Multiple Host Factors Interact with the Hypervariable Domain
of Chikungunya Virus nsP3 and Determine Viral Replication in
Cell-Specific Mode
Chetan D. Meshram,aPeter Agback,eNikita Shiliaev,aNadya Urakova,aJames A. Mobley,b,c,dTatiana Agback,f Elena I. Frolova,a Ilya Frolova
aDepartment of Microbiology, University of Alabama at Birmingham, Birmingham, Alabama, USA
bComprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, Alabama, USA
cDepartment of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, USA
dDepartment of Surgery, University of Alabama at Birmingham, Birmingham, Alabama, USA
eDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden
fA&A Structure & Dynamics AB, Uppsala, Sweden
ABSTRACT Alphaviruses are widely distributed in both hemispheres and circulate between mosquitoes and amplifying vertebrate hosts. Geographically separated al-phaviruses have adapted to replication in particular organisms. The accumulating data suggest that this adaptation is determined not only by changes in their glyco-proteins but also by the amino acid sequence of the hypervariable domain (HVD) of the alphavirus nsP3 protein. We performed a detailed investigation of chikungunya virus (CHIKV) nsP3 HVD interactions with host factors and their roles in viral replica-tion in vertebrate and mosquito cells. The results demonstrate that CHIKV HVD is in-trinsically disordered and binds several distinctive cellular proteins. These host fac-tors include two members of the G3BP family and their mosquito homolog Rin, two members of the NAP1 family, and several SH3 domain-containing proteins. Interac-tion with G3BP proteins or Rin is an absolute requirement for CHIKV replicaInterac-tion, al-though it is insufficient to solely drive it in either vertebrate or mosquito cells. To achieve a detectable level of virus replication, HVD needs to bind members of at least one more protein family in addition to G3BPs. Interaction with NAP1L1 and NAP1L4 plays a more proviral role in vertebrate cells, while binding of SH3 domain-containing proteins to a proline-rich fragment of HVD is more critical for virus repli-cation in the cells of mosquito origin. Modifirepli-cations of binding sites in CHIKV HVD allow manipulation of the cell specificity of CHIKV replication. Similar changes may be introduced into HVDs of other alphaviruses to alter their replication in particular cells or tissues.
IMPORTANCE Alphaviruses utilize a broad spectrum of cellular factors for efficient formation and function of replication complexes (RCs). Our data demonstrate for the first time that the hypervariable domain (HVD) of chikungunya virus nonstructural protein 3 (nsP3) is intrinsically disordered. It binds at least 3 families of cellular pro-teins, which play an indispensable role in viral RNA replication. The proteins of each family demonstrate functional redundancy. We provide a detailed map of the bind-ing sites on CHIKV nsP3 HVD and show that mutations in these sites or the replace-ment of CHIKV HVD by heterologous HVD change cell specificity of viral replication. Such manipulations with alphavirus HVDs open an opportunity for development of new irreversibly attenuated vaccine candidates. To date, the disordered protein frag-ments have been identified in the nonstructural proteins of many other viruses. They may also interact with a variety of cellular factors that determine critical as-pects of virus-host interactions.
Received11 May 2018Accepted6 June 2018
Accepted manuscript posted online13 June 2018
CitationMeshram CD, Agback P, Shiliaev N, Urakova N, Mobley JA, Agback T, Frolova EI, Frolov I. 2018. Multiple host factors interact with the hypervariable domain of chikungunya virus nsP3 and determine viral replication in cell-specific mode. J Virol 92:e00838-18.
https://doi.org/10.1128/JVI.00838-18.
EditorSusana López, Instituto de Biotecnologia/UNAM
Copyright© 2018 American Society for
Microbiology.All Rights Reserved.
Address correspondence to Ilya Frolov, [email protected].
C.D.M. and P.A. equally contributed to this study.
crossm
on November 6, 2019 by guest
http://jvi.asm.org/
KEYWORDS BIN1, CD2AP, Eilat virus, G3BP1, G3BP2, NAP1L1, viral RNA replication, alphavirus, chikungunya virus, virus-host interactions
T
heAlphavirusgenus in theTogaviridaefamily contains a wide variety of human and animal pathogens (1). Based on their geographical distribution, they are separated into New World (NW) and the Old World (OW) alphaviruses. In natural circulation, most of the currently known alphaviruses are transmitted by mosquito vectors between vertebrate hosts, in which they induce diseases of different severity (2). The NW alphaviruses, exemplified by Venezuelan (VEEV), eastern (EEEV), and western (WEEV) equine encephalitis viruses, cause a highly debilitating disease. In a wide variety of vertebrate species, including humans, it often results in meningomyeloencephalitis with a frequently lethal outcome (3). Most of the OW alphaviruses are less pathogenic, and their human-associated diseases are characterized by rash, arthritis, and fever (3). Despite a presence on essentially all continents and a significant public health threat, the molecular mechanisms of alphavirus replication and interactions with host cells are insufficiently investigated, and critical aspects of the viral biology remain to be better understood. The importance of the OW alphaviruses was underappreciated for a long time until the recent outbreak of chikungunya fever in both hemispheres with millions of people involved. Chikungunya virus (CHIKV) induces severe polyarthritis character-ized by excruciating pain that frequently continues for several years (4–8).The alphavirus genome is a single-stranded RNA of positive polarity of⬃11.5 kb. It mimics cellular mRNAs in that it has a cap at the 5=terminus and a poly(A) tail at the 3= terminus (9). Upon delivery into the cell, the genome is translated into P123 and P1234, the polyprotein precursors of viral nonstructural (ns) proteins (2). The subse-quent sesubse-quential processing of both ns polyproteins into individual nsPs, nsP1, nsP2, nsP3, and nsP4, differentially regulates the synthesis of the negative-strand RNA intermediates, new viral genomes (G RNA) and subgenomic (SG) RNA (10, 11). The latter RNA is encoded by the 3=one-third of the genome and translated into viral structural proteins, which ultimately form viral particles (2).
The initially synthesized ns polyproteins are targeted to the plasma membrane (PM). This binding to the internal surface of the PM (12) is mediated by specific alpha-helical peptide and palmitoylated amino acids (aa) of nsP1 (13, 14). After the first cleavage event mediated by nsP2-associated protease activity, the initially formed replication complexes (RCs) contain P123 and nsP4. They are capable of synthesis of the negative-strand RNA on the G RNA template to form the double-negative-stranded RNA (dsRNA) repli-cation intermediates (11, 15). The dsRNA synthesis induces the formation of the membrane spherules, the size of which correlates with the length of the original RNA template (16). The subsequent processing of P123 into nsP1⫹P23⫹nsP4 and ultimately into nsP1⫹nsP2⫹nsP3⫹nsP4 transforms spherule-associated RCs into their mature form and makes them active in G and SG RNA synthesis (11, 17, 18). Viral components of RCs, nsPs, exhibit enzymatic activities which are required for viral RNA synthesis. nsP1 and nsP2 facilitate RNA capping, in that nsP2 exhibits RNA triphosphatase activity (19) and nsP1 mediates a cascade of enzymatic reactions which result in the formation of m7GMP, termed Cap(0), at the 5= termini of viral G and SG RNAs (20, 21). nsP2 also
functions as a nucleoside triphosphatase (NTPase) and helicase during replication of viral genomic RNA and transcription of SG RNAs (22–24) and performs all of the steps of P123 and P1234 processing into individual nsPs (18, 25). nsP4 is an RNA-dependent RNA polymerase, the core subunit of viral RC (26–28). Functions of nsP3 in alphavirus replication are not well understood. However, it is absolutely essential for viral RNA replication. nsP3 protein contains three structural domains: two conserved amino-terminal domains that include the macro domain (29) and the Zn-binding alphavirus unique domain (AUD) (30) and the hypervariable carboxy-terminal domain (HVD) (31–33). It is becoming more evident that alphavirus nsP3 proteins, and their HVDs in particular, serve as hubs for assembly of RCs and other large cytoplasmic complexes that are composed of both cellular and viral proteins (34, 35). The initially formed
on November 6, 2019 by guest
http://jvi.asm.org/
nsP3-containing RCs also mediate recruitment of viral G RNA into the replication process (34).
HVDs of the members of different alphavirus serocomplexes demonstrate essentially no identity at the aa level. These domains were proposed to be disordered and were shown to interact with specific host factors (34, 35). The experimental data suggest that HVDs may play critical roles in the adaptation of alphaviruses to replication in different hosts and mosquito vectors (34). Among other host factors, G3BP proteins (G3BP1 and G3BP2) were found to be the most abundantly binding to HVD of all of the currently investigated OW alphaviruses, such as Sindbis (SINV) and Semliki Forest (SFV) viruses (36–39). G3BP-HVD interactions are particularly important for CHIKV replication, be-cause knockout (KO) of bothG3bpgenes in murine cells (G3bpdKO cells) makes them no longer capable of supporting CHIKV replication (34).
G3BP-HVD interaction is mediated by a short repeating peptide located in the carboxy-terminal fragment of CHIKV HVD (34, 38). In this study, we demonstrate that despite the absolute requirement of this interaction for CHIKV RC function, G3BP binding alone is insufficient for driving CHIKV RNA replication. Other fragments of nsP3 HVD determine replication of CHIKV in a cell-specific mode. Genetic manipulations with CHIKV HVD differentially affect virus replication in vertebrate and mosquito cells, and this opens an opportunity for designing cell-specific CHIKV mutants.
