0022-538X/91/084359-11$02.00/0
CopyrightC)1991, AmericanSociety forMicrobiology
Analysis of
the Herpes Simplex Virus Type 1
Oris
Sequence:
Mapping
of
Functional
Domains
DAVID W. MARTIN, SWATI PALIT DEB, JENNIFER S. KLAUER, AND SUMITRADEB* Department ofMicrobiology, University of Texas Health Science Center,
7703 Floyd CurlDrive, San Antonio, Texas 78284-7758 Received25January 1991/Accepted7 May 1991
The herpes simplex virus type 1 (HSV-1)
Oris
region resides within a 90-bp sequence that contains two bindingsitesfortheorigin-binding protein (OBP),designatedsites IandII.AthirdpresumptiveOBP-binding site(III)withinOris
hasstrongsequence similarity to sites I and II, but nosequence-specificOBPbindinghas yetbeendemonstratedatthissite. Wehavegenerated mutations in sitesI, II,and III anddetermined their replication efficienciesin atransientin vivo assay in thepresence ofahelper virus. Mutationsinany one ofthe sites reduced DNA replication significantly. To study the role ofOris
sequence elements in site I and the presumptive site III in DNA replication, wehave also generated a seriesof mutations that spanfrom site I acrossthe presumptive bindingsite Ill. These mutants were tested for their ability toreplicate and for the ability to bind OBP by using gel shift analyses. The results indicate thatmutations across site I drastically reduce DNA replication. Triple-base-pair substitution mutations that fall within the crucial OBP-binding domain, 5'-YGYTCGCACT-3' (whereYrepresents C or T), show a reduced level of OBPbindingandDNA replication. Substitution mutations in site I that are outside this crucial binding sequence show a more detrimentaleffectonDNAreplicationthan on OBPbinding. This suggests that these sequences are required for initiation ofDNA replication but are not critical for OBP binding. Mutations across the presumptive OBP-bindingsite III also resulted in a loss in efficiency of DNAreplication. Thesemutations influencedOBP binding toOris
in gel shift assays, even though the mutated sequences are not contained within known OBP-bindingsites.Replacement ofthewild-type site III with a perfectOBP-bindingsite Iresults inadrastic reduction ofDNAreplication.Thus, our DNAreplicationassays and in vitro DNA-binding studies suggest that thebinding oftheoriginsequencebyOBP is not the onlydeterminingfactor forinitiation of DNAreplication invivo.Thegeneral modelof initiationof DNAreplication
emerg-ing from studies on prokaryotic and eukaryotic systems indicates that most sequence-specific initiations start with therecognitionof and bindingtotheorigin ofreplicationby an initiatorprotein (2). Thisbinding signals the subsequent steps whichinvolvelocalizationof other protein factors such ashelicases and DNApolymerases, etc. The recognitionof the origin sequence by the initiator and the subsequent protein-protein interactions are of very high precision and result in aspecialized nucleoprotein structure. Herpes
sim-plex virus type 1 (HSV-1) is an attractive model system in
which to study the initiation of viral DNA replication.
Presumptive origins of DNA replication have been mapped on the viral genome, and the virus encodes many of the
proteins required for initiationofDNA replication. Thus, a study ofthe mechanism of initiation can be undertaken by
usinggenetic andbiochemical approaches.
HSV-1 has alinear,double-stranded DNAgenome of 152
kbp(21). Definitivemapping of viral DNA replication origins was accomplished by studying the replication ofdefective interfering particles (12-14, 16, 17) and cloned virus DNA segments in the presence of helper virus (23, 25, 27, 29). ThreepresumptiveoriginsareontheHSVgenome: two
Oris
are present as one copy in each c-repeat of the viral chromosome (27), and a third origin, designated OriL, is associated with theunique long (UL) region (29).
Sevenvirally encoded genesarenecessaryfor viral DNA
synthesis (22, 31). Elias et al. (11) identified a factor (the
* Correspondingauthor.
origin-binding protein or OBP) from infected cell extracts that could sequence specifically bind to
Oris.
OBP wassubsequentlyshownbyOlivoetal.(24)tobeencodedbythe UL9 gene and is one of the seven virally encoded genes essential for DNA replication. We (3) and later others(15,
28) have shown thatoriginbindingofOBPcanbecorrelated
to origin function. Because of its specific origin-binding
function, OBP may represent the initiator protein for viral DNAreplication.
The
Oris
sequencecontainsa45-bpimperfectpalindromewith a central A-T-rich stretch. Previous analyses have
demonstratedthe existenceoftwo OBP-bindingsites in this
region(10, 24).Byusing site-directed mutagenesis(3,9) and
methylationinterference (18), it has beendemonstrated that the OBP recognition site is included within the sequence
5'-YGYTCGCACT-3' (whereY representsCorT). The
Oris
region alsocontains asequence which hasstrong sequence
similarityto OBP-binding sitesI and II, designated siteIII.
No
sequence-specific
OBP binding, however, has yet beendemonstrated to this site (9, 28). Thus, the role of this sequence in any
Oris-OBP
interaction is not established.Analysis oforiginsequences inherpes simplex virus type 2 (HSV-2) has shown that deletion of part of a sequence
correspondingtosite III in HSV-1resulted inadramaticloss in DNA replication (20). Recent analysis in HSV-1, how-ever, has shown that deletion of site III affects
replication
efficiency onlymoderately (28).
We have mutagenized sites
I,
II,
and III.Replication
efficiencies of these mutated
origins
demonstrate that foroptimal
replication
all the three sites arerequired.
We have 4359on November 10, 2019 by guest
http://jvi.asm.org/
4360 MARTIN ET AL.
A. |AGCTTOOCCGCCGOOTAAAAGAAGTGAGAACGCO E CGTTCGCACTTCGTCCCAATATATATATTAT |TAGOCGAAGTGCGAOCACTOGCGCCGGCCCCGGGC
IACCGGCGGCCCATTTTCTTCACTCTTGCGCTTCGCA AGCOTGAAGCAGGGTTATATATATATAATAATCCCO ICTTCACGCTCGTGACCGCGGCCGOOOCCCGGTAC
PAUNDROME
B.
