This is a repository copy of A Mechanistic Study of the Lewis Base-Directed Cycloaddition of 2-Pyrones and Alkynylboranes.
White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/90396/
Version: Accepted Version
Article:
Crepin, D.F.P., Harrity, J.P.A., Jiang, J. et al. (3 more authors) (2014) A Mechanistic Study of the Lewis Base-Directed Cycloaddition of 2-Pyrones and Alkynylboranes. Journal of the American Chemical Society, 136 (24). 8642 - 8653. ISSN 0002-7863
https://doi.org/10.1021/ja501805r
[email protected] https://eprints.whiterose.ac.uk/ Reuse
Unless indicated otherwise, fulltext items are protected by copyright with all rights reserved. The copyright exception in section 29 of the Copyright, Designs and Patents Act 1988 allows the making of a single copy solely for the purpose of non-commercial research or private study within the limits of fair dealing. The publisher or other rights-holder may allow further reproduction and re-use of this version - refer to the White Rose Research Online record for this item. Where records identify the publisher as the copyright holder, users can verify any specific terms of use on the publisher’s website.
Takedown
If you consider content in White Rose Research Online to be in breach of UK law, please notify us by
1 A Mechanistic Study of the Lewis Base Directed Cycloaddition of 2-Pyrones and
Alkynylboranes
Damien F. P. Crépin,† Joseph P. A. Harrity,*,† Julong Jiang,† Anthony J. H. M. Meijer,*,† Anne-Chloé M. A. Nassoy† and Piotr Raubo‡
†Department of Chemistry, University of Sheffield, Sheffield, S3 7HF, U. K.
‡Research and Development, AstraZeneca, Alderley Park, Macclesfield, SK10 4TG, U.K.
Abstract: Significant rate enhancements in the Diels-Alder reaction of alkynes and
2-pyrones bearing a Lewis basic group are observed when a combination of
alkynyltrifluoroborates and BF3.OEt2 are used. This process generates functionalized
aromatic compounds with complete regiocontrol. The origin of the observed rate
enhancement has been studied by DFT methods, and they appear to originate for coordination
of the diene substrate to a mixture of alkynylborane intermediates, followed by a Lewis acid
mediated product equilibration step. Evidence for this mechanism is presented, as is the
2 Introduction
The Diels-Alder cycloaddition of 2-pyrones represents an efficient method of generating
functionalised cyclohexenes,1 and was first described by Diels and Alder over 80 years ago.2
This reaction also offers an effective method for the synthesis of highly substituted aromatic
compounds when alkynes are employed, whereby the intermediate cycloadduct undergoes
rapid retro-cycloaddition and expulsion of carbon dioxide.3 A range of alkynes are known to
participate in this process including those bearing hydrocarbon, ester, ketone,3 boronate,4
stannane5 and silyl6 groups. A significant challenge in this chemistry is the requirement of
high reaction temperatures over extended time periods, and variable reaction regiocontrol.
Such limitations are addressed only by the use of very reactive dienophiles such as
ynamides,7 thereby restricting the scope of products that can be generated by this strategy.
Recent studies in our labs have sought to address the high temperatures required in [4 + 2]
cycloaddition reactions of aromatic dienes by virtue of a Lewis acid-base complex induced
promotion of this process.8 This approach has the added advantage of generating compounds
with complete regiocontrol. We have developed a mild and regioselective synthesis of
aromatic difluoroboranes via the cycloaddition of 2-pyrones with in situ generated
alkynyldifluoroboranes.9 As outlined in Scheme 1, our optimization studies highlighted some
unexpected trends; the reaction required the use of 3 equivalents of both the alkyne substrate
and the Lewis acid fluorophile for optimal yields. Moreover, alkynylated by-products were
observed when the reaction was conducted under ambient conditions. In this context, the mild
Diels-Alder cycloaddition of 1,3-dienes with vinyl- and alkynylboranes has also been
established by Singleton and co-workers. These processes take place at ambient temperature
and with excellent levels of regiocontrol via a [4 + 3] transition structure.10
3 The directed cycloaddition reaction raised some interesting questions with regard to the
reaction mechanism: 1) Is Lewis acid activation of the 2-pyrone operating as well as, or
instead of, the directed cycloaddition? 2) Does the directing group control reaction
regioselectivity or does the reaction proceed via Singleton [4 + 3] mechanism? 3) How are
the alkynylated by-products formed? In order to understand this reaction more clearly, we
undertook theoretical DFT calculations of the cycloaddition reaction in order to establish a
clearer picture of this unusually rapid cycloaddition process. Theoretical studies of
Lewis-acid promoted Diels-Alder reactions have been reported in the literature,11 though not on
these systems. We report herein our findings and their application in improving the Lewis
acid promoted cycloaddition reaction.