RESULTS
CHIKV HVD is a completely disordered protein domain.Recent studies strongly suggested that alphavirus HVDs play critical roles in virus replication by mediating the assembly of protein complexes that facilitate RNA replication and possibly have other activities in virus-host interactions (34). Further understanding of the mechanism of their function implied introduction of wide-range modifications into HVDs and analysis of their effects on binding of specific proteins and on RNA and virus replication. Development of the rationale of mutagenesis and unambiguous interpretation of the data required the detailed knowledge of the HVD structure. This information helps to minimize the effect of the mutations on the overall protein folding and disruption of the potentially critical elements of the secondary structure.
CHIKV HVD is a 199-aa-long C-terminal fragment of nsP3 (Fig. 1A). It is enriched in disorder-promoting amino acids, such as proline, serine, lysine, glutamine, and glutamic acid (Fig. 1A). Computational prediction using IUPred software (40, 41) suggested that CHIKV HVD is likely intrinsically disordered but may have short structured fragments (Fig. 1B). To unambiguously identify the structural state of CHIKV HVD, we analyzed it by nuclear magnetic resonance (NMR) spectroscopy. The15N-labeled or15N- and13
C-double-labeled CHIKV HVD was produced inEscherichia coliand purified as a fusion protein with the carboxy-terminal Twin-Strep tag. Figure 1C presents the1H-15N correlation spectrum of
the region, where the cross peaks arising from the one-bond interaction between proton and nitrogen of the amide bond are expected to be observed. It exhibits a reduced chemical shift dispersion, especially in the1H dimension between 8.5 and 7.5 ppm (Fig. 1C),
which is a characteristic of the intrinsically disordered proteins (42). Since the15N,13C nuclei
are better dispersed even for the intrinsically disordered proteins, the sets of the three-dimensional (3D) experiments involving nitrogen and different types of carbon nuclei were performed (see Materials and Methods). Ultimately, we were able to assign 73% of N, 91% of C␣, 67% of C, and 73% of C=signals. The detailed analysis of CHIKV HVD structure and full assignment will be published elsewhere.
The deviation of chemical shift for C␣, C, and C=from the random coil values allows an estimation of the presence of secondary structure elements. The presence of 3 to 4 consecutive values of above 2 ppm is interpreted as the presence of␣-helix (positive value) or-strand (negative value). The majority of carbon signals in CHIKV HVD did not deviate more than 0.7 ppm from the random coil values, and no consecutive signals with a chemical shift more than 2 ppm were detected (Fig. 1D). These experimental data unambiguously demonstrated that CHIKV HVD is a disordered protein domain,
on November 6, 2019 by guest
http://jvi.asm.org/
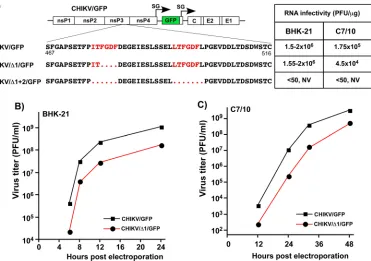
and its modifications in the following experiments performed in this study could change only the linear protein-binding motifs but not the protein secondary structure. Deletion of even one element of the repeat in HVD negatively impacts CHIKV replication.Our previous studies and those of other groups demonstrated that cellular G3BP1 and G3BP2 proteins interact with the short repeating peptides located at the carboxy termini of the OW alphavirus nsP3s (34, 38). KO of bothG3bpgenes (dKO) in the NIH 3T3 cells had a strong negative effect on replication of SFV and SINV while rendering CHIKV not viable in this cell line (34). To further evaluate the role of nsP3 HVD-G3BP interaction in virus replication, two recombinant CHIKV 181/25-based mu-tants were generated (Fig. 2A). The first mutant, CHIKV/Δ1/GFP, had one of the elements of the repeat deleted, and the second, CHIKV/Δ1⫹2/GFP, had both G3BP-binding sites removed. Since lack of cytopathic effect in cultured cells is not necessarily an indication of the absence of RNA replication, both mutants and the control CHIKV/
FIG 1CHIKV HVD is an intrinsically disordered domain. (A) Amino acid sequence of CHIKV 181/25 nsP3 HVD. The binding motif for BIN1 is depicted in green. The binding motifs for G3BP1 and G3BP2 are depicted in red. (B) Computational prediction of CHIKV nsP3 disorder probability by IUPred. The known binding motifs for BIN1 and G3BPs are marked by blue lines. (C) 2D best-TROSY spectra of CHIKV nsP3 HVD showing cross peaks corresponding to the one-bond interaction between proton,1H, and nitrogen,15N, of the amide bonds. (D) Results of chemical shift analysis of13C=,13C␣, and13C
nuclei in CHIKV HVD. The gray bars indicate unassigned carbons.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:4.585.39.428.68.498.2]GFP genomes contained the green fluorescent protein (GFP) gene under the control of an additional SG RNA promoter (Fig. 2A). The analysis of GFP expression was used as another means for indirect assessment of G RNA replication and transcription of SG RNA. The designed mutants were evaluated in terms of their ability to replicate in the cells of vertebrate and mosquito origins.
Equal amounts of thein vitro-synthesized RNAs of CHIKV/GFP, CHIKV/Δ1/GFP, and CHIKV/Δ1⫹2/GFP were electroporated into BHK-21 and C7/10 cells. In the infectious center assay (ICA) performed in BHK-21 cells, CHIKV/Δ1/GFP RNA demonstrated essen-tially the same infectivity as did the parental CHIKV/GFP, but in the repeated experi-ments, the rates of infectious CHIKV/Δ1/GFP virus release were reproducibly lower (Fig. 2B). CHIKV/Δ1⫹2/GFP, which lacked both G3BP-binding motifs in nsP3 HVD, failed to replicate in both cell lines with no detectable infectious virus release or cellular GFP expression (Fig. 2B and data not shown). In the subsequent follow-up experiments, we monitored for possible appearance of viable second-site revertants that could develop spreading infection but failed to detect any.
Similarly, in C7/10 cells, the in vitro-synthesized CHIKV/GFP- and CHIKV/Δ1/GFP-specific RNAs exhibited essentially the same infectivity patterns, but at any time posttransfection, titers of CHIKV/Δ1/GFP were between 1 and 2 orders of magnitude lower (Fig. 2A and C). The double deletion mutant CHIKV/Δ1⫹2/GFP failed to replicate in C7/10 cells. From these experiments, we concluded that deletion of even one G3BP-binding site in CHIKV HVD had a detrimental effect on viral replication rates, and deletion of both of them is lethal for CHIKV growth in both cell types.
Other HVD fragments, besides the G3BP-binding aa repeat, are also critical determinants of CHIKV replication.The above experimental data demonstrated that the presence of both HVD-specific, G3BP-binding sites is the prerequisite for efficient
FIG 2Deletions of G3BP-binding peptides in CHIKV nsP3 HVD have deleterious effects on viral replication. (A) Schematic presentation of CHIKV/GFP genome, sequences of the carboxy-terminal fragments of nsP3 HVD in the parental CHIKV/GFP and in the designed deletion mutants, and infectivities of thein vitro-synthesized RNAs in ICA performed on BHK-21 and C7/10 cells. The repeating element is indicated in red. Dots indicate deleted aa in the designed mutants. NV, not viable. The numbers given are the aa numbers in the original full-length CHIKV nsP3. (B and C) BHK-21 and C7/10 cells, respectively, were electroporated with 3g of the indicatedin vitro-synthesized RNAs, and samples of the media were collected at the indicated time points. Virus titers were determined by plaque assay on BHK-21 cells. The experiments were performed twice using different amounts ofin vitro-synthesized RNA and demonstrated similar differences in the rates of infectious virus release. The results of one of the experiments are presented.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.53.424.71.332.2]CHIKV replication in insect and vertebrate cells. However, this does not necessarily mean that the indicated repeating element is the only critical determinant of the HVD function in viral replication. Therefore, to continue the investigation of the role(s) of CHIKV HVD, we designed a variety of viral mutants that contained strong modifications in the HVD fragments other than the G3BP-binding repeat. The CHIKV/artHVD/GFP mutant encoded an artificial HVD in which the carboxy-terminal, repeat-containing fragment remained intact. The amino acid positions in the rest of the upstream-located HVD fragment were changed to generate essentially random peptide sequence (Fig. 3A). Thus, the newly designed HVD was similar to its natural counterpart in attributes such as length, aa composition, and hydrophobicity profile and, based on the computer predictions, remained intrinsically disordered (Fig. 3B). However, it exhibited a very low level of identity with the sequence of wild-type (wt) CHIKV HVD (Fig. 3A).
Electroporation of thein vitro-synthesized RNA of the CHIKV/artHVD/GFP variant did not initiate its replication in BHK-21 cells, despite the latter variant containing the intact carboxy-terminal G3BP-binding repeat in its HVD (Fig. 3A and C). This mutant also failed to replicate in mosquito C7/10 cells (data not shown). The above data strongly indicated that (i) the G3BP-binding motifs are not the only indispensable determinants of CHIKV HVD function in RNA replication and (ii) other HVD motifs play an essential role(s) in RC assembly and function similar to the carboxy-terminal G3BP-interacting repeat.