III
I
II5'-CGGGT AAGAAGTGAGAACGC X GCGTTCGCACTTCGTC AATATATATATATTATT
AGGCGAAGTGCGAGCACGrGCGCCGGCCCCGG-3'wildt
3'-GCCCA TTCTTCACTCTTGCGqCGCAAGCGTGAAGCAGGTTATATATATATAATAA CCGCTTCACGCTCGTG^CCGCGGCCGGGGCC-5 YP
TTT bs-7
TAC bs-8
TCC bs-9
CCT bs-1O
ATA bs-11
GG bs-12
ATA bs-13
GGT bs-14
AGA bs-15
GAG bs-17
GCCCATTTTCTT ACTCTTGCGCTTCGCAAGCGTGAAGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC 16 del
A
GCAACTTTTCTTCACTCTTGCGCTTCGCAAGCGTG AGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC bs-18del
AAA A
FIG. 1. Construction ofOris and its mutants. (A) Wild-type Oris constructed from 6 cassettes of overlapping oligonucleotides. The synthetic origin is cloned in the pOR vehicle (8). (B) Mutations were introduced by using cassette-directed mutagenesis. Overlapping oligonucleotidescontaining the desired sequences werehybridized, and theresultingcassettewasligated into a pOR vector. Forsubstitution mutants, Aresidues were exchanged for C and G for T. A single strand of the mutations is shown thatcorrespondstothe bottom strand(3'
to5') ofthewild-typeOris sequence. For 16del and bs-18del, thepositions of mutationsareindicatedbyarrowheads.
also performed base substitution mutagenesis of the region spanning site I through site III. These mutants have been assayed for their ability to support DNAreplication and to bind OBP. Our results indicate that the crucial OBP-binding domain at site I is required for both DNA replication and OBP binding; flanking nucleotides are essential for the initiation of DNA replication but are not critical for OBP
binding. Sequences in site III are essential for efficient replication and can modulate OBP bindingat
Oris.
MATERIALS AND METHODS
Plasmidcloneconstruction and mutagenesis. Construction ofwild-type and mutant
Oris-containing
plasmids has been describedpreviously (6). Origin sequences have been cloned between the HindIII and NcoI sites of a pOR vehicle (8) which consists of pML2 nucleotides 651 to 4361 with aHindlIl linker added to the 651 site and a polylinker (NcoI,
Sall, BamHI, andXmaI) added to theEcoRI site at nucle-otide 4363. Recombinant origins were constructed by using cassettes ofoverlapping oligonucleotides as described
pre-viously (5-7) (Fig. 1). Cassettes ofoverlapping
oligonucleo-tideswerehybridizedand thenligatedinto thepORvehicle. This mixture was usedto transform Escherichia coli DH5.
Ampicillin-resistant colonies were picked and screened for the mutations by Sanger dideoxy sequencing of both strands. Deletion (del) mutant 16 del and base substitution (bs) mutant bs-18 del were fortuitous isolates encountered
whilescreening for other mutants. For substitution mutants, A residues were exchanged for C and G residues were exchanged for T. The desired plasmids were purified by
cesium chloride-ethidium bromidedensitygradient centrifu-gation.
DNAreplication assay.Transient in vivoreplicationassays were performed to assess the function of mutant origins. Briefly, subconfluent Vero cells in 100-mm2 plastic dishes weretransfected with1,ugofsupercoiled testplasmid by the calciumphosphate precipitation techniqueasdescribed ear-lier (6, 7). At 4 h posttransfection, cells were shocked for2 minwith15% glyceroland washed with Hanksbalanced salt
solution, and the medium was replaced. Infection was started 6 h posttransfection with 5 to 10 PFU per cell of
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.66.557.71.434.2]4361
PALUNDROME
III I 11
CGCGTTAGAACGMTAGAAC IWCG¶ICGCA G-CCAATATATATATAICATT =CGAG5WGA^CA >cGcccG
GCCCAIX7lCA AAG AGlTATATATATATAATAACCCGC ACGCGTGAI:;CGCGGCCGC wildtype
CGGGTAATAGAAGTGAGAACGCG
GCCCAITA¶ICACTC'IGCGC
TCGCACGCxGTCCCAATATATATATATTATTAGGGCGCrGKAGC GAGACrGCGGCCGGCCCCGC;
AGCGUGAAGCAGGATATATATATAATAATCCCGCTTCACGCTCGTI ACCGGCCGGGGCC
CGGGrAAAAGAAGTGAGAACGCGAAGCIGCACrGTCCCAATATATATAT TrrA GCCCAimI-iACTTACITCGCCGCAAGCGTCAAGCAGGGTTATATATATATAATAAT
CmGrGAAAAGA
AGlfi>tCGCGAAGCGTTCGCACTTCGTCCCcAATATATATATATATTAsGCGCGAAGWCrG
AWACTGGCGCCGGCCCCGG GCCCAn:C q)CGCTCCAAGCGTGAACEAGGGrr TTTATATATATATATiU,"-CCGC-1- U.A,CA!C'{ 1rC;.M CCC(C; CECCGCGTICGCACTTCGTCCCAATATATATATATTATTA
CGCAAGCGIGAAGCAGGGTTATATATATATAATAAT
A.
*0
1-1
LOm
1-1
ir-- I
-CL) cl) CL) -0 .0 'D
>- I
-=0
4 Progeny l w
9.
B.