Computational methods
All calculations were performed at the DFT level of theory, by means of the hybrid B3LYP 12-13 functional as implemented in Gaussian0914. The 6-311G(d,p) basis set was used for the
atoms H, C, N, O, B, F and Cl. All structures of reactants, intermediates, transition states and products were fully optimized without any symmetry restrictions. Transition states were located using the QST3 algorithm15-17 or the Berny algorithm18.
A frequency calculation was carried out to characterize all optimized structures as local minima or transition states. Transition states were identified by having one imaginary frequency. An intrinsic reaction coordinate (IRC)19 calculation was performed for each of the
transition states to ensure that a given transition state connected the correct reactants and products. Solvent effects were included in all calculations through the IEF-PCM20-22 model implemented in Gaussian0914 with dichloromethane (DCM) as solvent. The atomic charges
were fitted to the electrostatic potential energy (ESP) following the Merz-Kollman scheme.23,24 All enthalpies and free energies quoted below were evaluated at 298.15 K
Theoretical Results
Our first objective was to attempt to explore possible mechanisms for the formation of the alkynylated aromatic boranes such as 3b,c shown in Scheme 1. Our initial hypothesis involved alkyne disproportionation prior to cycloaddition. However, Frohn and co-workers had shown that alkynyl difluoroboranes can be isolated and characterised as single compounds upon the treatment of alkynyl trifuoroborates with boron trifluoride.25
4 was considered. Following Ref. 27a, the enthalpy profile for the ligand exchange reaction between two alkynylborane IM monomers is shown in Scheme 2.
Scheme 2 The energy profile of the ligand exchange between two alkynylborane (monomer
IM) molecules in DCM. The inset shows the transition state TS1M structure, where the bonds forming are dashed.
In non-polar solvents like dichloromethane, the alkynylboranes will form a loosely coordinated dimer IIM, which results in a small enthalpy change of -2.6 kJ mol-1. This pre -reaction complex then undergoes ligand exchange to generate bis(alkynyl)borane ID and a molecule of BF3. Inspection of the transition state (TS1M) of this process shows that this
reaction involves a concerted mechanism via a four-membered ring involving two three -centre two-electron bonds (Scheme 2). The enthalpic barrier for the ligand (phenylacetylide moiety) exchange between two alkynylborane molecules is calculated to be +65.9 kJ mol-1
from the loosely coordinated dimer IIM. The product of this process IIIM is again a loosely bound complex. However, this complex is a dimer of the diphenylacetylide ID and BF3,
5 formation of bis(alkynyl)borane from two alkynylborane molecules is an endoergic process, since the Gibbs energy increases overall by 13.2 kJ mol-1 compared to two individual
alkynylborane molecules. Therefore, for this process to be viable it must be coupled with the Diels-Alder cycloaddition step (vide infra).
Following a similar pathway, the tris(alkynyl)borane IT can be formed from either an alkynylborane IM/bis(alkynyl)borane ID disproportionation, or by ligand exchange from two bis(alkynyl)borane ID molecules. The likely relative concentrations of IM and ID suggest that ligand exchange is more likely to happen via the reaction of IM and ID. Thus, the intermediates and transition states along this reaction pathway were optimized. The energy profile for the generation of tris(alkynyl)borane is shown in Scheme 3.
6 The enthalpy barrier for this disproportionation reaction is +65.5 kJ mol-1. It again delivers a
loosely coordinated product. Dissociation of this complex provides free tris(alkynyl)borane
IT, which is available to participate in the subsequent Diels-Alder reaction. We note that in this case the overall reaction Gibbs energy is negative (-2.5 kJ mol-1). Thus, our calculations show that equilibration of a series of alkynylboranes can take place, and that this equilibrium favours the formation of trisalkynylborane IT and its corresponding BF3-complex IIID. This needs to be taken into account when studying the subsequent Diels-Alder reaction.