Interestingly, in one of the repeated RNA transfection experiments on BHK-21 cells, we detected selection of a viable second-site revertant of CHIKV/artHVD/GFP. A single adaptive mutation, K258I, was identified in the Zn-binding domain of nsP3. When introduced into the nsP3 sequence of the original CHIKV/artHVD/GFP, it made this variant viable and capable of developing spreading infection in BHK-21 cells (Fig. 3C). However, despite the stimulatory effect of the K258I mutation, viral replication in BHK-21 cells and particularly in other tested cell lines remained low (Fig. 3D), and no further experiments with this second-site revertant were performed in this study.
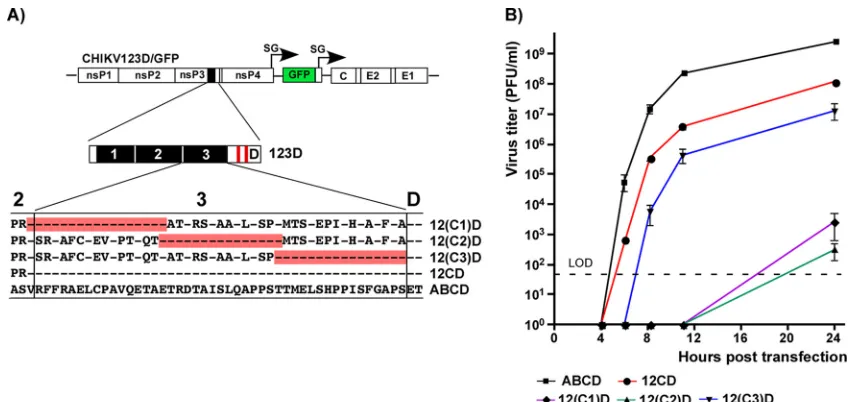
Fragments of CHIKV HVD differentially determine virus replication in different cell types.The designed CHIKV/artHVD/GFP mutant was further exploited as a basis for dissecting the functions of natural HVD sequences in CHIKV replication. The fragment of artHVD located between the G3BP-binding sequence and AUD was divided into 3 parts of similar lengths (Fig. 4A). In the cDNA of the CHIKV/artHVD/GFP genome, each of them and their combinations were replaced by the wt counterparts (Fig. 4B and C). For the sake of clarity in presentation of the results, the mutated fragments are labeled 1, 2, and 3, and their wt counterparts are labeled A, B, and C (Fig. 4A, B, and C). The in vitro-synthesized recombinant viral genomes were transfected into BHK-21 and mosquito C7/10 cells to assess the RNA infectivity and viral growth rates. These experiments revealed an interesting interplay of different HVD fragments in determin-ing the level of CHIKV replication in both cell types. The overall dependence of replication rates on the HVD sequence was reproducible in multiple repeating exper-iments and can be summarized as follows. (i) As described above, the intact G3BP-binding fragment D alone in CHIKV/123D/GFP nsP3 could not drive virus replication in both cell types, and this variant was not viable. (ii) The addition of adjacent, immedi-ately upstream-located fragment C made the CHIKV/12CD/GFP variant viable and capable of replication in BHK-21 cells, albeit not to the wt level (Fig. 4B and D). However, it remained poorly replicating in mosquito C7/10 cells (Fig. 4C and E). In the latter cells, infectivity of CHIKV/12CD/GFP RNA remained essentially at wt level, but at any time postelectroporation, titers of the released virus were 4 to 5 orders of magnitude lower (Fig. 4E). Accordingly, CHIKV/12CD/GFP formed smaller than wt but readily visible plaques in BHK-21 cells and only small foci of GFP-positive cells, but not plaques, in the mosquito cell line (Fig. 4C). (iii) An addition of wt fragment B had a more positive impact on replication of CHIKV/1B3D/GFP variant in C7/10 than BHK-21 cells (Fig. 4C and E). In contrast to CHIKV/123D/GFP, the B fragment-containing CHIKV/1B3D/ GFP was viable in BHK-21 cells, but at any time, its titers remained 3 orders of magnitude below those of the virus with natural, wt HVD. (iv) The replacement of the
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 3CHIKV nsP3 HVD fragments other than the carboxy-terminal, G3BP-binding repeat determine virus replication. (A) Schematic presentation of the parental CHIKV/GFP genome and its derivative, CHIKV/artHVD/GFP, with artificially designed HVD, and alignment of the wt and mutated HVD sequences. The repeating element is indicated in red. Dashes indicate identical aa. (B) Computational prediction of disorder probability of CHIKV nsP3 with artHVD (shown in blue) and wt CHIKV nsP3 (shown in red) by IUPred. The mutated HVD fragment is indicated by a green line. (C) Replication rates of the indicated viruses, CHIKV/GFP, CHIKV/artHVD/GFP, and its derivative CHIKV/artHVD(K258I)/GFP, which contained an adaptive mutation, K258I, in nsP3. BHK-21 cells were electroporated with 3 g in vitro-synthesized RNAs. Viral samples were harvested at the indicated time points, and titers were determined by plaque assay on BHK-21 cells. LOD, limit of detection. (D) Titers of CHIKV/GFP and CHIKV/artHVD(K258I)/GFP at 24 h after infection of the indicated cell lines at an MOI of 1 PFU/cell. Titers were determined by plaque assay on BHK-21 cells. The results of one of the reproducible experiments performed in triplicates are presented. Means and standard deviations (SDs) are indicated.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.41.480.85.591.2]FIG 4Fragments of CHIKV nsP3 HVD differentially regulate viral replication. (A) Alignment of amino acid sequences of wt ABCD HVD and mutated 123D HVD counterpart. Fragments A, B, and C in wt HVD and the corresponding mutated fragments 1, 2, and 3 are indicated by black boxes. Red boxes indicate positions of the G3BP-binding sites. Dashes indicate identical aa. (B) Schematic presentation of the original CHIKV/GFP genome and modified HVDs in the genomes of the designed viruses. Equal amounts (3g) of thein vitro-synthesized RNAs of the indicated constructs were electroporated into BHK-21 cells, and RNA infectivities and plaque sizes were compared in the ICA. (C) Schematic presentation of HVDs of the same viruses as in panel B, shown for clarity of presentation. Equal amounts of thein vitro-synthesized RNAs of the indicated constructs were electroporated into C7/10 cells, and RNA infectivities and plaque sizes were compared in the ICA performed on C7/10 cells. NV, not viable. (D) Replication rates of the indicated variants were determined in the experiment presented in panel B. Samples were harvested at the indicated time points, and viral titers were determined by plaque assay on BHK-21 cells. The experiments were repeated multiple times using different sets of
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.44.503.71.639.2]most amino-terminal fragment 1 by its counterpart A (CHIKV/A23D/GFP variant) did not yield noticeable positive effect on infectious virus release and development of spread-ing infection and remained not viable in both cell types (Fig. 4B and C). (v) However, the same fragment A had detectable positive impact on virus replication in mosquito cells when combined with B and D sequences in CHIKV/AB3D/GFP (Fig. 4E). The latter variant replicated in C7/10 cells to the levels of wt CHIKV/GFP that encoded the entire ABCD-containing HVD (Fig. 4E). (vi) The addition of fragment C to B- and D-containing HVD in CHIKV/1BCD/GFP made the latter variant capable of replication in BHK-21 cells at essentially wt level (Fig. 4D). However, in C7/10 cells, growth rates of CHIKV/1BCD/ GFP remained below wt (Fig. 4E) and were similar to those of CHIKV/1B3D/GFP mutant. Thus, the experimental data strongly suggested that fragment C of CHIKV HVD plays a critical role in viral replication in mammalian BHK-21 cells, and fragment B is more important for CHIKV replication in the mosquito C7/10 cell line.
Dissection of functional elements in HVD fragments C and B. Comparing the infectious titers of harvested viral samples on vertebrate and mosquito cell lines provided additional information about the replication step determined by HVD se-quences. Stocks of viruses that encoded fragment 3 (mutant) instead of wt fragment C demonstrated 10- to 20-fold-lower infectious titers on BHK-21 than on C7/10 cells (Fig. 4F). This was an indication that in addition to the repeat-containing fragment D, the upstream-located fragment C plays a major role in CHIKV replication in the cell line of vertebrate origin. Lower infectivity of the viruses suggested that the function of C fragment is particularly important at the early step of virus/RNA replication, likely during the formation of viral RCs.
To further dissect the HVD sequences that have a stimulatory effect on CHIKV replication in vertebrate cells, the above-described HVD-specific fragment C was addi-tionally divided into three smaller segments, and each of them was cloned into CHIKV/123D/GFP variant to replace the corresponding mutated counterpart in frag-ment 3 (Fig. 5A). Equal amounts of thein vitro-synthesized recombinant viral genomes were transfected into BHK-21 cells, and replication rates of the viruses were compared (Fig. 5B). The addition of only-16-aa-long wt peptide, which is adjacent to G3BP-binding-site-containing fragment D, made the designed CHIKV/12(C3)D/GFP variant viable. Thus, the short wt HVD sequence located immediately upstream of G3BP-binding-repeat-containing fragment, but not the other two tested aa sequences, efficiently stimulated virus replication. However, the positive effect of the 16-aa-long peptide was not as strong as that of the entire fragment C (Fig. 5B). The detected stimulatory effect was not specific just to BHK-21 cells. The CHIKV/123(C3)D/GFP variant showed similar replication trends in other tested cell lines, such as HEK 293 and NIH 3T3 cells (data not shown).