(2) "'
.0
1.0_
0.j-a
C)
.C32
0CY)
z0
HSV-1 strain KOS. Incubations were at 37°C. Cells were
harvested 16 hpostinfection. The resulting cell pellet was
lysed in 10 mM Tris (pH 7.0)-100 mM EDTA-0.5% sodium dodecyl sulfate(SDS)-proteinase K (100 ,ug/ml) at 50°C for 12 h. This was followed bytwo phenol-chloroform extrac-tions and onechloroform extraction. Theresultingaqueous
fractionwasethanolprecipitated and digested with RNase A
(10 ,ug/ml) for 1 h at 37°C, followed by a second ethanol
precipitation. Samples of the resulting DNA were digested
with BamHI and DpnI. BamHI linearizes the plasmid, while DpnIcleaves5'-GATC-3' sequences onlywhen theadenine
ismethylated. Since plasmids aregrownindam-positiveE. coli DH5, the adenine is methylated, and the resulting
sequences are cleaved by DpnI. Replicated DNA is not methylatedatthesepositionsandisnotcleavedby DpnI.As
aresult, replicated DNA remainsasunit-length monomers,
while input DNA is digested into many small fragments. DigestedDNAswereelectrophoresedina0.8%agarosegel.
Gels wereblottedonto GeneScreen Plus (DuPont) by alka-line transfer and then hybridized with nick-translated pBR322DNA.The blotwaswashed, dried,andexposedfor autoradiography. Autoradiograms exposed with no
intensi-FIG. 2. Replication of site I, site II, and site III mutants. Wild-type and mutant plasmids were transfected into Vero cells, followed by infection with HSV-1 strain KOS. The DNA was
isolated and digested with DpnI and BamHI, runonanagarosegel, Southern blotted, andprobed with nick-translated pBR322. Muta-tions tested are shown above. (A) Autoradiogram of replication assay. The progeny(replicated DNA) and input DNA are shown. Notetheloss in replication in del I (site I mutant), pORS-1 (deletion of siteII), bs-15(triple-base-pair substitution in site III), and del 31 (deletion of sites II and III).(B) The histogram shows the relative
replication efficiencies of themutantsversus wild-typeOris. Auto-radiograms of the replicationassay werescannedby laser
densitom-etry,andthesignalwasnormalizedtowild-type levelsonthe basis
ofinput (the largestDpnI-generated fragment).
fiers wereused for laserdensitometric scanningtocompare
therelativeefficiencyofreplication of the variousconstructs after normalization of theinput relative towild-type levels. Asa measure of the input DNA,availableas atemplate for
DNA replication, wehave used the largest DNAfragment arising by DpnI digestion. This serves as a control for our
transfection.
Generation of invitro-synthesizedOBPderivative.Deb and Deb (3a) recently demonstrated that the DNA-binding domain ofOBP is contained within a 269-amino-acid
seg-ment near the C terminus. A C-terminal 318-amino-acid
fragment of OBP covering the DNA-binding domain was,
therefore, used for DNA-binding studies. This fragment is designated del 1-534 and retains the sequence-specific DNA-binding activity of wild-type OBP (3a). To generate the protein, a BamHI (21655)-EcoRV (21463) fragment of the OBP gene was used. A NcoI linker was added to construct an initiator codon at the BamHI site. This frag-ment wascloned into a pGEM3 vector downstream of the SP6promoter. Ethidium bromide-cesiumchloride gradient-purified plasmid DNA was linearized by EcoRI, phenol-chloroformextracted, andethanolprecipitated. Transcripts
weresynthesized bySP6 polymerase followingtheprotocol
supplied by Promega. The DNAtemplate wasremoved by treatmentwith RNase-free RQ1 DNase (Promega), phenol-chloroform extracted, and ethanol precipitated. The
syn-thetic RNA was then translated in vitro by using rabbit reticulocyte lysates (Promega) in the presence of
[35S]me-thioninetolabel thenascentprotein accordingto the
manu-facturer'sprotocol.
DNA-bindingassays.(i) Filter-bindingassay. Filter-binding competition experiments were performed to determine the specificity of the binding of in vitro products with origin
sequences. The in vitro translation mixture was incubated with 32P-labeled
Oris
probe on ice for 30 minin a bindingdel-1
pOR-SI
bs-15
del-31
'i,
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.83.288.238.488.2]AL.
PAUNDROME
III
I
II
5-CGGGTAAAGAAGTGAGAACGCG GCGTTCGCACTTCGTCC AATATATATATATTATT
GGCGAAGTGCGAGCACT8GCGCCGGCCCCGGw3
t 3GCCCA TTCTTCACTCTTGC CGCAAGCGTGAA TTATATATATATAATAAQCCCGCTTCACGCTCGTGCCGCGGCCGGGGCC-CCT ATA GG ATA
TTT TAC TCC
bs-7 bs-8
bs-9 bs-10
bs-1l bs-12 bs-13
1.0
0)
.5
w
0.5
0
0)
cc
CN '0" )M N
1L
I I IrO C
co) C)l)m -0
n0 .0 .0
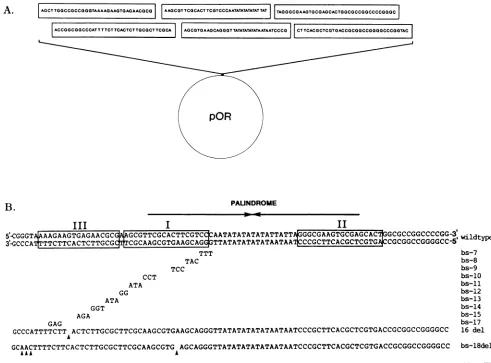
FIG. 3. Replication of site I mutants. Wild-type and mutant
plasmids were transfected into Vero cells, followed by infection with HSV-1 strainKOS. The DNA wasisolated and digested with DpnI and BamHl, run on an agarose gel, Southern blotted, and probed with nick-translated pBR322. Mutations tested are shown above. Histogram shows relative replication efficiencies of site I
mutants versus wild-type Oris. Autoradiograms of the replication assay were scanned by laser densitometry, and the signal was
normalizedtowild-type levelsonthe basis ofinput.
III
I
CGGGTAAAAGTGAGACGCCfAGCGTICGCACTrCGTIC4AATATATATATATTATTA
GCCCAI mACr T TATAATAAT
CCT ATA
rrI TAC
TCC
0) D.,
C) C) C)
I I I
0c
0.0.0.
