since the ORF29 protein would be synthesized prior to the onset of DNA replication (52, 53). Based on this, gel shifts were performed with protein extracts derived from PAA-treated infected cells, and as predicted, the ORF29 anti-sera supershifted the complex formed with these extracts, which migrated in the position of the major complex formed with non-PAA-treated extracts (Fig. 7D).
DISCUSSION
The function or functions of the partial OBP binding site downstream of the AT-rich stretch within the VZV OriS re-mained unknown prior to the work presented here. Since the VZV OBP did not bind to this sequence (7, 59), which we have designated Box D, it was not clear if the sequence was involved in origin-dependent replication. The plasmids used in the orig-inal identification of the minimal VZV OriS, however, con-tained Box D (Fig. 1). We therefore wished to determine if this sequence was involved in origin-dependent replication and/or in the expression of the ORF62 and ORF63 genes which flank the VZV OriS. The data obtained from DpnI replication as-says using plasmids containing VZV OriS sequences showed that Box D acted as a negative regulator of origin-dependent replication in both the absence and presence of transcribable flanking genes. The levels of increase of replication also were very similar in both cases. Thus, the mechanism of DNA rep-lication inhibition appears to be independent of transcription. A second interesting finding from these experiments is that
based on comparing the ratios of replicated to unreplicated plasmids shown in Fig. 2D and 3B, the presence of transcrib-able flanking genes appeared to slightly decrease the level of replication efficiency observed. This is true for both the wild-type and Box D mutant reporter plasmids.
This finding could be attributed to several factors. For ex-ample, the OriS structure within the pVO2 plasmid, which lacks the flanking gene promoter elements, is from the Dumas strain, while the pLitmus plasmid contains the pOka OriS. In addition, there are structural differences between the two or-igins in terms of the TA and GA repeats, which also could affect the replication efficiency of the two plasmids (18, 50, 57). The TA repeat ranges from (TA)16in Dumas strain to (TA)12 in pOka and (TA)9 in vOka. Stow and Davison (57) showed
that the deletion of (TA)6in the Dumas strain resulted in a 55% decrease in replication efficiency. The presence of shorter TA repeats in vOka and pOka could explain attenuation com-pared to the Dumas strain in Stow’s experiment, although the difference in the length of the GA repeats is another variable factor. The Dumas strain has (GA)6, whereas Oka strains have
longer GA repeats, (GA)10in pOka and (GA)9in vOka. The function of the GA repeat in origin-dependent DNA replica-tion still is unknown, but the presence of longer GA repeats in vOka and pOka may compensate for the presence of short TA repeats compared to that of the Dumas strain. However, the DNA sequences of both Box A and Box D sites and their proximal flanking regions are identical in Dumas, pOka, and vOka (18). In addition, in the four clades used by Peters et al. (50), the nucleotide differences at or near the minimal origin are not present in the Box A or Box D sequence. Finally, these data suggest that the presence of active gene transcription has a negative effect on DNA replication efficiency.
We expected to have lower gene expression of both ORF62 and ORF63 reporters, by about 25 to 30% in the presence of PAA, because of less available total plasmid in its presence compared to that in its absence. However, the presence of PAA caused 8- and 5-fold reductions in reporter gene expres-sion with the wild-type plasmid for the Renilla (ORF62) and firefly (ORF63) activities compared to expression in the ab-sence of PAA. Thus, the decrease in reporter gene expression is not a simple linear correlation with the decrease in the amount of replication of the reporter plasmid. These results suggest that in the model system used here, origin-dependent DNA replication and flanking gene transcription are coupled. These data also reinforce and add additional validation to the findings of Jones et al. (26). In that study, the ORF62/63 intergenic region pLitmus reporter cassette, including the OriS sequences, was inserted into a nonnative site in the VZV genome. Thus, it was not clear if replication was initiated at the OriS within the pLitmus cassette and if the apparent influence of OriS or elements proximal to OriS was influenced by repli-cation. However, it was shown that the presence of elements within or near the minimal VZV origin sequences had a sig-nificant effect on flanking gene expression. Here, we have clear proof of replication or the lack thereof and the fact that OriS-dependent DNA replication influences flanking gene expres-sion. These findings are in contrast to those with HSV-1, where the presence of the DNA replication origins had no influence on flanking gene expression (43, 60).