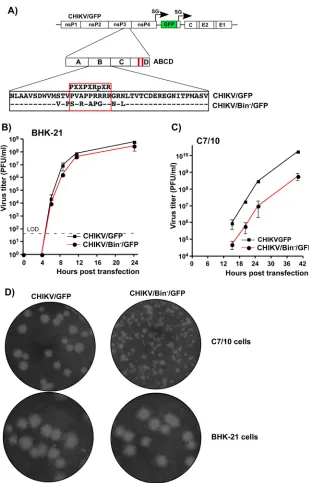
Similarly, CHIKV nsP3 HVD-specific fragment B was additionally investigated in terms of defining the critical element that determines virus replication. The previously pub-lished data demonstrated that the HVDs of the studied OW alphaviruses, including CHIKV, contain a proline-rich, SH3-binding motif that can bind with high affinity to BIN1 (amphiphysin II) protein (43). In our study, we introduced a combination of point mutations into this motif of CHIKV HVD (CHIKV/Bin⫺/GFP) (Fig. 6A) and analyzed their effect on viral replication in BHK-21 and C7/10 cells. Equal amounts of in vitro -synthesized CHIKV/Bin⫺/GFP and CHIKV/GFP RNAs were electroporated into both cell lines, and viral samples were harvested at different times postelectroporation for assessment of infectious titers. In repeated experiments, these viruses replicated with equal efficiency in BHK-21 cells (Fig. 6B). However, in C7/10 cells, the replication rate of
FIG 4Legend (Continued)
RNAs with consistent results. (E) Replication rates of the indicated variants were determined in the experiment presented in panel C. At the indicated time points, viral titers were determined by plaque assay on C7/10 cells. The experiments were repeated multiple times using different sets of RNAs with consistent results. The experiments presented in panels D and E show the data of the most representative experiments that used all the indicated variants together. (F) Titers of multiple, randomly selected samples of the indicated viruses harvested from BHK-21 and C7/10 cells were determined in parallel on both BHK-21 and C7/10 cells. The ratios of the determined titers are presented as means and SDs.
on November 6, 2019 by guest
http://jvi.asm.org/
CHIKV/Bin⫺/GFP was almost 100-fold lower than that of CHIKV/GFP, which encoded wt HVD (Fig. 6C). In addition, the decrease in replication in mosquito cells was further corroborated by a difference in plaque sizes recorded for CHIKV/Bin⫺/GFP compared to CHIKV/GFP (Fig. 6D). The designed mutant showed smaller plaques in C7/10 and the same-size plaques in BHK-21 cells. Thus, in agreement with the above data on the mapping of the critical aa sequences in CHIKV HVD (Fig. 4), these experiments showed that the previously described BIN1-SH3-binding motif (44) plays an essential role in CHIKV replication in insect cells rather than in the cells of vertebrate origin.
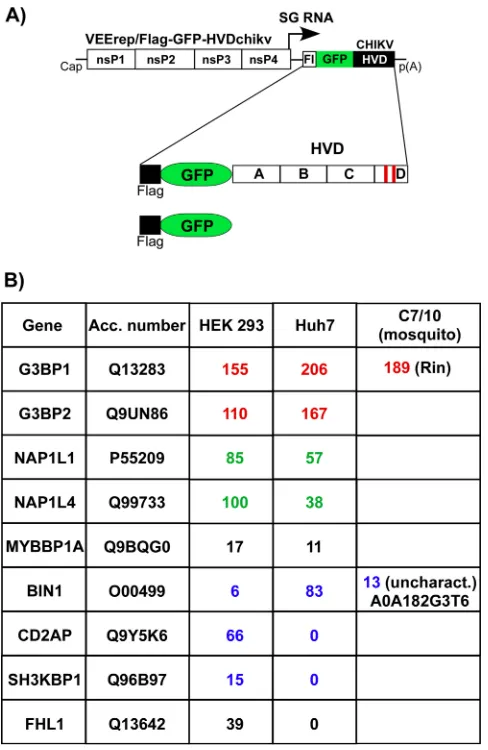
Fragments of CHIKV HVD interact with specific cellular proteins.The above data suggested that CHIKV HVD may interact with a variety of host factors, and these interactions determine virus replication in different cell types. To identify the interact-ing host proteins, we expressed CHIKV nsP3 HVD from VEEV replicon as Flag-GFP-HVD fusions (Fig. 7A). This expression system was applied based on the previous data that VEEV and CHIKV HVDs utilize different host factors for building higher-order complexes involved in virus replication (34). Thus, VEEV nsP3 that is encoded by the replicon itself does not compete with expressed Flag-GFP-HVDchikv fusion for binding factors, par-ticularly at early times postinfection (p.i.). VEEV replicons were packaged into infectious viral particles and used to infect both human and mosquito cells. Complexes of HVD-interacting cellular proteins were isolated at ⬃2 h p.i., when expression of Flag-GFP-HVDchikv remained very low and thus physiologically relevant, using mag-netic beads with Flag-specific monoclonal antibody (MAb) (see Materials and Methods for details). The coisolated cellular proteins were separated using SDS-PAGE and identified by mass spectrometry. To determine only proteins that interact with HVD with high affinity, we have used a highly stringent cutoff. In agreement with our previously published data (34, 35), Flag-GFP-HVDchikv coprecipitated large amounts of G3BP1 and G3BP2 from any vertebrate cells used and Rin protein from C7/10 cells. In vertebrate cells, CHIKV HVD also coimmunoprecipitated cellular SH3-binding proteins BIN1, CD2AP, and SH3KBP1. In mosquito C7/10 cells, it interacted with A0A182G3T6, the mosquito BIN1 homolog (Fig. 6B). Interestingly, isolation of CD2AP/SH3KBP1 and BIN1 was strongly dependent on the cell type. In human Huh7 and mouse NIH 3T3 cells (35), BIN1, but not CD2AP or SH3KBP1, efficiently interacted with CHIKV HVD. However, in another human cell line, HEK 293 cells, CHIKV HVD preferentially interacted with CD2AP
FIG 5The carboxy terminus of fragment C determines CHIKV replication in vertebrate cells. (A) Schematic presentation of CHIKV/ 123D/GFP genome and aa sequences of fragment 3 that contained the peptides of mutated HVD restored to wt sequence. The restored fragments are indicated in red, and dashes indicate aa that are identical to those in wt CHIKV nsP3 HVD. (B) BHK-21 cells were electroporated with 3g of thein vitro-synthesized RNAs of the indicated constructs. Media were replaced at the indicated time points, and viral titers were determined by plaque assay on BHK-21 cells. The experiments were repeated three times with highly reproducible results. LOD, limit of detection.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.38.464.69.270.2]and SH3KBP1, but not with BIN1. The extended list of CHIKV HVD-interacting host proteins in different cell lines is presented in Data Set S1 in the supplemental material. The coimmunoprecipitation (co-IP) samples from both human cell lines also con-tained high levels of NAP1L1 and NAP1L4 proteins, which we previously found to uniquely interact only with CHIKV HVD and not HVDs of other alphaviruses in mouse NIH 3T3 and hamster BHK-21 cells (34, 35). No NAP1 homolog was found in the co-IP
FIG 6Proline-rich, SH3-binding motif in CHIKV HVD is more important for viral replication in mosquito than in vertebrate cells. (A) Schematic presentation of CHIKV/GFP genome and its derivative CHIKV/ Bin⫺/GFP with mutated SH3-binding motif. Dashes in the alignment indicate aa that are identical in wt
and mutated CHIKV nsP3 HVD. (B and C) BHK-21 and C7/10 cells, respectively, were electroporated with 3g ofin vitro-synthesized RNAs. At the indicated time points, media were replaced and viral titers were determined by plaque assay on BHK-21 cells. LOD, limit of detection. These experiments were repeated twice using different amounts of RNA and produced consistent results. (D) Size of plaques formed by parental CHIKV/GFP and its CHIKV/Bin⫺/GFP derivative on BHK-21 and C7/10 cells. For both cell lines,
plaques were stained by crystal violet at 3 days postinfection.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:11.585.49.358.67.550.2]samples derived from mosquito cells. Another coisolated cellular protein was FHL1, but it was present only in the samples isolated from Huh7 cells, not from HEK 293 cells. Interestingly, we have previously found its homolog FHL2 in the co-IP samples from mouse fibroblasts (35).