I >%
03
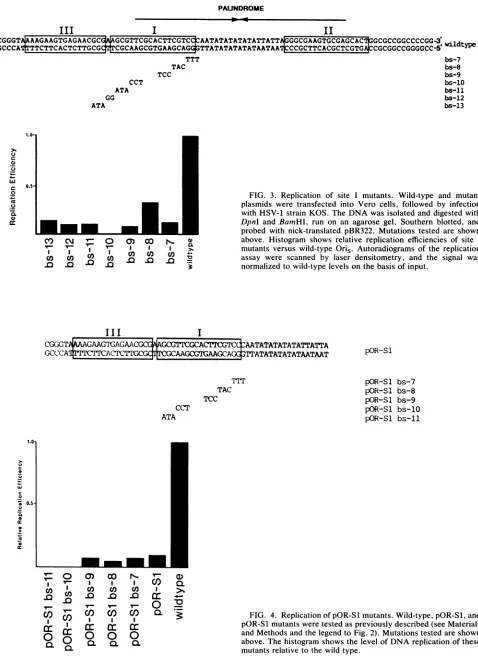
FIG. 4. ReplicationofpOR-Slmutants.Wild-type,pOR-Sl,and
pOR-Slmutantsweretestedaspreviously described (seeMaterials andMethods andthelegendtoFig. 2). Mutations testedareshown
above. The histogramshows thelevel of DNA replication ofthese
mutants relativetothewild type. pOR-Sl
pOR-Sl pOR-Sl pOR-Sl pOR-Sl
pOR-Sl bs-7 bs-8 bs-9 bs-10 bs-11
v
O
CO I Q
c 0
z
LU
c 0
1;1
.2
OL
cc11 T10 Ici
cc11
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.84.562.64.729.2]Oris
A.
Wild-type OBP
Del 1-534
(A
0
CL 0:
cr c C)
tO
ar a
CL et
I O. Oa
CI.
E
I I I
Sf.Lew
1 13 1 2 12 364534 686 804 851
1 534
4,
FIG. 5. (A) Lines showing schematically the wild-type and del 1-534genes. The solid bar represents amino acid sequences remain-ing; the hatched bar represents amino acid sequences deleted. Some ofthe amino acid positions are indicated by numbers. (B) Filter-binding competition experiments showing Oris-specificbinding by thein vitro-synthesized del 1-534. The in vitro translation mixture wasincubatedwith32P-labeledOrisprobe as described inMaterials andMethods. Afterincubation, the mixtures were passed through nitrocellulose filter papers and washed. The bound DNA was countedby Cerenkov countingandthen eluted fromthefilter and loadedonto a5% nativepolyacrylamide gel. DNA was visualized by autoradiography. Each lane contained 3 ,u1 of in vitro-translated product,labeledOris,and theindicated amount of competitor DNA. Note that the inhibition by self-competitor (2- to 10-fold) was significantlygreater than thatbynonself-competitor(0- to2.5-fold).
PAUNDROME
III
I
II
5-CGGGT AAAGAAGTGAGAACGC GCGGTTCGCACTTCGTCt AATATATATATATTATTA
GGCGAGTGCGAGCACGGCGCCGGCCCCGG-3,
wildtype
3'-GCCCA TTCTTCACTCTTGCG CGCAAGCGTGAAGCAG TTATATATATATAATAA CCGCTTCACGCTCGTGA CGCGGCCGGGGCC-5
CCT ATA GG ATA
TTT TAC TCC
bs-7 bs-8 bs-9
bs-10 bs-11 bs-12
bs-12
aL a
0
DO CO CO DCO U)D Ca = .0 .0 .0 .0 .0 .0 .0 3' .
I, + + t + + + + -I Protein
e`e 4Complex I
4Complex 11
4 Free DNA
B. *Complex I
7Complex I I
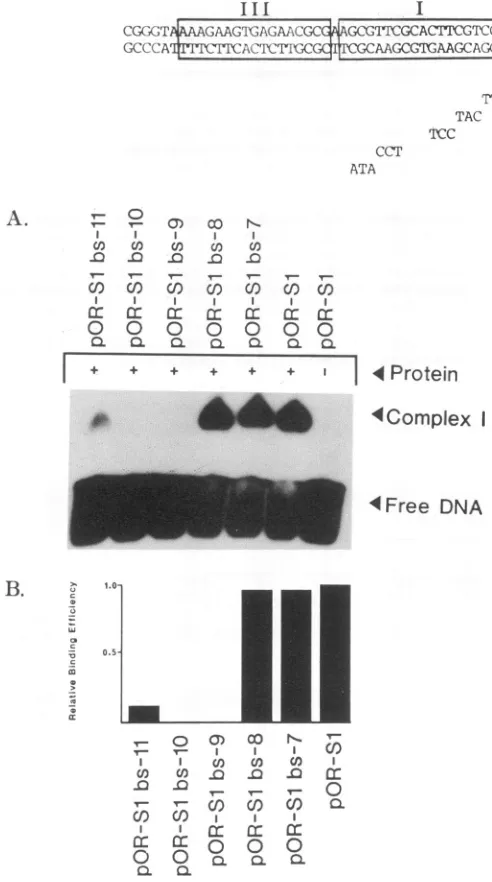
FIG. 6. Gel shift analysis of site I mutants. del 1-534 was
incubated withwild-typeormutantOrisprobefor 30minat4°Cas
described in Materials and Methods. Theresulting complexeswere
resolvedon anative 5%polyacrylamide gelasdescribedin Materi-alsand Methods. Mutants testedare shown above.(A) Autoradio-gram ofgelshiftanalysis. NotecomplexI (OBPbindingtosites I
andII)andcomplexII(OBP bindingtosites IorII)formation in the wild-type lane. Note alterations in theintensityof thesecomplexes with mutantorigins. (B)HistogramshowinglevelsofcomplexIand II formation ofmutantorigins relativeto wild-typeOris. Autorad-iogramsofgelshiftanalysiswerescannedbyusinglaser densitom-etry.Resultingvalueswere normalizedtowild-typelevels. B.
bs
*a
_u _ec
-c
CD
m
CD
CC
c)C ' 0o )cn 0D .. ) II
a) CO J 9 COn
DO QO
_0 .0 .0 J
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.62.540.322.714.2]4364 MARTIN ET AL.