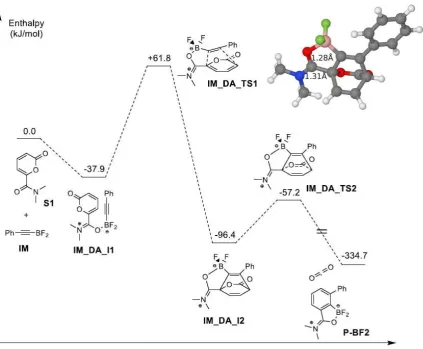
7 The mapped energy profile for the Diels-Alder reaction between alkynylborane IM with S1 is shown in Scheme 4. The first step in this regio-directing Diels-Alder reaction is the coordination of the amide carbonyl directing group of the 2-pyrone to the boron atom of the alkynylborane. This step lowers the enthalpy by -37.9 kJ mol-1. The subsequent Diels-Alder reaction therefore proceeds via this pre-reaction complex IM_DA_I1. Inspection of the cycloaddition transition state (IM_DA_TS1) shows that the Diels-Alder reaction can be described as asynchronous. Thus, the two bonds C1-CA, C2-CB do not form simultaneously. In the optimized TS structure, the bond length of C1-CA is 1.81 while the distance between C2 and CB is still as large as 2.97 as is clear from Figure 1(a). Thus, our calculations clearly indicate that the reaction via a directed [4 + 2] mechanism rather than the Singleton [4 + 3] mechanism.
8
Figure 1 The three transition states of the Diels-Alder reaction. Panel (a): reaction of IM
with S1. Panel (b): reaction of ID with S1. Panel (c): reaction of IT with S1.
[image:9.595.96.520.71.420.2]9 cycloadditions of 2-pyrones are known and have been studied by DFT methods in the past.27 Finally, the product of this cyclisation reaction is a bridged compound IM_DA_I2 which is shown in the inset of Scheme 4. It is worth noting that the O-C bond length of the carbonyl group in this bridged compound extends to 1.28 and that the N-C bond on the other hand shortens to 1.31 , which is quite similar to a C=N double bond. However, this bridged compound IM_DA_I2 is not stable towards release of CO2. The barrier for the CO2 release and rearrangement of the aromatic ring is +39.2 kJ mol-1 while the enthalpy change for this
step is -238.2 kJ mol-1.
We have also studied the related pathways of this regioselective Diels-Alder reaction starting with the bis(alkynyl)borane ID or the tris(alkynyl)borane IT. The corresponding enthalpy profiles are shown in Scheme 5 and Scheme 6, respectively. Comparing Scheme 5 and 6 to each other and to Scheme 4 shows some similarities and some differences between the three reactions. For Scheme 5, like for Scheme 4, the initial step is the formation of a pre-reaction complex, whereby it needs to be noted that the enthalpy change for Scheme 4 is about twice as large as for Scheme 5. It is therefore not surprising that the formation of this pre-reaction complex is actually endothermic for IT, as is evident from Scheme 6. The transition states for each of the three schemes on the other hand shows that the Diels-Alder reaction can be described as asynchronous in each case forming one C-C bond before the second one. In all three cases the bridged product of the DA reaction is not stable and should lose CO2 quite easily, given the relative barrier of ~40 kJ mol-1 in each case with a large reaction enthalpy. To be able to compare these reactions quantitatively, it is necessary to be able to define an effective barrier in each case. This can be less than straightforward, particularly when there are multiple transition states and minima to consider. Thus, in order to assign a barrier consistently, we use the Energetic Span (ES) model developed by Kozuch. 28-30 Within the ES model, the crucial states (i.e. stationary points on the potential energy surface) of the system are those that give the maximum energy difference between a minimum and a subsequent transition state, i.e. give you the maximum effective barrier or energetic span for the reaction.28-30 Thus, for Scheme 4, those states are IM_DA_I1 and IM_DA_TS1, which are referred to as the turnover determining intermediate (TDI) and turnover determining transition state (TDTS), respectively. As a result, the effective barrier for this reaction is 99.7 kJ mol-1.
10 bis(alkynyl)borane (ID) and 2-pyrone (S1).
Scheme 6 The energy profile of the regio-directing Diels-Alder reaction between
11 For Scheme 5 and Scheme 6, it is clear that the transition state (TS) of the cyclisation step, i.e. ID_DA_TS1 and IT_DA_TS1, respectively, is the TDTS. For Scheme 5, the pre-reaction complex is the TDI, whereas for Scheme 6, the separated reactants form the TDI. Therefore, the effective barrier, i.e. the energetic difference between TDTS and TDI, for Scheme 5 and Scheme 6 are calculated to be +94.0 and +95.7 kJ mol-1, respectively.