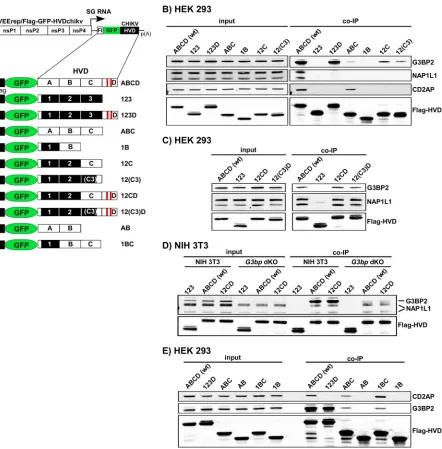
To confirm the results and to further explore the binding sites, samples from the independent co-IP experiments were analyzed by Western blotting. For this study, we used VEEV replicons expressing various combinations of wt and mutated CHIKV HVD fragments fused with Flag-GFP (Fig. 8A). The specific Abs detected the presence of G3BP in the samples isolated from the cells infected with the constructs encoding the D fragment, VEErep/Flag-GFP-ABCD and VEErep/Flag-GFP-123D (Fig. 8B). Unexpectedly, low levels of G3BP were also found in the co-IP samples from the cells infected with VEErep/Flag-GFP-ABC and VEErep/Flag-GFP-12C that lacked CHIKV HVD fragment D, which contained the canonical G3BP-binding repeat. The low-level binding of G3BP to HVD in the absence of binding repeats indicated the existence of an additional G3BP-binding site(s) in fragment C. Therefore, we then focused on the carboxy-terminal 16-aa-long sequence of fragment C, located immediately upstream of fragment D,
FIG 7Host factors binding to CHIKV HVD in different cell types. (A) Schematic presentation of VEEV replicons that encode Flag-GFP-HVDchikv fusion or Flag-GFP. (B) Indicated cell lines were infected with packaged VEErep/Flag-GFP-HVDchikv replicon or the control replicon expressing only Flag-GFP at an MOI of 20 IU/cell. Cells were harvested at 2 h postinfection, and complexes were immunoprecipitated as described in Materials and Methods. Proteins were separated by SDS-PAGE and identified by mass spectrometry. Total spectra for the most abundant proteins are presented. Groups of functionally similar proteins are indicated by different colors. None of the presented proteins were detected in the control samples generated using Flag-GFP. Acc. number, accession number for UniProt database.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:12.585.85.329.70.445.2]which was found crucial in the replication earlier in this study (Fig. 5). We expressed Flag-GFP-12(C3) fusion and analyzed the presence of G3BP in the co-IP sample. The results presented in Fig. 8B demonstrated that the carboxy-terminal element of the C fragment is indeed capable of G3BP binding, despite that its sequence does not contain the canonical G3BP-binding elements (34, 38). However, the presented Western blot analysis suggested that the efficiency of this binding is relatively low.
The members of an NAP1 family, NAP1L1 and NAP1L4, were reproducibly precipi-tated with CHIKV HVD from vertebrate HEK 293, Huh7, and NIH 3T3 cells (Fig. 7B) (34, 35). Western blot analysis with NAP1L1-specific Abs also definitively detected the latter
FIG 8Mapping of G3BP-, NAP1-, and CD2AP-binding sites in CHIKV nsP3 HVD. (A) Schematic presentation of VEEV replicon and expressed Flag-GFP-HVD cassettes that contained wt and mutated CHIKV HVD fragments. Fragments with wt aa sequences are indicated by open boxes and labeled A, B, C, and D, and mutated counterparts of A, B, and C are indicated by black boxes and labeled 1, 2, and 3. (B, C, D, and E) Indicated cell lines were infected with packaged replicons encoding indicated fragments of CHIKV HVD. At 2 h postinfection, protein complexes were isolated using magnetic beads with Flag-specific MAbs as described in Materials and Methods. The presence of studied cellular proteins was analyzed by Western blotting using specific Abs and corresponding secondary Abs labeled with different infrared dyes. Membranes were scanned on a Li-Cor imager. The following primary antibodies were used: anti-G3BP2 rabbit polyclonal antibodies (A302-040; Epitomics), anti-CD2AP rabbit polyclonal antibodies (sc-9137; Santa Cruz), anti-NAP1L1 rabbit polyclonal antibodies (14989-1-AP; Proteintech), and anti-Flag mouse monoclonal antibodies (F1804; Sigma).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:13.585.56.498.68.517.2]protein in the co-IP samples generated on the cells infected with VEErep/Flag-GFP-ABCD but not with any other constructs, including VEErep/Flag-GFP-ABC and VEErep/ Flag-GFP-123D (Fig. 8B). The most plausible explanation for the lack of NAP1L1 binding to CHIKV HVD deletion mutant constructs was that NAP1L1 binding requires the presence of intact aa sequences of both fragments C and D. Indeed, two isoforms of NAP1L1 were readily detectable in the samples generated from the cells infected with replicons expressing Flag-GFP-12CD and Flag-GFP-12(C3)D fusions (Fig. 8C). This was an indication that NAP1L1 interacts with HVD on the border between C and D fragments (see Fig. 11). Taken together, the data suggested that C3 fragment takes part in interaction with G3BP and/or NAP1 proteins, and thus, a possibility remained that NAP1 proteins bind to CHIKV HVD indirectly through interaction with G3BPs. Therefore, we infected NIH 3T3 G3bp dKO cells, which do not express G3BP1 and G3BP2, with VEErep/Flag-GFP-12CD and VEErep/Flag-GFP-12(C3)D replicons and coimmunoprecipi-tated HVD-binding proteins (Fig. 8D). NAP1L1 was readily identified in the samples generated from G3bp dKO cells, suggesting that this protein directly interacts with CHIKV HVD.
Another vertebrate protein identified by mass spectrometry as interacting with CHIKV HVD (ABCD construct) was CD2AP. Further analysis of its interaction with CHIKV HVD revealed an interesting pattern of its binding to the HVD aa sequences. CD2AP was found only in the co-IP samples from the cells expressing either the full-length ABCD or ABC or BC fragments of HVD (Fig. 8B and E). This was an indication that it likely interacts with the peptide that includes the carboxy-terminal sequence of fragment B and amino terminus of fragment C (see Fig. 11). The most recently published study confirmed the location of this binding site (45).
HVD determines cell specificity of alphavirus replication.The above data (Fig. 4) suggested that nsP3 HVD interactions with different host factors determine cell spec-ificity of CHIKV replication. In an attempt to additionally examine this possibility, we introduced into this study another alphavirus, Eilat virus (EILV) (46). The latter virus replicates exclusively in mosquito cells. One of the reasons for such cell specificity is that its nsPs facilitate viral RNA replication only in the cells of insect origin. Despite its predisposition to mosquito cell lines, EILV shares genome strategy and nsP identity with other, better-studied alphaviruses, such as SINV, SFV, and VEEV. The sequence align-ment of CHIKV and EILV nsP3 HVDs showed that they have essentially no similarity at the aa level with the exception that both domains are enriched in proline-rich motifs that can potentially bind SH3 domain-containing proteins (Fig. 9A). Another important characteristic of EILV relevant to this study is that the carboxy terminus of its nsP3 HVD contains the same canonical repeating aa elements that have been shown to interact with G3BPs in CHIKV HVD (Fig. 9A). In the context of EILV, the latter repeat mediates interaction with the insect-specific homolog of G3BP, Rin (data not shown). In addition, EILV HVD is more than 60 aa shorter than the CHIKV-specific counterpart.
Based on CHIKV and EILV sequences, we designed a set of recombinant alphavirus genomes and investigated their replication. One of the plasmids contained the genome of the previously designed CHIKV/GFP and encoded wt HVD (Fig. 9B). The second plasmid contained cDNA of the chimeric EIL/CHIKV/GFP (Fig. 9B). Its G RNA encoded the replication machinery of EILV, and the structural genes were derived from CHIKV 181/25, because the natural EILV glycoproteins do not facilitate viral entry into verte-brate cells (47, 48). However, replication of this chimeric virus remains restricted to mosquito-derived cell lines (see below). On the basis of CHIKV/GFP, we designed a CHIKV/HVDeilv/GFP chimeric construct, in which the natural HVD in CHIKV-specific nsPs was replaced by that derived from EILV. Another chimeric viral genome, EIL/HVDchikv/ CHIKV/GFP, encoded heterologous CHIKV HVD in EILV ns polyprotein instead of the natural, EILV-specific counterpart.
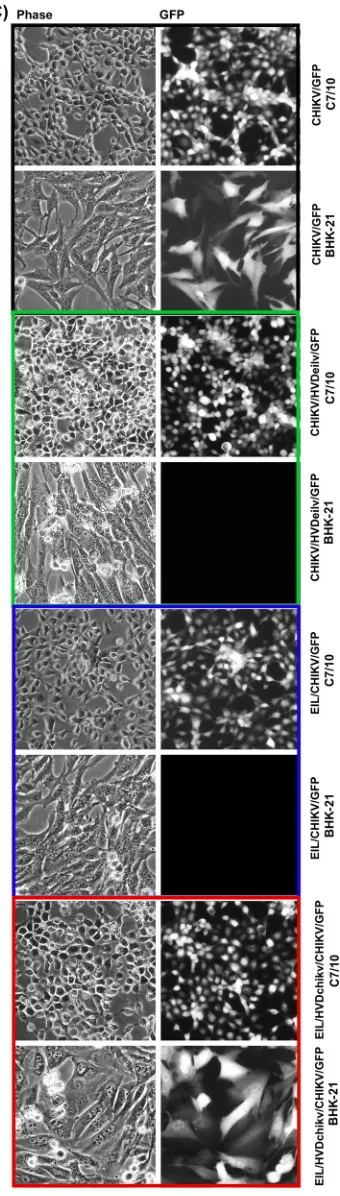
G RNAs of these 4 constructs were synthesizedin vitroand transfected into both BHK-21 and mosquito C7/10 cells. The control CHIKV/GFP RNA efficiently initiated replication in both cell types (Fig. 9B and C), while electroporation of EIL/CHIKV/GFP
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 9CHIKV and EILV HVDs determine cell specificity of viral replication. (A) Alignment of the aa sequences of CHIKV and EILV HVDs. Dots indicate gaps introduced for better alignment. Dashes indicate identical aa. Red boxes indicate positions of the carboxy-terminal, G3BP/Rin-binding repeats. (B) Schematic presentations of recombinant alphavirus CHIKV and EILV/CHIKV genomes with homologous and heterologous nsP3 HVDs. The white and black boxes indicate CHIKV and EILV genomes, respectively. Titers of rescued viruses after electroporation of 3g of thein vitro-synthesized RNAs into indicated cell lines. Titers of the stocks were determined by plaque assay on both BHK-21 and C7/10 cells. NV, not viable. Titers of plaque-forming
(Continued on next page)
on November 6, 2019 by guest
http://jvi.asm.org/
[image:15.585.268.438.72.670.2]RNA induced virus replication only in mosquito cells. In repeating experiments, after electroporation of EIL/CHIKV/GFP RNA into BHK-21 cells, no infectious virus was recov-ered. Within 24 h postelectroporation, very few cells became GFP positive, but no spreading infection was detected during further incubation at either 37°C or 28°C.