III I
CG(k;tP?r AAGTlGAGAACGCcGCGFA ICGCAC¶[TCGr2 'AATATATATATATTATTA
GCCCA ItI'II\CruGlC?r rCaAAGCGCTAAACA TTATATATATATAATAAT
CCT ATA
TAC TCC
I
co co
-0 0
I I
Q Q
a
- L
CY)
I .0
CO
0.+L
00 N c) X) in
'r- 1rl*
CO CO
I
0 0
.L
+ + + I
0 0
0.0.L
1 4
Protein
AComplex
I4Free
DNAB. r_
-2
m
m
-Z1
v- 0
'r-I V-I
0)
nl
COI~ I
co cc
0 0
CL
Ca) OCo
I,,-l I
0f) 0)
nfn n
co) cO cn
I I
CEz
0.r- 0.CL 0.r
v-0.
buffer (50 mM
N-2-hydroxyethylpiperazine-N'-2-ethane-sulfonic acid [HEPES] adjusted topH 7.5 with NaOH, 0.1 mM EDTA, 0.5 mM dithiothreitol, 10% [wtlvol] glycerol, and 100 mMNaCl) in thepresenceof different cold
compet-itor DNAs. After incubation, the mixtures were passed
through nitrocellulose filter papers and washed with the
samebuffer. The bound DNAwaseluted from thefilter and
loaded onto a 5% native polyacrylamide gel. DNA was
visualized by autoradiography. Eachlanecontained 5 ,ulof in vitro-translated product, labeled
Oris,
and the indicated amountofcompetitor DNA.(ii) Gel shift assay. The DNA binding ofOBPwith
wild-type and mutant origins wasanalyzed by gel shift analysis.
Bindingwascarriedoutby the method describedby Deb and Deb(3). For each bindingassay, about0.3pmol of synthetic
proteinwasused. Bindingassayswererepeatedtwice. Care
was takento use the same batch of proteinwhen different
base-pair-substitution mutants in the same context were
assayed for protein binding. The synthetic protein was
FIG. 7. Gel shift analyses ofpOR-Sl mutants. del 1-534 was
incubated withpOR-SlorpOR-Slmutantprobesasdescribedinthe
text. Mutations testedare shown above. (A) Autoradiogramofgel shiftanalysis.Notediminishedcomplex formationinpOR-Slbs-11
and loss ofcomplex formationinpOR-Slbs-9andpOR-Slbs-10. (B) Histogram showing levels of complex I formation ofpOR-Sl
mutantsrelativetopOR-Sl.
analyzed by an SDS-polyacrylamide gel electrophoresis as
described by Deb and Deb (3a). A single bandcorresponding to themolecular weight of del 1-534 wasobserved in each
case. For the DNAbinding, thein vitrotranslationmixture
was incubated with 32P-labeled wild-type or mutant
Oris
probe on ice for 30 min in a buffer composed of 50 mMHEPESadjustedtopH 7.5 with NaOH, 0.1 mMEDTA,0.5 mMdithiothreitol, 10%(wt/vol) glycerol,and100 mM NaCl. Loading dye was added up to a volume of 10% of the
incubatedsample. The mixturewasloadedontoanative 5%
polyacrylamide gel made with 0.5x TBE (3a) and run at
roomtemperatureat30 mA. Thegelwasfixedinasolution
of 10% acetic acid and 2%glycerol, dried, and autoradio-graphed.
RESULTS
ReplicationanalysisofmutantsinsitesI, II,and III of
Oris.
To determine the role of sites I, II, and III in DNA
replication, we tested replication efficiencies of four
Oris
derivatives depicted in Fig. 2. del 1 deletes part of site I; pOR-Sl deletes site II; bs-15 substitutes three consecutive basepairs in siteIII;del31 deletesboth sitesIIandIII.Test plasmids were transfected into Vero cells, followed by
infection with HSV-1 (strain KOS). The cells were
har-vested, and DNAwas extracted at 16 hpostinfection. The isolated DNA wascutwithBamHI andDpnI andanalyzed by Southern blothybridizationasdescribed in Materialsand Methods. Replication analysis depicted in Fig. 2 shows that
noneof themutantsreplicatedasefficientlyasthewildtype. LongerexposuresindicatebothpOR-Sl and bs-15 replicate at10to 15%efficiencycompared with that of the wildtype. A number of different experiments (not shown) indicate a
decreased level of origin function for pOR-Sl and bs-15. Deletion in site Iordeletion in site IIaswellas site III has moredrastic effects onreplication. No significant difference
in replication efficiencies was observed previously (6)
be-tweenthe wildtypeandpOR-Sl with BHK-C21 cells anda
particular isolate of HSV (F). These experiments have been repeated several times with closer attention toquantitation
ofreplication data with the sensitive DpnIassayprocedure.
The importance of sites I, II, and III has also been
demon-strated by others (15, 28).
Replicationanalysis ofmutationsinsite Iof
Oris.
To havea closer look at the functionally important nucleotides in
pX)R-S1
pOR-S1 bs-7
pOH-SI
bs-8pOR-Sl bs-9
pOR-Si bs-10 pOR-Sl bs-l1
VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.74.320.74.515.2]PAUNDROME
III
I11
5-CGGGT A AAGAAGTGAGAACGCG AGCGTTCGCACTTCGTC AATATATATATATTATT GGCGAAGTGCGAGCAC
GGCGCCGGCCCCGG-3_
3-GCCCAT TTCTTCACTCTTGCG trCGCAAGCGTGAAGCAGG TTATATATATA-AATAA CCGCTTCACGCTCGTG CGCGGCCGGGGCC-5
GGT bs-14
AGA bs-15
GAG bs-17
GCCCATTTTCTT AC2CTTGCGCTTCGCAAGCGTGAAGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC 16 del
A
GCAACTTTTCTTCACTCTTGCGCTTCGCAAGCGTG AGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC bs-18d(&
AAA A
A.