The question of whether Box D is aciselement influencing
on November 7, 2019 by guest
http://jvi.asm.org/
transcription in a DNA replication-independent manner was addressed by the examination of the effects of the mutation of the CGC core motif on flanking gene expression in the pres-ence and abspres-ence of PAA concentrations which inhibit DNA replication. This strategy was chosen to allow the expression of immediate-early and early gene products without the disrup-tion of potentialcis-acting transcription regulatory elements, which might occur in the case of the deletion or site-specific mutagenesis of OriS sequences essential for replication. The mutation of the core CGC motif had a major effect on the expression of the flanking reporter genes in the pLitmus plas-mid (Fig. 4B and C). Some of the change likely is attributable to the increase in DNA replication efficiency observed in the DpnI replication assays, which would reduce the ability of the cellular transcription apparatus and VZV transcription pro-teins to gain access or remain bound to the DNA. This finding suggests that the Box D site is acting as a second coupling point between VZV origin-dependent replication and flanking gene expression. Also, Box D mutation resulted in small but repro-ducible and statistically significant decreases in the expression of both reporter genes in the presence of PAA compared to results of the experiment done with the wild-type reporters under the same conditions. Thus, Box D may play a role in flanking gene expression which is distinct from its role in DNA replication during the immediate-early and early phases of VZV gene expression. However, the major role of Box D appears to be the downregulation of origin-dependent DNA replication efficiency.
Four possibilities existed for proteins which bind to the Box D sequence and, through that interaction, influence VZV DNA replication: (i) cellular proteins, (ii) virus-encoded pro-teins, (iii) cellular proteins modified by virus infection, or (iv) a combination of cellular and VZV-encoded proteins. A sec-ond consideration was that sequences flanking Box D also were important for the effects observed. Of particular interest was whether the GA-rich stretch characteristic of the VZV origin and immediately downstream of the AT-rich sequence was involved. The EMSAs indicated that a distinct infected-cell complex was formed with duplex DNA probes containing Box D. Thus, only the first possibility regarding the nature of the proteins binding to Box D was eliminated. The data from the competition assays (Fig. 6B and Table 2) clearly indicated that the GA-rich stretch played no role in the formation of the complex. In contrast, the data strongly suggested that the downstream 18 bp which contained two additional CGC motifs contributed equally to the formation of the infected cell com-plex. Since additional competition assays indicated that oligo-nucleotides lacking the more distal of the two sites did not compete, it appeared that the first downstream CGC motif was equally important for complex formation. This was confirmed by the mutation of this CGC motif. Thus, at least one protein which binds to sequences containing CGC appears to be in-volved in the formation of the infected cell complex.
Time-course analysis of the formation of the infected cell complex showed that it begins to appear at 12 h postinfection and persists and increases in amount throughout the remain-der of infection. This indicates that a VZV function expressed during the immediate-early or early phases of infection either binds directly to Box D or interacts with or modifies a cellular factor, resulting in cellular factor binding. The EMSA
experi-ments performed with extracts from PAA-treated infected cells confirmed this and indicated that DNA replication is not required for the formation of the complex.
Due to the high level of homology of Box D with Box A, the obvious candidate was the VZV ORF51 OBP. However, no interaction of the recombinant OBP fragments with Box D was seen in previous work (7, 59). Work from the Schaffer labora-tory found that variant C-terminal cleavage products of the HSV-1 UL9 OBP produced in infected cells bind to origin sequences. These variants affect HSV-1 pathogenesis in mice and may be involved in the switch from origin-dependent to origin-independent DNA replication (34, 35). However, super-shift experiments using antibodies directed against VZV ORF51 and the C terminus of HSV-1 UL9 (with which ORF51 shares 44% homology) failed to demonstrate the interaction of ORF51 or ORF51 fragments present in VZV-infected cell extracts with the Box D-containing probe.