In contrast to EIL/CHIKV/GFP, the EIL/HVDchikv/CHIKV/GFP chimera, which encoded CHIKV HVD in EILV nsP3, became viable in both cell types. In BHK-21 cells, it was producing readily detectable levels of GFP and was developing a spreading infection (Fig. 9B and C). Of note, this chimeric virus replicated in BHK-21 cells only at 28°C, a temperature that is used for the cells of mosquito origin, but not at the vertebrate cell-specific 37°C temperature. The most plausible explanation for this temperature-dependent replication is that EILV nsPs are highly adapted to mosquito cell-specific low incubation temperatures. Another distinguishing characteristic of EIL/HVDchikv/GFP/ CHIKV was its inability to cause a cytopathic effect (CPE) in BHK-21 cells (data not shown). Even after a 4-day-long incubation of cells under agarose cover at 28°C, we detected only the large foci of GFP-positive cells, but not plaque formation. Most likely, EILV-specific nsP2 does not have nuclear functions, which are exhibited by the OW alphaviruses and are the critical contributor to CPE development and inhibition of the innate immune response (49, 50). The reciprocal replacement of CHIKV HVD by that derived from EILV in CHIKV/HVDeilv/GFP restricted replication of the latter virus to mosquito cells only (Fig. 9B and C). In contrast to parental CHIKV/GFP, the chimeric virus was no longer viable in BHK-21 cells, which are normally highly permissive for CHIKV replication.
These experimental data strongly suggested that HVDs play critical roles in the adaptation of CHIKV and EILV to replication in particular cell types. Despite having very similar G3BP-binding repeating peptides, heterologous HVDs changed cell specificities of replication for both viruses. This was a strong support for the hypothesis that HVD fragments located upstream of the carboxy-terminal G3BP/Rin-binding repeat play an indispensable role in CHIKV replication.
The above data strongly suggested that fragment C of CHIKV HVD, whose presence restores the NAP1-binding aa sequence, plays a critical role in defining virus replication in vertebrate cells. To additionally test this hypothesis, the CHIKV-specific fragment C was cloned upstream of the heterologous EILV-specific fragment, containing the G3BP/ Rin1-binding aa repeat, into CHIKV/HVDeilv/GFP (Fig. 10A). The addition of this small fragment made the resulting recombinant CHIKV/HVDeilv(C)/GFP viable in BHK-21 cells. Thein vitro-synthesized RNA of this chimeric virus demonstrated the same infectivity as that of CHIKV/GFP. This was an indication that in contrast to parental CHIKV/HVDeilv/ GFP, the C-fragment-containing chimera is viable and does not require additional adaptive mutations for initiation of replication. However, at any time postelectropora-tion, infectious titers of CHIKV/HVDeilv(C)/GFP were 30- to 50-fold below those of CHIKV/GFP. Thus, growth rates of CHIKV/HVDeilv(C)/GFP generally mimicked those of CHIKV, encoding 12CD HVD (Fig. 4D). This was an indication that despite having the C fragment, the chimeric HVD cannot mediate interaction with the complete set of host factors essential for CHIKV replication in vertebrate cells. These results additionally supported our hypothesis that nsP3 HVD is a critical determinant of specificity of CHIKV replication for different cells, with multiple binding host factors being involved.
DISCUSSION
In recent years, alphavirus nsP3 has emerged as a key player in the assembly and function of viral RCs that serves as a hub for recruitment of numerous host proteins required for viral RNA replication. The amino-terminal macro domain of nsP3 (Fig. 11) exhibits a high level of conservation between alphaviruses and other positive-strand
FIG 9Legend (Continued)
viruses are shown as PFU/ml, and titers of noncytopathic viruses were measured in GFP-positive focus-forming units (FFU/ml). (C) BHK-21 and C7/10 cells were infected with the indicated viruses, which were rescued by electroporation of permissive cells. Images were taken on a fluorescence microscope.
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 10Addition of CHIKV HVD fragment C to EILV HVD makes the resulting chimeric CHIKV/HVDeilv(C)/GFP capable of replication in vertebrate cells. (A) Schematic presentation of recombinant CHIKV genomes with wt or heterologous HVDs, infectivity of thein vitro-synthesized RNA in ICA on BHK-21 cells, and titers of viruses harvested 24 h after electroporation of BHK-21 cells. (B) Replication rates of the indicated viruses in BHK-21 cells after electroporation of thein vitro-synthesized RNAs. Viral titers at the indicated time points were determined by plaque assay on BHK-21 cells. (C) GFP expression by replicating viruses in BHK-21 and C7/10 cells. Cells were infected with the indicated viruses at an MOI of 5 PFU/cell. Stocks of CHIKV/GFP and CHIKV/HVDeilv(C)/GFP used for infection were generated on BHK-21 cells, and the stock of CHIKV/HVDeilv/ GFP was generated on C7/10 cells. Images were acquired on a fluorescence microscope at 8 h and 16 h after infection of BHK-21 and C7/10 cells, respectively.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:17.585.46.380.74.622.2]RNA viruses. The macro domain has been demonstrated to bind ADP-ribose, poly(ADP)-ribose, and RNA and has a detectable level of mono(ADP-ribosyl)hydrolase activity (29, 51, 52). The earlier reports have shown that the macro domain can acquire adaptive mutations in response to strong modifications in RNA promoter elements in the alphavirus genome (32, 53). This implied its potential role in viral G RNA replication.
The next conserved Zn-binding domain (Fig. 11), termed AUD, appears to be present only in alphavirus nsP3 and does not have homologs in the nonstructural proteins of other viruses (30). There is no clear information about its function in the literature, except that this and a few other studies have shown that some adaptive mutations in AUD compensated for changes in the downstream-located hypervariable domain, HVD (Fig. 3) (32, 54).
HVD (Fig. 11) exhibits an exceptionally low level of identity between different alphavirus species, and before this study, the definitive information about the CHIKV nsP3 HVD folding was lacking. We carried out detailed characterization of a 199-aa-long CHIKV HVD by NMR spectroscopy at atomic resolution. The results revealed the absence of stable secondary structures in this domain and clearly demonstrated that CHIKV HVD was present in solution in a disordered conformation. Consequently, all of the modi-fications generated in CHIKV HVD as part of this study could not have affected its secondary structure.
Within recent years, alphavirus HVDs have attracted a lot of attention. The studies of the HVD function in VEEV, EEEV, SFV, SINV, and CHIKV replication strongly suggested that this domain mediates assembly of prereplication complexes at the plasma and endosomal membranes in vertebrate and mosquito cells (32, 34, 35, 38). These evi-dences implied a critical role of nsP3 HVD in early stages of the alphavirus life cycle. At later times postinfection, it is also involved in the formation of large protein complexes, but their function at this time remains unclear (14, 36, 55). One of the intriguing characteristics of nsP3 HVDs is their ability to interact with distinct sets of cellular proteins, which are specific for virus species and cell types (34–36). Another important feature is a high level of redundancy in the function of these HVD-binding cellular factors in RC assembly and RNA replication. Instead of single host proteins, alphaviruses utilize entire families, such as G3BP or FXR protein families (34–36). Moreover, the interaction of HVDs with particular cellular proteins can be destroyed by either specific mutations in the HVDs or KO of corresponding cellular genes (34, 56). Such changes usually strongly affect replication rates of most alphaviruses but do not make them nonviable. This points to another level of redundancy in that other binding partners can to some extent facilitate readily detectable levels of RC assembly and function. How-ever, the results of this and our previous study (34) demonstrate that compared to other alphaviruses, CHIKV exhibits a unique characteristic. The host factors that belong to different protein families do not have the redundant function(s) in CHIKV RNA replication, and lack of CHIKV HVD interaction with individual cellular proteins or protein families can make virus nonviable. Thus, CHIKV represents a very convenient experimental system for studying functions of different HVD-binding proteins in viral replication.