12 These results are shown in the third column of Table 1. If we assume that changes in entropy are similar for all three reactions, then we can define a relative rate based on the enthalpy alone. These relative rates are given in the fifth column of Table 1. It is clear from Table 1 that either calculation gives qualitatively the same results, namely that the reaction rates for
ID and IT are similar and much larger than the rate for IM.
Table 1 The effective barriers (energetic spans) for the Diels-Alder reaction with different dienophiles Dienophile kJ mol Relative rate kJ mol Relative rate
Alkynylborane (IM) 109.5 1.0 99.7 1.0
Bis(alkynyl)borane (ID)
100.7 34.8 94.0 10.0
Tris(alkynyl)borane (IT)
100.4 39.3 95.7 5.0
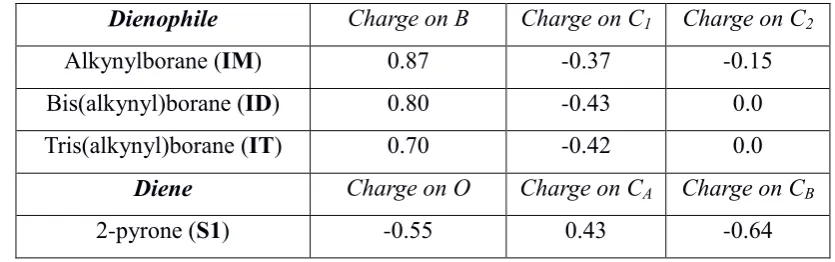
[image:13.595.66.536.226.375.2]Inspection of the energy profiles in Schemes 4-6 shows a clear dependency of the enthalpy of activation on the nature of the ligands around boron. In order to rationalize this result, an ESP (Merz-Kollman) charge analysis was conducted, which is given in Table 2 for each of the four reactants.
Table 2: Merz-Kollman (ESP) charges for each of the reactants involved in Schemes 4-6.
Dienophile Charge on B Charge on C1 Charge on C2
Alkynylborane (IM) 0.87 -0.37 -0.15
Bis(alkynyl)borane (ID) 0.80 -0.43 0.0
Tris(alkynyl)borane (IT) 0.70 -0.42 0.0
Diene Charge on O Charge on CA Charge on CB
2-pyrone (S1) -0.55 0.43 -0.64
[image:13.595.86.504.507.638.2]13 profile. At the same, the increasing charge on C1 (and decreasing charge on C2) with increasing degree of substitution would lead to more effective nucleophilic attack of C1 onto CA, which is apparent in the lowering of the barrier, as is clear from comparing Schemes 4-6. In fact, notwithstanding the fact that one cannot assume these charges to be accurate to 0.01, the slightly lower charge on C1 for IT compared to ID suggests a slightly higher barrier, as is clear from Table 1.
The calculations shown in Table 1 suggest that the cycloadditions should provide a mixture of products favouring mono- and dialkynylated boranes. However, experimental studies show that aryldifluoroboranes are the major products. We speculated therefore that the final stage of the cycloaddition process required a further equilibration of arylboranes, and therefore undertook a study into potential ligand exchange processes in the products. Interestingly, as shown in Scheme 8, calculations show that the boron trifluoride promoted conversion of P
-BR2viaP-BFR to P-BF2 is exothermic. For the first transformation from P-BR2 to P-BFR and are -16.1 kJ mol-1 and -22.0 kJ mol-1, respectively. For the final transformation into P-BF2, and are -26.1 kJ mol-1and -31.5 kJ mol-1, respectively.
Scheme 7 The ligand (phenylacetylide group) exchange reaction between P-BR2, P-BFR
and BF3
14
Scheme 8 The energy profile of the ligand exchange reaction between P-BR2 and BF3.
Please note that dissociation of the final complex is endothermic by 10.7 kJ mol-1.
Scheme 9 The energy profile of the ligand exchange reaction between P-BFR and BF3.
15 Our calculations clearly show that the formation of P-BF2 should indeed be quite facile, given the low barriers, which are significantly lower than the corresponding barriers to formation of ID and IT. Thus, our calculations clearly explain and confirm the experimental observations. However, the above discussion does not consider the role of the directing group and the effect it is has on the formation of the final products. Unfortunately, the directing group for 2-pyrone is in such a position, that the study of a non-directed reaction leading to the same products is not feasible. However, one of us has previously reported a non-directed Diels-Alder reaction as a route to synthesize functionalized aromatic boronic esters, as shown in Scheme 10.31
Scheme 10 Alkynylboronate Diels-Alder reaction, where BPin is pinacolborane.