The VZV ORF29 single-stranded DNA-binding protein was another candidate for inclusion in the major infected cell com-plex. The kinetics of the expression of the ORF29 protein are reminiscent of the formation of the major complex (52, 53). Further, while the ORF29 protein interacts with single-stranded DNA and is involved in viral DNA replication, it also has been shown to modulate and interact with the VZV ORF67 (gI) promoter in a cell type- and sequence-specific manner. The presence of the ORF29 protein in transient-transfection assays increased the IE62-dependent activation of the promoter in T cells (4, 21). These results were validated using recombinant virus strains containing mutations in an ORF29-responsive element (29RE) within gI promoter se-quences in the SCID mouse model of VZV pathogenesis (24). Our results clearly show that the ORF29 protein is a compo-nent of the major infected cell complex formed with the Box D-containing probe. It is present within the complex at both late times during infection and in complexes formed using PAA-treated infected cells. Thus, proteins produced or mod-ified during or following DNA replication are not required for complex formation.
The analysis of the Box D sequence for specific cellular transcription factor binding sites (Ali Baba 2 and TransFac; BIOBASE Biological Databases) showed that Box D has 70% homology with the consensus site for the E2F family of tran-scription factors. The identical portions of the sequence in-clude the CGC motif and the CGC-to-AAA mutation that resulted in the loss of more than 80% of the infected-cell complex also was predicted to ablate the binding of the E2F family factors. E2F1 has been shown to be required for the productive infection of bovine herpesvirus 1 (63). E2F family members also are modified during HSV-1 infection (1) and influence HSV-1 replication (17, 23, 44). However, no evi-dence for the interaction of E2F1, E2F2, E2F3, E2F4, or E2F5 with the Box D-containing probe was found via supershift experiments or in competition assays. Computer analysis iden-tified the cellular factor Zfp64, which is involved in cellular differentiation (54), as a strong candidate, since the Zfp64 consensus binding site is CGCG. However, as was the case with the E2F family members, no evidence of the presence of Zfp64 in the infected-cell complex was detected.
We cannot exclude the presence of cellular factors or cellu-lar factors modified by infection with the ORF29 protein
on November 7, 2019 by guest
http://jvi.asm.org/
within the infected cell complex based on our results. Since there is no sequence homology between the Box D probe and the VZV gI 29RE, it is possible that cellular factors interacting with the ORF29 protein can influence its duplex DNA-binding activity and confer sequence specificity either in certain cell types or at various stages of the viral replication cycle. Thus far, attempts to identify other potential components of the major Box D complex by enriching for the infected cell extract Box D-binding activity using ammonium sulfate precipitation and affinity chromatography have resulted in the loss of binding, further suggesting that a complex rather than a single protein species is involved in the binding.
VZV origin-dependent DNA replication currently is be-lieved to involve a classic theta structure following the activa-tion of the origin (Fig. 8). The VZV ORF62 and ORF63 genes flank the OriS and are transcribed in opposite directions from the same strand of DNA (14). Thus, collisions would occur between the DNA replication and transcription machineries. These collisions, based on the VZV genome arrangement, would be codirectional rather than head on. It has been shown in bacteria that codirectional collisions are less disruptive than head-on collisions (39, 40, 51); however, these collisions can result in the disruption of both replication and (primarily) transcription complexes. The VZV ORF62 and ORF63 genes, while expressed at immediate early times are, unlike their
HSV-1 homologues, also expressed throughout the viral repli-cation cycle and are incorporated into the viral particle (12, 29, 52). Therefore, one possible function of the ORF29 protein and the other component(s) of the Box D binding complex during lytic infection could be to slow down replication fork movement, allowing more efficient and extended expression of the IE62 and IE63 proteins during the viral replication cycle (Fig. 8). Such a model would be in agreement with the data presented here showing that the mutation of Box D results in increased DNA replication and decreased reporter gene ex-pression. Alternatively, since the amount of Box D complex increases with time, it could sufficiently shut down or could stall replication fork movement to allow the switch from origin-dependent to origin-inorigin-dependent DNA replication.