-o oo uI
-n
0L)
-C v
io
(o Ul)
.0v 0 .0
_-. :.-.m.jhhl -Progeny
N
InputB.
1.0-0
c)
C)
c 0.5-0 CO C)
Is
_L)f (D
.0
T- CL
>
U) (/) :
cn cn
-J n 3:
site I and site III, we generated a series of mutations
acrossthisregion.Thesewerethentested for DNA
replica-tionand OBPbinding. Mutant bs-7 resulted inadrastic loss
inDNAreplication (Fig. 3).Partof thismutation fallswithin the OBP footprint but is outside of the critical binding domain (3, 9, 18). The mutation introduced by bs-8 also markedly reduced DNA replication. This mutation falls completelywithin the OBPfootprintin site I (3, 10, 11, 18,
24) and is immediately adjacent to the critical binding domain. Mutants bs-9 and bs-11 had very low levels of replication. These mutants are within the critical OBP-bindingdomain(3, 18). Themost drastic loss inreplication
was seen withbs-10. This mutation isinthecentral part of the critical binding sequence. No replication signal was
noted, even in overexposed autoradiograms (not shown).
bs-12andbs-13 also hadverylow levels ofreplication. Thus,
theintegrityof thissetof residuesacrosssite I isabsolutely
critical in order to maintain an efficient level of DNA replication. The residues most crucial are represented by
mutants bs-9, bs-10, and bs-11. These correlate to the
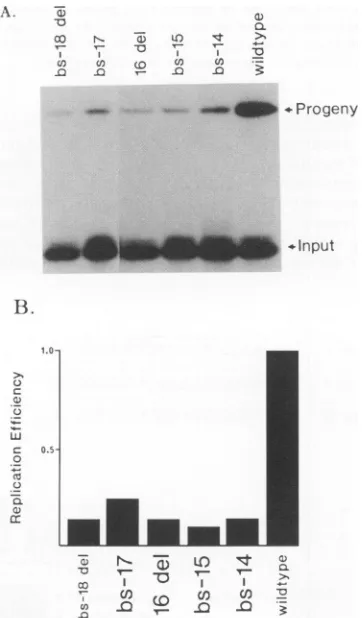
FIG. 8. Replication of site III mutants. Wild-type and mutant origins were tested as previously described. Mutations tested are shown above.(A)Autoradiogram of replicationassay. Theposition ofthe progeny and input DNA is shown. Note loss in replication efficiency of mutant originsrelative to wild-type levels. (B) Histo-gram showing relative replication efficiencies of site III mutants versuslevelof wild-type Oris replication.
sequencespreviously shown to represent critical sequences for OBP binding at this site (3, 18). Residues outside the critical binding domain are also required for efficient DNA replication.
Figure 4 shows a histogram obtained after replication analysis of pOR-Sl mutants with mutations in the OBP-binding site I. None of the mutants replicated significantly. This is not unexpected because, even in the presence of site II, site I mutants have very low levels of origin acti-ity.
Gelshiftanalysis of OBP-binding siteImutants. Totestthe binding of OBPto mutantorigins, theDNA-binding domain of OBP, a C-terminal 318-amino-acid fragment, was gener-atedby usinginvitrotranscriptionand translation systems.
Figure5 demonstrates the sequencespecificity of bindingof
the wild type and del 1-534.Competitionbinding assays were carriedout withspecific (pOR-S) andnonspecific (pBR322) DNAsascompetitors. Theautoradiogramwasalso scanned by laser densitometer to quantitate the extent ofbinding.
Binding was inhibited more efficiently by pOR-S (2- to
10-fold) than by pBR322 (0- to 2.5-fold) under a 50 molar excessofcompetitoroverthe probe. This suggests that the DNA-binding observed is highly sequence specific. Addi-tionalobservationswithcold
Oris
mutantDNAsas compet-itors suggest that del 1-534 recognizes that same critical residues in site Ias does intact OBP(3b). Thisprotein (del 1-534) was usedfor gel shift analysis with 32P-labeled wild-typeandmutantoriginsasprobes.OBP haspreviouslybeen showntobindto atleasttwositesonOris,
site I and site II. SiteIhas been showntobe the strongestbindingsite(10). A deletion in site I abolished OBPbindingaswellasreplication(3). To determine the roleofsiteI residues in OBPbinding,
substitutionmutants were tested for theirbindingefficiency
by using gel shift analysis. As shown in anearlier section,
these substitution mutants show a considerable loss in replication efficiency. The results of the gel shift analysis with the invitro-synthesized protein are showninFig.6.The
figureshowsformation oftwocomplexes,suggesting binding
at one (complex II) or both (complex I) sites. The binding experimentswererepeatedtwice. On eachoccasion, similar
patterns were observed, with about 10 to 15% variation in
binding when laser densitometry was done on the
autorad-iograms (datanotshown).
These resultsindicatethatsubstitutions within thecrucial
binding domainof OBP are detrimental to OBP
binding
ason November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.58.547.70.172.2] [image:7.612.79.260.191.500.2]4366 MARTIN ET AL.