16 understand the role of the directing group and to be able to explain the difference between the non-directed Diels-Alder reaction and the novel mild directed Diels-Alder reaction, we decided to re-examine the MEP for this previously reported non-directed Diels-Alder reaction instead. The enthalpy profile for this reaction was calculated and is shown in Scheme 11.
Scheme 11 The energy profile for the non-directed Diels-Alder reaction as shown in Scheme 10.
17 time, leading to a bridged intermediate, which will evolve CO2 to yield the final product.
Comparison of Scheme 11 to Schemes 4-6 shows that the effective barrier for the non -directed Diels-Alder reaction pathway is much higher than for the directed Diels-Alder reaction. For the non-directed DA reaction, the TDI is non-d_DA_I1 for the formation of both products, whereas the TDTS is the cycloaddition transition state in both cases. Thus, the effective barrier to generate the more stable product Pb is +118.3 kJ mol-1 whereas it is
+138.5 kJ mol-1 for generating the product Pa. From Scheme 12, the product Pb is predicted to be the dominant product due to the significantly lower barrier, which is also confirmed by the experimental observations.
By considering the effective barriers in the three directed Diels-Alder reactions which were discussed above (varying from 94.0 kJ mol-1 to 99.7 kJ mol-1), it is clear that the non
-directed reaction will require harsher conditions. Moreover, it seems therefore unlikely that this mechanism could be in operation in parallel to the directed Diels-Alder reaction.
[image:18.595.66.529.537.754.2]In order to ensure that the above analysis was not specific to the amide substrate S1, all calculations were repeated with the pyridine-pyrone 1. The complete reaction profiles for these reactions are given in the supporting information. However, the most important data is summarized in Table 3.
Table 3: Comparison of the most relevant enthalpies for the reaction of the three boron-alkynyl species with the amide (S1) and pyridine (1) substrates. All enthalpies quoted are in kJ mol-1
amide (S1) pyridine (1)
(P-BF2) -334.7 -335.0
(P-BFR) -318.2 -323.8
(P-BR2) -298.6 -310.5
(P-BF2) 99.7 101.4
(P-BFR) 94.0 91.2
(P-BR2) 95.7 92.2
(P-BR2 P-BFR) -16.1 -18.5
(P-BFR P-BF2) -26.1 -30.3
18
(P-BFR P-BF2) 33.2 62.9
It is clear from the comparison of the enthalpies for the reaction of the boron-alkynyl species with either the 2-pyrone amide (S1) or the 2-pyrone pyridine (1), that the enthalpic parameters for both reactions are very similar, providing reassurance that the analysis of the reaction with S1 is applicable to other substrates as well. The calculations also suggest that the reaction with 1 should be faster than the reaction with S1 under the same conditions. However, the higher barriers for the subsequent disproportionation process back from P-BR2 to P-BF2 should lead to higher conversion to difluoroborane in the case of S1 versus 1.32
Overall therefore, our calculations have highlighted three key processes in the boron trifluoride promoted cycloaddition of alkynyltrifluoroborates and 2-pyrones: (1) rapid pre -equilibration of alkynylboranes through ligand exchange. (2) Lewis acid/base complexation of the dienophile/diene, respectively, followed by cycloaddition via a [4 + 2] mechanism. (3) Equilibration of cycloadducts to a single product, thereby avoiding the formation of product mixtures. It is clear that the precise Lewis acid used has a significant effect on the effective barriers. Thus, it should be possible to use alternative Lewis acids to modulate these effects, and our investigations towards this end are described later in the next section.
Experimental Results
Our computational investigations highlighted the potential of a mechanism that involved
rapid and reversible formation of tris(alkynyl)borane, followed by cycloaddition and
disproportionation. We wanted to confirm the viability of this mechanism experimentally. In
this context, Siebert and co-workers reported the synthesis and characterization of
tris(t-butynyl)borane 4 as a Lewis acid-base complex with pyridine,33 and it occurred to us that we
could employ this chemistry to probe the key cycloaddition step. More specifically, if the
tris(alkynyl)borane was unreactive with pyrone 1, then we should be able to observe the
resulting complex and compare data with that reported by Siebert. However, if the alkyne
was reactive, then we would expect to recover the corresponding cycloadduct 5c. In the event
that 5c was isolated, we could confirm the possibility of the final disproportionation process
by subjecting this compound to a boron Lewis acid.
19 We prepared tris(t-butynyl)borane 4 according to the literature procedure and found that it
underwent cycloaddition with 1 to generate bis(alkynyl)borane derivative 5c in 95% yield.