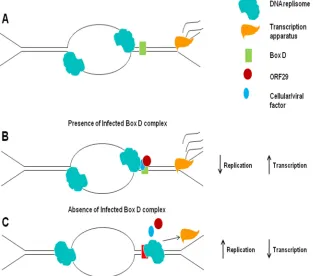
[image:11.585.137.450.76.352.2]Additional functions of the ORF29 protein (as a component of the infected Box D complex) and of the Box D sequence may be in the establishment of latent infection. This suggestion follows from the findings of Cohen et al. (11), who found that the overexpression of the ORF29 protein impaired the estab-lishment of latency in a rodent model. Some limited replication of VZV is believed to occur in DRG neurons prior to the establishment of latency, resulting in the presence of multiple copies of the viral genome per neuron (27). Based on the model presented here, the overexpression of the ORF29 pro-tein could result in a premature shutdown of origin-dependent FIG. 8. Model for the role played by the VZV ORF29 protein in origin-dependent DNA replication and flanking gene expression. (A) Sche-matic of the theta structure. (B) In the presence of the infected Box D-containing complex, the stalling of the replication fork would occur, which would result in a physical barrier to fork movement in one direction and likely changes in the DNA structure resulting from the inhibition of the OriS opening at the fork moving in the opposite direction. This would result in increased transcription and decreased replication and could be enhanced by higher concentrations of the ORF29 protein. (C) In the absence of the infected Box D complex, there is the removal of the physical protein barrier formed at Box D, resulting in increased replication, followed by the head-to-tail collision of the DNA replisome and transcription machinery, resulting in decreased transcription. The frequency of these collisions would be enhanced by mutations in Box D that eliminated the binding of the infected cell complex.
on November 7, 2019 by guest
http://jvi.asm.org/
replication and therefore insufficient copies of the genome being replicated prior to other steps in the establishment of latency. Zerboni et al. (65, 66) have shown that ORF62 and ORF63 transcription persists in human fetal neurons infected with VZV in the SCID mouse model of VZV pathogenesis. The presence of the Box D-binding complex in neurons, as described above, slows down but does not completely stop DNA replication while facilitating the expression of ORF62 and ORF63 gene products. This slowing down of DNA repli-cation might allow chromatin association with the viral DNA, leading to the ultimate shutdown of productive viral replica-tion.
ACKNOWLEDGMENTS
This work was supported by grant AI18449 from the National Insti-tutes of Health and grants from the John R. Oishei Foundation and the National Shingles Foundation.
REFERENCES
1.Advani, S. J., R. R. Weichselbaum, and B. Roizman.2000. E2F proteins are posttranslationally modified concomitantly with a reduction in nuclear
bind-ing activity in cells infected with herpes simplex virus 1. J. Virol.74:7842–
7850.
2.Ambagala, A. P., T. Krogmann, J. Qin, L. Pesnicak, and J. I. Cohen.2010. A varicella-zoster virus mutant impaired for latency in rodents, but not
impaired for replication in cell culture. Virology399:194–200.
3.Balliet, J. W., and P. A. Schaffer.2006. Point mutations in herpes simplex virus type 1 oriL, but not in OriS, reduce pathogenesis during acute infection
of mice and impair reactivation from latency. J. Virol.80:440–450.
4.Boucaud, D., H. Yoshitake, J. Hay, and W. T. Ruyechan.1998. The varicella-zoster virus (VZV) open-reading frame 29 protein acts as a modulator of a
late VZV gene promoter. J. Infect. Dis.178:S34–S38.
5.Chattopadhyay, S., and S. K. Weller.2006. DNA binding activity of the herpes simplex virus type 1 origin binding protein, UL9, can be modulated by sequences in the N terminus: correlation between transdominance and DNA
binding. J. Virol.80:4491–4500.
6.Chattopadhyay, S., and S. K. Weller.2007. Direct interaction between the N-and C-terminal portions of the herpes simplex virus type 1 origin binding
protein UL9 implies the formation of a head-to-tail dimer. J. Virol. 81:
13659–13667.
7.Chen, D., and P. D. Olivo. 1994. Expression of the variella-zoster virus origin-binding protein and analysis of its site-specific DNA-binding
proper-ties. J. Virol.68:3841–3849.
8.Cohen, J. I., E. Cox, L. Pesnicak, S. Srinivas, and T. Krogmann.2004. The varicella-zoster virus open reading frame 63 latency-associated protein is
critical for establishment of latency. J. Virol.78:11833–11840.
9.Cohen, J. I., D. Heffel, and K. Seidel.1993. The transcriptional activation domain of varicella-zoster virus open reading frame 62 protein is not
con-served with its herpes simplex virus homolog. J. Virol.67:4246–4251.