PAUNDROME
III I II
5'-CGGGTAIAAAGAAGTGAGAACGC AGCGTTCGCACTTCGTC AATATATATATATTATTAGCGAAGTGCGAGC ACGCGCCGGCCCCGG-3,wildt
3-GCCCAI1TTCTTCACTCTTGCG CGCAAGCGTGAAGCAG TTATATATATATAATAA CCGCTTCACGCTCGTG CGCGGCCGGGGCC-5 yp
GGT bs-14
AGA bs-15
GAG bs-17
GCCCATTTTCTT ACTCTTGCGCTTCC,CAAGCGTGAAGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC 16 del
A
GCAACTTTTCTTCACT-CTTGCGCTTCGCAAGCGTG AGCAGGGTTATATATATATAATAATCCCGCTTCACGCTCGTGACCGCGGCCGGGGCC bs-18del
A AA A
A. a,
o r-, )D I I +
co0
co~
.0a)
OL
+1
0)
Q
>1
--I Protein
FIG. 9. Gel shift analysis of site III mutants. del 1-534 was incubated with wild-type or mutant Oris probe as previously de-scribed. Mutations tested are shown above. (A)Autoradiogram of gelshift analysis.(B)Histogramshowing levelsofcomplexIand II formationofmutantoriginsrelative towild-typeOris.
4 Complex
4 Complex 11
4 Free DNA
B.
*CoI1plcx
I
C(omnplex
II10~
C
0 -.
0.5
-;
-0 T
v
Trl
Tl
r-.0 Ir
substitutionmutantswere testedfor their abilitytoreplicate in thetransient in vivo replication assay. Figure 8 shows the results of this assay, alongwith ahistogramto comparethe relative replication efficiencies. Mutants bs-14 and bs-15 replicated poorly. These mutants map entirely outside of site I. Furthermore, they map to a region that by sequence similarity would represent the critical nucleotide sequence for the OBP recognition within site III. Interestingly, a
S.5
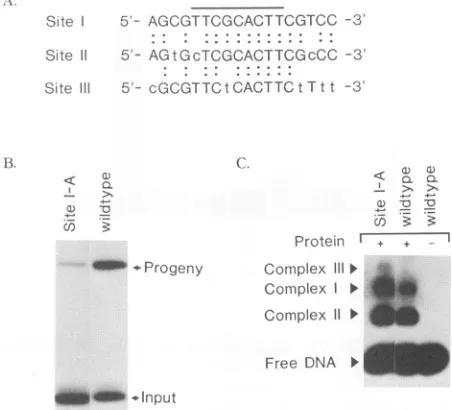
Site 5- AGCGTTCGCACTTCGTCC -3
Site 11 5'- AGtLGcTCGCACTTCGcCC -3!
Site ll 5- cGCGTTCtCACT TCtT t t -3
I. C.
Il >1
a) -D
U) BC
NOW .Progeny
<a) a)
-L >, >
a) V -o
Protein ri++
Cormiplex Ill E ..
Complex 1
-Complex 11 1 _
well as to DNA replication. The substitutions outside this crucial sequence (bs mutants 7, 8, 12, and 13) are more tolerant for OBP binding than for replication. This suggests a primary role of OBP binding for the residues inside the crucial domain. Residues outside the crucial domain, al-though essential for efficient replication, may not be directly involved in binding OBP. An involvement of these se-quences, however, in attaining the right structure for a preinitiation complex cannot be ruled out.
Todetermine the effects of the absence of siteII on OBP
binding to site I mutants, we used pOR-Sl derivatives as shown in Fig. 7. Clearly, the effect on OBP binding observed with these mutants is similar to that seen with site I mutants
havinganintact site II. Itseems, however, the reduction in
bindingcapability is more drastic when siteIIisabsent. This mayindicate cooperativity between the two sites as reported
elsewhere (2a, 9).
DNAreplication analysis of site IIImutants. To determine the role of site III residues in DNA replication, several
Free DNA F
UEUb -lnput
FIG. 10. Effect of an additional site I in Oris. (A) Sequence comparisonof sitesI, II,andIII. The overline indicatesthe critical OBP-bindingdomain identifiedearlier(3).SiteIsequenceis shown incapitalletters. SitesIIand III are from thestrand opposite to that used for siteI.Theyareshownthis way to demonstrate similarity of their nucleotide residues with those of site I.Connecting dots and capitallettersin sitesIIand IIIindicate identicalresiduesbetween thesites. (B)Replicationof site I-A. Mutant site I-A was constructed by usingcassette-directed mutagenesis. The site III sequence was replaced by a site I sequence in the context of the whole origin. This sequence, site I-A, is in the same relative orientation as site III. The replicationassay wasperformed as previously described. The posi-tions of progeny andinput bands are shown. Mutant site I-A did not replicateasefficientlyasthe wild-type origin. (C) Gel shift analysis of siteI-A.Thegelshift assay wasperformedaspreviously described. Note enhancedcomplex I andIIformation in the site I-A lane and the formationofathirdcomplex,III.
:.:
owl
.0
a
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.100.266.192.519.2] [image:8.612.332.558.355.560.2]Ons
III
I
II
CGGGTAAAAGAAGTGAGAACGCGAAGCGTTCGCACTTCGTCCCAATATATATATATTATTAGGGCGAAGTGCGAGCACTGGCGCCGG
attaTAAAAaAAGTGAGAACGCGAAGCGTTCGCACTTtGTCCtAATAatatatataTtaTtaGGacAAagtgcgaaCgCttCGCgtt
aGGGTAAAAGAAGTGAGAACGCGAAGCGTTCGCACTTCGTCCtAATAgtatatataTtaTtaGGgcAAagttGcGgCactGgCgCcc CttcTAAAAaAAGTGAGAACGCGAAGCGTTCGCACTTtGTCCtAATAgtatatataTtaTtaGGacAAagtgcgaaCgCttCGCgtt CctGcAccctgAtTGgcccaGaGgccCGTTCGCACcaatcaCCAATAagTtTtaATaATaAttattgcaacaaAGtgugaaCaCtac tcatgttttGgcaTGtGtcCaaccAcCGTTCGCACTTtcTttCtATATATATATATataTAtatatAtaTatatagAgaaagagaGa
HSV-1 oris
HSV-1 oriL
HSV-2 oris HSV-2 oriL
EHV ori
VZV ori
FIG. 11. Comparison ofOrissequence. The HSV-1 Orissequence was compared with severalherpesvirusDNAreplicationorigins to determine conserved residues. Nucleotides in HSV-1 OriL, HSV-2OrisandOriL,varicella-zoster virus (VZV)Ori, and an equine herpesvirus (EHV) origin identical to HSV-1 Oris are shown in capital letters. OBP-binding sites I and II and the presumptive siteIIIare shown for reference on HSV-1Oris.
single-base-pair deletion, represented by16del,alsoshowed a dramatic loss in DNA replication. bs-17 also had dimin-ished replication efficiency, although the level of replication was slightly higher than seen for bs-14, bs-15, and 16 del. One residueof this mutant (bs-17) falls into what may be, by sequencecomparison to site I, a critical sequence in site III. bs-18 del also had a low level of DNA replication. Thus, sequences in site III of
Oris
areessential forOris-mediated
DNAreplication.