Notably, we did not observe any chloroborane derived cycloadducts (Scheme 13). This result
supported our computational calculations in which the tris(alkynyl)borane derivative
functions as a 2 component in the [4 + 2] cycloaddition between 2-pyrones and potassium
(alkynyl)trifluoroborate salts.
Scheme 13 Synthesis and cycloaddition of a trialkynylborane
Further control experiments were carried out to validate the possibility of the final
disproportionation step (Scheme 14). We first verified that difluoroborane complex 5a was
obtained upon exposure of bis(alkynyl)borane derivative 5c to boron trifluoride (scheme 14).
The observation that this process takes place smoothly at room temperature matches the
results of our theoretical study that predict the barrier of ligand exchange between BF3 and
products to be quite small (c.f. Scheme 7).
Scheme 14 Equilibration of dialkynylborane to difluoroborane
In order to demonstrate the generality of the disproportionation process, complexes 6b and 6c
were also prepared (Scheme 15). Treating pyrone S1 with trifluoroborate salt 2 in the
presence of boron trifluoride for 4 minutes led to the isolation of 6b and 6c in 11% and 17%
20 when a 1:1 mixture of 6b and 6c was treated with boron trifluoride, further validating the
computational results (Scheme 15b).
Scheme 15 Equilibration of alkynylboranes to difluoroborane.
Both theoretical and experimental results allow us to propose a mechanism accounting for the
transformation (Scheme 16). A rapid equilibrium between (alkynyl)dihalogenoborane A,
bis(alkynyl)halogenoborane B and tris(alkynyl)borane C followed by the [4 + 2]
cycloaddition between the 2-pyrone and each borane derivative affords cycloadducts D, E
and F, respectively. Finally, intermediates E and F are converted into dihalogenoborane
cycloadduct D by reacting with BX3.
Scheme 16 Overall mechanistic scheme for the directed cycloaddition reaction.
Extension of the methodology using BCl3
Our theoretical studies indicated that the precise Lewis acid used has a significant effect on
21 16, it seemed logical that other boron trihalides (BX3) could act as suitable Lewis acids to
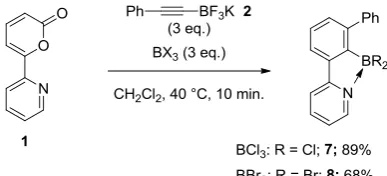
promote the cycloaddition process. Indeed, we found that both BCl3 and BBr3 can function as
effective promoters of the cycloaddition (Scheme 17). Using the reaction conditions initially
reported in our previous study (i.e. 3 equivalents of potassium (alkynyl)trifluoroborate 2, 3
equivalents of Lewis acid in refluxing CH2Cl2),9 cycloadducts 7 and 8 were isolated in 89%
and 68% yield, respectively.
Scheme 17 Use of alterative BX3 promoters
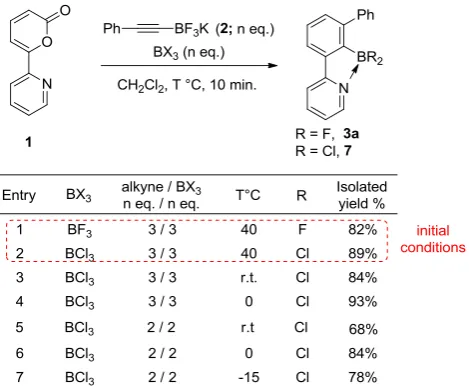
As BCl3 appeared to be more promising, an optimization study was carried out on the
cycloaddition of 1 and 2 (Table 4). Interestingly, we found that the temperature of reaction
could be reduced to rt or 0 °C without affecting the yield of isolated product (entries 3 and 4).
Furthermore, the number of equivalents of alkyne and BCl3 could be reduced without
significantly reducing the yield of 7 when the reaction was carried at 0 °C (entry 6). This
temperature was found to be optimal since higher or lower reaction temperature (entries 5 and
7) led to the decrease of isolated yield of 7. Further reduction of the number of equivalents of
alkyne and BCl3 gave incomplete conversions after prolonged reaction times. Overall, this
study demonstrated that higher reactivity could be achieved when BCl3 was used as the
[image:22.595.199.393.237.325.2]reaction promoter.