10.Cohen, J. I., T. Krogmann, S. Bontems, C. Sadzot-Delvaux, and L. Pesnicak.
2005. Regions of the varicella-zoster virus open reading frame 63 latency-associated protein important for replication in vitro are also critical for
efficient establishment of latency. J. Virol.79:5069–5077.
11.Cohen, J. I., T. Krogmann, L. Pesnicak, and M. A. Ali.2007. Absence or overexpression of the varicella-zoster virus (VZV) ORF29 latency-associ-ated protein impairs late gene expression and reduces VZV latency in a
rodent model. J. Virol.81:1586–1591.
12.Cohen, J. I., S. E. Straus, and A. M. Arvin.2007. Varicella-zoster virus, p.
2773–2818.InD. M. Knipe et al. (ed.), Fields virology, 5th ed. Lippincott
Williams & Wilkins, Philadelphia, PA.
13.Davison, A. J., and J. E. Scott.1985. DNA sequence of the major inverted
repeat in the varicella-zoster virus genome. J. Gen. Virol.66:207–220.
14.Davison, A. J., and J. E. Scott.1986. The complete DNA sequence of
varicella-zoster virus. J. Gen. Virol.67:1759–1816.
15.Deb, S., and M. Doelberg.1988. A 67-base-pair segment from the Ori-S region of herpes simplex virus type 1 encodes origin function. J. Virol.
62:2516–2519.
16.Desloges, N., M. Rahaus, and M. H. Wolff.2005. The varicella-zoster virus-mediated delayed host shutoff: open reading frame 17 has no major function, whereas immediate-early 63 protein represses heterologous gene expression.
Microbes Infect.7:1519–1529.
17.Ehmann, G. L., H. A. Burnett, and S. L. Bachenheimer.2001. Pocket protein p130/Rb2 is required for efficient herpes simplex virus type 1 gene expression
and viral replication. J. Virol.75:7149–7160.
18.Gomi, Y., et al.2002. Comparison of the complete DNA sequences of the
Oka varicella vaccine and its parental virus. J. Virol.76:11447–11459.
19.Grose, C., et al.2004. Complete DNA sequence analyses of the first two varicella-zoster virus glycoprotein E (D150N) mutant viruses found in North America: evolution of genotypes with an accelerated cell spread phenotype.
J. Virol.78:6799–6807.
20.Hay, J., and W. T. Ruyechan.2007. Alphaherpesvirus DNA replication, p.
138–143.InA. Arvin et al. (ed.), Human herpesviruses: biology, therapy, and
immunoprophylaxis, vol. 1. Cambridge University Press, Cambridge, MA. 21.He, H., D. Boucaud, J. Hay, and W. T. Ruyechan.2001. Cis and trans
elements regulating expression of the varicella zoster virus gI gene. Arch.
Virol. Suppl.17:57–70.
22.Hernandez, T. R., R. E. Dutch, I. R. Lehman, C. Gustafsson, and P. Elias.
1991. Mutations in a herpes simplex virus type 1 origin that inhibit
interac-tion with origin-binding protein also inhibit DNA replicainterac-tion. J. Virol.65:
1649–1652.
23.Hilton, M. J., et al.1995. Induction by herpes simplex virus of free and
heteromeric forms of E2F transcription factor. Virology213:624–638.
24.Ito, H., et al.2003. Promoter sequences of varicella-zoster virus glycoprotein I targeted by cellular transactivating factors Sp1 and USF determine
viru-lence in skin and T cells in SCIDhu mice in vivo. J. Virol.77:489–498.
25.Jackers, P., et al.1992. Characterization of regulatory functions of the
varicella-zoster virus gene 63-encoded protein. J. Virol.66:3899–3903.
26.Jones, J. O., M. Sommer, S. Stamatis, and A. M. Arvin.2006. Mutational analysis of the varicella-zoster virus ORF62/63 intergenic region. J. Virol.
80:3116–3121.
27.Kennedy, P. G., and R. J. Cohrs.2010. Varicella-zoster virus human
gangli-onic latency: a current summary. J. Neurovirol.16:411–418.