FIG 11Locations of to-date identified binding sites of cellular proteins in CHIKV nsP3 HVD. CHIKV nsP3 is structurally divided into three domains, which include macro domain, AUD, and HVD. In this study, HVD was further divided into the indicated four fragments, A, B, C, and D. The identified locations of binding sites for cellular proteins are indicated in different colors. A question mark indicates that a possibility remains that CD2AP requires interaction with more than one HVD motif for efficient binding. “3” indicates a position of a noncanonical G3BP-binding site in C3 peptide of CHIKV HVD.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:18.585.48.365.71.148.2]G3BP proteins of vertebrate cells and their Rin homolog in mosquito cells have been shown to interact with HVDs of the OW alphaviruses and that of EEEV, the NW alphavirus representative (34, 35, 38). For most of the viruses, except EEEV, this interaction is mediated by short aa repeats located at the carboxy termini of their HVDs (38). In the case of CHIKV, G3BP binding is particularly critical. In contrast to the results of the studies performed on SINV and SFV, deletion of even a single element of the G3BP-binding HVD repeat had a strong negative effect on CHIKV replication in both vertebrate and mosquito cells (Fig. 2) (34). Deletion of both elements, in turn, com-pletely abrogated virus replication. This lack of G3BP interaction with HVD-specific repeats may potentially affect virus ability to block the formation of stress granules, which have been proposed to have an antiviral function (57, 58). However,G3bpdKO cells do not support replication of the OW alphaviruses as efficiently as the wt cell lines, although the stress granules cannot be formed (34). Moreover, the NW alphavirus VEEV does not induce redistribution of G3BPs into virus-specific complexes (31, 32). Taken together, these data imply that G3BP-HVD interactions are mainly important for the OW alphavirus RC assembly, and the role of this interaction in stress response to viral infection has to be reevaluated.
Our data have demonstrated that G3BP/Rin binding to HVD is indispensable for CHIKV replication in both vertebrate and mosquito cells. Nevertheless, this interaction alone is insufficient for supporting CHIKV replication. The extensive mutagenesis of CHIKV HVD that eliminated all of the potential binding sites, except those specific to G3BP, made CHIKV/artHVD/GFP virus nonviable (Fig. 3). Similarly, the replacement of natural CHIKV HVD by the EILV-derived counterpart, which also contains a canonical G3BP-binding aa repeats, made the chimeric virus incapable of replication in vertebrate cells (Fig. 9). Sequential restoration of wt sequences in the artificially designed artHVD (Fig. 4) clearly demonstrated that at least one additional binding site for other cellular proteins can restore CHIKV/artHVD/GFP replication, albeit not to the wt level.
Interestingly, further restoration of HVD sites that bind specific host proteins differ-entially stimulated CHIKV replication in mosquito and vertebrate cells (Fig. 4). The binding site that is important for CHIKV replication in the cells of vertebrate origin was located in the HVD immediately upstream of the G3BP-binding repeats (Fig. 5). Addi-tion of this aa sequence to either carboxy-terminal, repeat-containing CHIKV HVD fragment (Fig. 5 and 8) or to EILV HVD that contained a similar Rin-binding repeat (Fig. 10) restored the ability of CHIKV to replicate in vertebrate cells. This critical HVD-specific peptide was found to interact with cellular NAP1L1 and NAP1L4 proteins. It also contained an additional binding site for G3BP (Fig. 8). However, the latter G3BP-HVD interaction was less efficient than that determined by the downstream-located, canon-ical repeating peptide, and this G3BP-binding element alone was incapable of driving CHIKV replication (Fig. 2). Therefore, the biological significance of this additional interaction is unlikely but remains a possibility. We have not investigated it further in this study.
Interaction with NAP1L1 is independent of the G3BP binding (Fig. 8), and NAP1L1 appears to be the primary candidate for promoting CHIKV replication in vertebrate cells. Similar to the results of our previous study of G3BP and FXR functions in alphavirus replication (34), here we found that two members of the NAP1 protein family likely facilitate CHIKV replication redundantly. Both NAP1 proteins were reproducibly coimmunoprecipitated with CHIKV HVD, but not with HVDs of other alphaviruses, such as EEEV, VEEV, and SINV (35) (Fig. 8). Therefore, their function in virus replication appears to be specific to CHIKV. Mutations in the NAP1-binding HVD fragment, frag-ment C, had a stronger effect on CHIKV RNA infectivity and viral replication rates in BHK-21 cells than in mosquito cells (Fig. 4). Importantly, we could not identify a mosquito homolog of NAP1 in immunoprecipitation experiments. This can be a plau-sible explanation for the poor stimulatory effect of intact fragment C on CHIKV/12CD/ GFP replication in C7/10 cells. Based on available data, we hypothesize that similarly to FXRs and G3BPs, NAP1 proteins stimulate the formation of CHIKV replication complexes in vertebrate cells, and this effect might be also determined by the ability of NAP1
on November 6, 2019 by guest
http://jvi.asm.org/
proteins to undergo multimerization (59). The exact mechanism of NAP1L1 and NAP1L4 function is now under investigation.
The histone chaperone NAP1 proteins are mostly localized in the cytoplasm. They serve as a carrier of histones during nuclear import, mediate nucleosome assembly and chromatin remodeling, and regulate critical processes in cell biology such as cellular transcription and cell cycle progression (60). Thus, KO or downregulation of the expression by RNA interference (RNAi) of both members of the NAP1 family will likely lead to cell death. To date, there are no published data that such NAP1L1 and NAP1L4 dKO cell lines can be viable. Despite this methodological limitation, the list of NAP1L1 functions is continuously growing. Recent studies have demonstrated interaction of NAP1L1 with the intrinsically disordered, carboxy-terminal fragment of HCV NS5A protein (61). However, the data about the role of this interaction in HCV biology are somewhat contradictory (61, 62).
The second important CHIKV HVD-specific peptide that is critical for viral replication in mosquito, but not in vertebrate BHK-21 or NIH 3T3, cells is represented by the proline-rich motif (Fig. 1A and 6A). This peptide has been previously implicated in interaction with BIN1 protein (44). The latter protein contains both Bin-Amphiphysin-Rus (BAR) and SH3 domains. The SH3 domain has been demonstrated to directly bind to CHIKV and SFV HVDs (43). The BAR domain was proposed to regulate membrane curvature formation during invagination of viral replication complex containing mem-branous spherules, but this function remains yet to be shown (44). To date, only BIN1, but not BIN2, has been found to interact with CHIKV HVD and HVDs of several OW alphaviruses (34, 35). Our new data suggest that the role of the proline-rich peptide interaction with host factors in viral replication is likely more complicated than was previously expected. In mosquito cells, the latter peptide indeed efficiently interacts with BIN1 homolog (Fig. 7), and the introduced mutations have a negative effect on CHIKV replication (Fig. 6). However, in hamster BHK-21 cells, the effect of the same mutations on virus replication was undetectable (Fig. 6). CHIKV HVD also showed interaction with BIN1 in murine NIH 3T3 and human Huh7 cells, but only very small numbers of BIN1-specific peptides were detected in CHIKV HVD co-IP samples derived from HEK 293 cells (Fig. 7) (35). No BIN1-HVD interaction was also found in human HOS cells (45). Instead, two other SH3 domain-containing proteins, CD2AP and SH3KBP1, coimmunoprecipitated with CHIKV HVD from HEK 293 cells, with CD2AP being the main HVD-interacting partner (Fig. 7 and 8). The amounts of CD2AP and SH3KBP1 in CHIKV HVD co-IP samples from NIH 3T3 cells were relatively small, and they were not coisolated from Huh7 cells (Fig. 7) (35). Thus, it appears that depending on the cell type, CHIKV HVD interacts with either BIN1 or CD2AP/SH3KBP1 proteins. Currently, it is not clear whether the difference in the efficiency of complex formation depends on the relative amount of these proteins in the specific cell lines or/and these proteins compete for binding to HVD. Our data suggest that the CD2AP binding site is located a few aa downstream of the BIN1-interacting peptide at the junction of B and C fragments (Fig. 8 and 11). Recently published data from another group also proposed this location (45). Interestingly, CD2AP and SH3KBP1 have three SH3 domains, and this suggests a possibility that to achieve high binding affinity, the latter proteins may require interaction with more than one HVD-specific proline-rich motif. Thus, a further structural analysis is needed for better understanding cell-specific interaction of BIN1 and CD2AP/SH3KBP1 with CHIKV HVD in different cell types. Using our experimental systems, we previously found that in NIH 3T3 cells, EEEV HVD interacted with four SH3 domain-containing host proteins that included CD2AP, SH3KBP1, and two SH3 and BAR domain-containing proteins, SNX9 and SNX33. However, in these cells, EEEV HVD did not bind BIN1 (35). In contrast to other alphaviruses, VEEV HVD did not detectably bind any BAR domain-containing proteins at all but instead interacted exclusively with CD2AP and SH3KBP1 in NIH 3T3 and BHK-21 cells. Hence, the above data and earlier reports suggest that different alphaviruses have evolved to exploit a wide variety of SH3 domain-containing proteins and appear to use them in a cell-specific mode.
The family of the SH3 domain-containing proteins is represented by⬃300 members
on November 6, 2019 by guest
http://jvi.asm.org/
(63). They are involved in a range of key processes of cell biology, such as endocytosis, membrane trafficking, and cytoskeletal organization (64). Their common characteristic is interaction with the proline-rich aa sequences, which demonstrate high diversity. The affinity of SH3 domains for the peptide ligand is relatively low, and selectivity is quite poor (63). Therefore, binding of a particular SH3 domain-containing protein is likely determined by both sequences of the binding motif and concentration of the protein relative to other SH3 domain-containing family members. Thus, despite the continuing progress in understanding the molecular mechanism of alphavirus replication, the functional role of cellular SH3 domain-containing proteins in this process remains elusive. Apparently, we are only at the beginning of understanding the complexity of CHIKV HVD interactions with SH3 domain-containing proteins in different cell types and hosts.