22 The reaction scope was next investigated using our optimized conditions (i.e. 2 equivalents of
potassium alkynyltrifluoroborate and 2 equivalents of BCl3). First, we prepared a series of
potassium alkynyltrifluoroborate salts and used them in the cycloaddition reaction with
2-pyrones 1 and (Table 4). In both cases, similar trends were obtained. Alkyne substrates
bearing phenyl-, nbutyl- and tbutyl-substituents reacted rapidly under mild conditions to give
the corresponding functionalised aromatic products 7, 12-19 in good to excellent yields
(entries 1-8). The combination of thiazole-substituted 2-pyrone 20 and TMS-alkyne 11
resulted in a slower reaction and this process was conducted at slightly elevated temperature,
[image:23.595.182.417.72.265.2]resulting in some protodesilylation of the product.
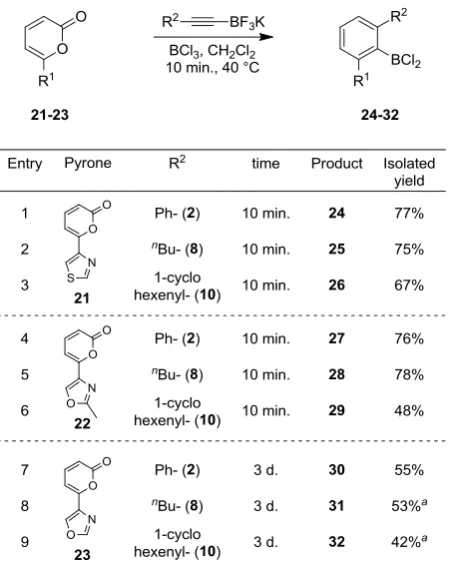
23 The cycloaddition of 2-pyrones attached at the 4-position of various 1,3-azoles groups was
found to be more challenging, and these reactions were generally conducted at elevated
temperature (Table 6). Nonetheless, these reactions allow a range of biaryl products to be
accessed in good yield. Pyrone 21, which is a regioisomer of pyrone 20 in Table 5, also
underwent the cycloaddition with alkynes 2, 8 and 10 (entries 1-3). Pyrone 22, having a
2-methyloxazole as directing group, was also efficient under our reaction conditions, affording
products 27-29 in good yields (entries 4-6). Interestingly, a small change in the
2-methyloxazole motif showed a dramatic change in reactivity; 2-pyrone 23 proved to be
reluctant to undergo the cycloaddition, requiring a reaction time of 3 days and affording
cycloadducts 30-32 in modest yields (entries 7-9). Notably however, attempts to mediate the
reaction of 23 and 2 with BF3.OEt2 failed to deliver any product and only starting 2-pyrone
[image:24.595.186.412.358.641.2]was returned in this case, highlighting the potential of BCl3 to offer enhanced reactivities.
Table 6 Cycloaddition scope
Conclusion
We have carried out theoretical and experimental studies of the mechanism of a Lewis acid
promoted cycloaddition of alkynyltrifluoroborates and 2-pyrones bearing a Lewis base
promoter. Calculations show that rapid equilibration of in situ generated alkynyl
24 trialkynylboranes. These latter two species undergo very rapid reaction via coordination of
the Lewis basic donor and a [4 + 2] cycloaddition, followed by disproportionation to generate
the observed aryl difluoroboranes products. The reactions can be further enhanced by the use
BCl3, allowing cycloaddition to take place within 10 minutes at 0 oC to generate a range of
functionalized aromatic products in high yield.
Supporting information
The supporting information contains general experimental protocols and NMR spectra.
Computational supporting data includes Cartesian coordinates (in XYZ format) and energies.
This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgements
We are grateful to AstraZeneca, EPSRC, and The University of Sheffield for financial
support. All calculations were performed on the “Jupiter” cluster of the Theoretical Chemistry Group at the University of Sheffield as well as the central “Iceberg” cluster of the University of Sheffield. The authors thank Mr. Christopher M. Parks for useful discussions
and help with the calculations.
References
1. Woodard, B. T.; Posner, G. H. Adv. Cycloadd. 1999, 5, 47. 2. Diels O.; Alder, K. Ann. Chem. 1931, 490, 257.
3. Afarinkia, K.; Vinader, V.; Nelson, T. D.; Posner, G. H. Tetrahedron 1992, 48, 9111.
4. (a) Delaney, P. M.; Moore, J. E. Chem. Commun. 2006, 42, 3323; (b) Delaney, P. M.; Browne, D. L.; Adams, H.; Plant, A.; Harrity, J. P. A. Tetrahedron 2008, 64, 866.