28.Khalil, M. I., J. Hay, and W. T. Ruyechan.2008. The cellular transcription factors Sp1 and Sp3 suppress varicella zoster virus origin-dependent DNA
replication. J. Virol.82:11723–11733.
29.Kinchington, P. R., J. K. Hougland, A. M. Arvin, W. T. Ruyechan, and J.
Hay.1992. The varicella-zoster virus immediate-early protein IE62 is a major
component of virus particles. J. Virol.66:359–366.
30.Kinchington, P. R., et al.1988. Identification and characterization of a varicella-zoster virus DNA-binding protein by using antisera directed against
a predicted synthetic oligopeptide. J. Virol.62:802–809.
31.Kinchington, P. R., J. P. Vergnes, P. Defechereux, J. Piette, and S. E. Turse.
1994. Transcriptional mapping of the varicella-zoster virus regulatory genes
encoding open reading frames 4 and 63. J. Virol.68:3570–3581.
32.Kinchington, P. R., J. P. Vergnes, and S. E. Turse.1995. Transcriptional
mapping of varicella-zoster virus regulatory proteins. Neurology45:S33–S35.
33.Kost, R. G., H. Kupinsky, and S. E. Straus.1995. Varicella-zoster virus gene
63: transcript mapping and regulatory activity. Virology209:218–224.
34.Link, M. A., and P. A. Schaffer.2007. Herpes simplex virus type 1 C-terminal variants of the origin binding protein (OBP), OBPC-1 and OBPC-2, coop-eratively regulate viral DNA levels in vitro, and OBPC-2 affects mortality in
mice. J. Virol.81:10699–10711.
35.Link, M. A., L. A. Silva, and P. A. Schaffer.2007. Cathepsin B mediates cleavage of herpes simplex virus type 1 origin binding protein (OBP) to yield OBPC-1, and cleavage is dependent upon viral DNA replication. J. Virol.
81:9175–9182.
36.Lungu, O., C. A. Panagiotidis, P. W. Annunziato, A. A. Gershon, and S. J. Silverstein.1998. Aberrant intracellular localization of varicella-zoster virus
regulatory proteins during latency. Proc. Natl. Acad. Sci. U. S. A.95:7080–
7085.
37.Lynch, J. M., T. K. Kenyon, C. Grose, J. Hay, and W. T. Ruyechan.2002. Physical and functional interaction between the varicella zoster virus IE63
and IE62 proteins. Virology302:71–82.
38.Martin, D. W., S. P. Deb, J. S. Klauer, and S. Deb.1991. Analysis of the herpes simplex virus type 1 OriS sequence: mapping of functional domains.
J. Virol.65:4359–4369.
39.Merrikh, H., C. Machon, W. H. Grainger, A. D. Grossman, and P. Soultanas.
2011. Co-directional replication-transcription conflicts lead to replication
restart. Nature470:554–557.
40.Mirkin, E. V., and S. M. Mirkin.2007. Replication fork stalling at natural
impediments. Microbiol. Mol. Biol. Rev.71:13–35.
41.Moriuchi, H., M. Moriuchi, and J. I. Cohen.1995. The varicella-zoster virus immediate-early 62 promoter contains a negative regulatory element that
binds transcriptional factor NF-Y. Virology214:256–258.
42.Moriuchi, M., H. Moriuchi, S. E. Straus, and J. I. Cohen.1994. Varicella-zoster virus (VZV) virion-associated transactivator open reading frame 62
protein enhances the infectivity of VZV DNA. Virology200:297–300.
43.Nguyen-Huynh, A. T., and P. A. Schaffer.1998. Cellular transcription factors enhance herpes simplex virus type 1 OriS-dependent DNA replication. J.
Vi-rol.72:3635–3645.
44.Olgiate, J., G. L. Ehmann, S. Vidyarthi, M. J. Hilton, and S. L. Bachen-heimer.1999. Herpes simplex virus induces intracellular redistribution of
E2F4 and accumulation of E2F pocket protein complexes. Virology258:257–
270.
45.Olsson, M., et al.2009. Stepwise evolution of the herpes simplex virus origin
binding protein and origin of replication. J. Biol. Chem.284:16246–16255.
46.Orlov, S. V., et al.2007. Novel repressor of the human FMR1