Interestingly, similarly to NAP1L1, BIN1 was shown to interact with the disordered fragment of hepatitis C virus (HCV)-specific NS5A protein (62, 65–67). Alteration of this interaction has a negative effect on HCV replication. Thus, NAP1L1 and BIN1 may have similar functions in replication of these two very different groups of positive-strand RNA viruses.
The results of this study demonstrate that CHIKV nsP3 HVD interacts with at least 3 families of cellular proteins. They include two members of the G3BP family, two members of the NAP1 family, and several members of the SH3 domain-containing protein family. The summary of our current knowledge of the location of the binding sites of these cellular factors on CHIKV HVD is presented in Fig. 11. All of these interactions are critically important for virus viability and determine the efficiency of its replication in a cell-specific mode. (i) Binding of G3BPs or mosquito homolog Rin is universally indispensable for viral replication in both vertebrate and mosquito cells. (ii) Interaction with NAP1 proteins with HVD is critical for CHIKV replication in vertebrate cells and is CHIKV specific. (iii) Binding of SH3 domain-containing proteins, which include BIN1, CD2AP, and SH3KBP1, to proline-rich motifs of HVD has a more proviral role in the cells of mosquito origin and plays a less important role during viral growth in vertebrate cells. Identification of different SH3 domain-containing proteins bound to CHIKV HVD in different vertebrate cells suggests that CHIKV and probably other alphaviruses have evolved to redundantly utilize very diverse members of this family for their replication. Manipulations with the set of binding sites in CHIKV HVD change the efficiency of CHIKV replication in different cell types. Similar modifica-tions can likely be introduced into HVDs of other alphaviruses to specifically alter their replication rates.
MATERIALS AND METHODS
Cell cultures.NIH 3T3, HEK 293, and Vero cells were obtained from the American Type Culture Collection (Manassas, VA). BHK-21 cells were kindly provided by Paul Olivo (Washington University, St. Louis, MO). These cell lines were maintained at 37°C in alpha minimum essential medium (␣MEM) supplemented with 10% fetal bovine serum (FBS) and vitamins. The G3bp dKO cell line has been described elsewhere (34). Mosquito C7/10 cells were obtained from Henry Huang (Washington Univer-sity, St. Louis, MO). They were propagated at 28°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated (HI) FBS and 10% tryptose phosphate broth (TPB).
Plasmid constructs.The original plasmids containing the infectious cDNAs of the attenuated strain CHIKV 181/25 and Eilat alphavirus (EILV) were kindly provided by Scott Weaver (University of Texas Medical Branch, Galveston, TX) (46, 68). Plasmids containing cDNAs of VEEV replicon encoding Flag-GFP or Flag-GFP fused with different fragments of CHIKV HVD, and plasmids carrying CHIKV and EIL/CHIKV genomes with mutated or heterologous HVDs were designed using standard PCR-based techniques. The detailed schematic presentations of HVDs and their aa sequences are shown in the corresponding figures. All of the introduced modifications were confirmed by sequencing. Plasmids carrying the genomes of helper constructs, which were used for packaging of RNA replicons into infectious viral particles, were described elsewhere (69). Sequences of the plasmids and details of the cloning proce-dures can be provided upon request.
In vitroRNA transcription and transfection.Plasmids were purified by ultracentrifugation in CsCl gradients. Then, they were linearized using unique restriction sites located downstream of the poly(A) of viral, replicon, and helper genomes. RNAs were synthesized by SP6 RNA polymerase in the presence of a cap analog (New England BioLabs). Quality of the synthesized RNAs was tested by agarose gel electrophoresis, and aliquots of the transcription reaction mixtures were used for electroporation without additional RNA purification. Electroporations of BHK-21 and C7/10 cells by
on November 6, 2019 by guest
http://jvi.asm.org/
in vitro-synthesized virus-specific RNAs were performed under previously described conditions (70, 71). VEEV replicon genomes were packaged by coelectroporation of theirin vitro-synthesized RNAs and homologous helper RNA. Viruses and packaged replicons were harvested at 20 to 24 h postelectroporation. Viral titers were determined by plaque assay on BHK-21 or C7/10 cells (72). The infectious titers of packaged replicons were determined by infecting BHK-21 cells (5⫻105cells/well)
in 6-well Costar plates with 10-fold dilutions of the samples and counting the number of GFP-positive cells at 6 h postinfection.
Analysis of virus replication.Because of the capability of alphaviruses for rapid evolution to a more efficiently replicating phenotype, most of the analyses of viral replication were performed using the samples of RNA-electroporated cells. One-tenth of electroporated BHK-21 and C7/10 cells was seeded into 35-mm dishes, and media were replaced at the indicated times posttransfection (see corresponding figure legends for details). Viral titers in the harvested samples were determined by plaque assay on BHK-21 (72) or C7/10 (47) cells (see corresponding figure legends for details).
Infectious center assay (ICA).Infectivities of viral RNAs were analyzed in parallel with analysis of viral replication. BHK-21 or C7/10 cells were electroporated with the same amounts ofin vitro -synthesized genomes of the designed viral mutants and control genome encoding wt nsP3 HVD. Tenfold dilutions of electroporated cells were seeded in 6-well Costar plates containing subconfluent monolayers of naive BHK-21 or C7/10 cells. After 2 h of incubation at 37°C, cells were overlaid with agarose supplemented with MEM and 3% FBS. C7/10 cells were incubated for 2 h at 28°C and then overlaid with DMEM supplemented with 0.6% tragacanth, 5% TPB, and 5% FBS. Plaques were stained with crystal violet after 3 days of incubation at 37°C or 28°C, and RNA infectivity was determined as PFU/g of electroporated RNA.
Isolation and identification of cellular proteins that interact with alphavirus HVDs.HEK 293 or other cells in the quantity of 8⫻106were infected with viral particles containing replicon genomes
encoding Flag-GFP fused with different fragments of CHIKV HVD at a multiplicity of infection (MOI) of 20 infectious units (IU)/cell. Cells were harvested at 2 h postinfection (p.i.), when GFP was at the very early stage of expression, and thus, concentrations of expressed fusion proteins remained biologically relevant. Protein complexes were isolated from the postnuclear fraction of the NP-40-lysed cells using magnetic beads loaded with the Flag-specific monoclonal antibody (MAb) (Sigma) as described elsewhere (34). Proteins were separated on 4 to 12% NuPAGE gels (Invitrogen), stained with Coomassie blue, and identified by mass spectrometry as previously described (34). Identified proteins were selected as specifically binding to particular HVDs if, in the repeated experiments, the specific spectra were 20- to 100-fold higher than those detected in the control samples derived from the cells infected with replicons expressing Flag-GFP.
Recombinant CHIKV HVD purification and NMR experiment.CHIKV HVD-coding sequence (aa 325 to 523, Fig. 1A), containing Twin-Strep tag at the carboxy terminus, was cloned into pE-SUMOpro-3 plasmid (LifeSensors Inc.) encoding the amino-terminal His-SuMO tag. The plasmid was transformed into
E. coliRosetta 2(DE3)pLacI (Novagen). The15N- and15N,13C-labeled proteins were produced by growing
cells in M9 minimal medium supplemented with [15N]NH
4Cl (2 g/liter) andD-[13C6]glucose (3 g/liter). The
recombinant CHIKV HVD was first purified on a HisTrap HP column (GE Healthcare). After cleavage of the SUMO tag, CHIKV HVD was purified on a Strep Tactin Sepharose column (GE Healthcare). Size exclusion chromatography in NMR buffer [25 mM N2HPO4, 50 mM NaCl, 2 mM tris(hydroxypropyl)phosphine] on
a Superdex 200 10/30 column (GE Healthcare) was used as a final purification step. The purified protein was concentrated to⬃0.3 to 0.5 mM, and NaN3was added to 1 mM.
NMR experiments were performed on Bruker Avance III spectrometers operating at 14.1 T equipped with a cryo-enhanced QCI-P probe at a temperature of 298 K. Gradient sensitivity enhanced 3D triple-resonance pulse programs HNCA, HNCO, HN(CO)CACB, H(NCOCA)NNH, and (H)N(COCA)NNH with transverse relaxation-optimized spectroscopy (TROSY) were acquired. In addition, the 2D version of MUSIC (SER, LAVIA, TAVI, and PRO) was applied as well as15N best-TROSY. All experiments came from the
Bruker standard library of pulse programs. The backbone resonances and Cwere assigned and used in this publication. Full assignment, including side chains, will be published elsewhere. To estimate the secondary structure prosperity of CHIKV HVD, random coil chemical shifts calculated with neighbor correction factor in POTENCI (73) were subtracted from the experimental13C␣,13C, and13C=chemical
shifts. In the figures and in the text, the standard nomenclature for amino acids of the carbon atoms was used, where C␣is the carbon next to the carbonyl group C=and Cis the carbon next to C␣(74).
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at https://doi.org/10.1128/JVI .00838-18.
SUPPLEMENTAL FILE 1,XLSX file, 0.1 MB.
ACKNOWLEDGMENTS
We thank Scott Weaver for providing infectious cDNA clones of CHIKV 181/25 and EILV. We also thank Valeriya Kuznetsova and Arpine Sokratian for technical assistance. This study was supported by Public Health Service grants AI118867 and AI133159 to E.I.F. and AI095449, AI119627, and AI127744 to I.F.
on November 6, 2019 by guest
http://jvi.asm.org/
REFERENCES
1. Griffin DE. 2001. Alphaviruses, p 917–962.InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed. Lippincott Williams & Wilkins