5. Evnin, A.; Seyferth, D. J. Org. Chem. 1967, 32, 952. 6. Seyferth, D.; White D. J. Organomet. Chem. 1972, 34, 119.
7. Kranjc, K.; Štefane, B.; Polanc, S.; Kočevar, M. J. Org. Chem. 2004, 69, 3190.
8. Vivat, J. F.; Adams, H.; Harrity, J. P. A. Org. Lett. 2010, 12, 160.
9. Kirkham, J. D.; Butlin, R. J.; Harrity, J. P. A. Angew. Chem. Int. Ed. 2012, 51, 6402.
10. (a) Singleton, D. A.; Martinez, J. P. J. Am. Chem. Soc. 1990, 112, 7423; (b) Singleton, D. A. J. Am. Chem. Soc. 1992, 114, 6563; (c) Leung, S.-W.; Singleton, D. A. J. Org. Chem. 1992, 57, 4796; (d) Leung, S.-W.; Singleton, D. A. J. Org. Chem. 1997, 62, 1955.
25 Soto-Delgado, J.; Saez, J. A.; Tapia, R. A.; Domingo, L.R. Org. Biomol. Chem. 2013, 11, 8357; (g) Arno, M.; Picher, M. T.; Domingo, L. R.; Andres, J. Chem. Eur. J. 2004, 10, 4742; (h) Sarotti, A. M. Org. Biomol. Chem. 2014, 12, 187; and references therein.
12. (a) Becke, A. D. Phys. Rev. A, 1988, 38, 3098; (b) Becke, A. D. J. Chem. Phys., 1993, 98, 1372. 13. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B, 1988, 37, 785.
14. Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D.J. Gaussian, Inc., Wallingford CT, 2009.
15. Peng, C.; Schlegel, H. B. Israel J. Chem., 1993, 33, 449.
16. Peng, C.; Ayala, P. Y.; Schlegel, H. B.; Frisch, M. J. J. Comp. Chem., 1996, 17, 49. 17. Cances, M. T.; Mennucci, B.; Tomasi, J. J. Chem. Phys., 1997, 107, 3032.
18. Schlegel, H. B. J. Comp. Chem., 1982, 3, 214. 19. Fukui, K. Acc. Chem. Res., 1981, 14, 363.
20. Mennucci, B.; Tomasi, J. J. Chem. Phys., 1997, 106, 5151.
21. Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Chem. Phys. Lett., 1998, 286, 253. 22. Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. J. Chem. Phys., 2002, 117, 43. 23. Sing, U. C.; Kollman, P. A. J. Comput. Chem., 1984, 5, 129.
24. Besler, B. H.; Merz, K. M.; Kollman, P. A. J. Comput. Chem., 1990, 11, 217. 25. Bardin, V. V.; Adonin, N. Y.; Frohn, H.-J. J. Fluor. Chem. 2007, 128, 699.
26. Negishi, E.-i.; Idacavage, M. J.; Chiu, K.-W.; Yoshida, T.; Abramovitch, A.; Goettel, M. E.;
Silveira, A.; Bretherick, H. D. J. Chem. Soc., Perkin Trans. 21978, 1225.
27. (a) Cao, Y.; Osuna, S.; Liang, Y.; Haddon, R. C.; Houk, K. N.; Braunschweig, A. B. J. Am. Chem. Soc.2013, 135, 9240; (b) Štefane, B.; Perdih, A.; Pevec, A.; Šolmajer, T.; Kočevar, M. Eur. J. Org.
Chem.2010, 5870; (c) Kranjc, K.; Kočevar, M. New J. Chem. 2005, 29, 1027.
28. Kozuch, S.; Shaik, S. Acc. Chem. Res.2011, 44, 101.
29. Kozuch, S.; Martin, J. M. L. ChemPhysChem.2011, 12, 1413. 30. Kozuch, S. WIREs Comput. Mol. Sci. 2012, 2, 795.
31. Kirkham, J. D.; Leach, A. G.; Row, E. C.; Harrity, J. P. A. Synthesis2012, 44, 1964.
26
temperature, 70% conversion is obtained with 80% P-BF2 and 20% P-BFR and only traces of P -BR2. If instead 1 is mixed with IM1 under the same conditions, then 92% conversion is achieved with 45% P-BF2, 27% P-BFR, and 28% P-BR2 (as estimated by 400 MHz 1H NMR spectroscopy of crude reaction mixtures).
33. Bayer, M. J.; Pritzkow, H.; Siebert, W. Eur. J. Org. Chem.2002, 2069.