Gel shiftanalysis of OBP bindingtositeIIImutants of
Oris.
Gel shift analyses were performed to test the ability of the site III mutants to interact with the DNA-binding domain of OBP. All of the mutants in this region of the origin had approximately the same level of complex II
forma-ion (Fig. 9). However, there was a difference seen in the
ability to bind the origin at two sites, since bs-14, bs 17, and bs-18 del all had lower levels of complex I
forma-ion. In bs-15, the ability to form complex I was actually enhanced to some extent. However, this mutant replicated
the least efficiently of all of the site III mutants. Thus, it seemsthatspecific mutations in the siteIIIregion markedly reduce the efficiency of DNA replication and alter overall OBP binding to the origin, even though these mutated sequences are well separated from defined OBP-binding
sites.
Analysis ofan
Oris
mutant with a second site I in placeof site III. No sequence-specific OBP binding has yet been demonstrated at site III, although we demonstrate above thatmutations in thisregionhaveaneffect on OBPbindingto
Ori..
To determine whether strong OBP binding at theposition of site III would facilitate
Oris
replication, we constructed a mutant origin (site I-Amutant) that changedsite III into an exact copy of site I, while keeping the orientation of site III unchanged. We then tested this
origin
for theabilitytoreplicateandbind OBP. The resultsof these two assays are shown in Fig. 10. The site I-A mutant
replicated, butat alowerefficiency thandidthe wild type. Gel shift analysis revealed that bothcomplex I and IIwere present and were enhanced relative to wild-type levels. In
addition,athird complex(III), whichran more slowlythan
complex I, was apparent. Presumably, this is due to OBP
bindingatall three siteson the mutantorigin. Thus, strong OBPbindingtothethird site in thiscontextisdetrimentalto
originfunction.
DISCUSSION
Oris
derivatives with mutations in sites I, II,andIIIwere tested for DNAreplicationinaninvivo transient assay. Ourresults (Fig. 2) show that mutations in any of these sites drastically reduce DNAreplication, signifying the functional importance of all three sites. The site I mutation seems to be themostdrastic. A decreased levelofDNAreplication was, however, observed with mutations in sites II and III. The
quantitativedifferences inreplication efficienciesof
Oris
andOris-1
reported here compared with those reported earlier(6) may be due to improved assay conditions, including
rigorous quantitationof DNA before and aftertransfection,
in addition to the use ofdifferent strains of virus and cell lines.
Wegeneratedaseries ofspecificmutationsacrossthe
Oris
region spanning OBP-binding site I through a presumptive
thirdbindingsite(siteIII) both in the presence and absence
of site II. The exact nucleotide requirements have not been defined in this region. We first assayed the ability of
mutants across site I toreplicateandtobindOBP(Fig. 3, 4, 6, and7). Similarresults were obtained with thetwo series of mutants. bs-7 was found to result in a low level of
replication. Thismutantdoes notcompletelymapwithin the OBP footprint at site I (3, 10, 11, 18, 24). It falls directly
between site I and the A-T stretch. It is possible that the mutations introduced result in some structuralchange in the DNA thatdoesnotallowfor efficientinitiation. Inaprevious
paper, Doelberg and Deb (6) showed that a mutant
desig-nated bs-1, which mutates the first three base pairs of the A-T stretch near binding site I, also lost its replicative ability. They proposed that this site might be required for initiation of origin melting. Thus, it is possible that the mutation introduced by bs-7 may also be affectingasimilar function.
Mutantbs-8 falls within the OBP
footprint
(3, 10, 11, 18,24). Thismutantshowsaloss in DNAreplication. Several of thesubstitution mutantsresideacross aregionwhose nucle-otide sequence elements were determined tobe critical for OBPbindingwithin thefootprint. bs-9 isan
example
ofone of these mutants. Here, we see that there is a direct correlation between a significant loss in OBP binding and DNA replication. Koffand Tegtmeyer (18) haveproposed
that the OBPrecognitionsite may berepresented
by
a setofpentanucleotides, inverted relative to each other on the different strands
(5'-GTTCG-3'/3'-GCGTG-5')
with a2-bp
overlap. The mutation introduced
by
bs-10 represents the most drastic mutation in this region, sincecrucial residues arechanged from both strands of theoverlap.
Thismutant did not replicate. Nosignal
was detected even when theautoradiogram was
overexposed.
Interestingly, this mutant had the lowest level of complex I formation. bs-11 is amutantthatresides within the
footprint
anddisrupts
asingle
on November 10, 2019 by guest
http://jvi.asm.org/
residue of thecritical
binding
domain identified earlier. Thismutantstill hasalow
replication efficiency,
eventhough
the level of OBPbinding
isslightly
increased. bs-13 isadouble-base-pair
substitutionmutation, essentially
atthejunction
ofsite I and site
III,
in whichasingle
residue is within the siteI
footprint
(3, 10, 11, 18, 24).
This mutant showed a slightincrease in
binding
relative to the other site I mutants butreplicated
poorly.
Base substitutions in the criticalbinding
domain showacorrelation between
binding
andreplication.
Basesubstitutionsoutside the critical
binding
domain donot correlateaswell. These sequences may berequired
todirectorientation of other
replication
factors or to allow for the formation of the correct structure ofanucleoprotein
com-plex.
A series of mutations were also created to allow us to determine critical residues in the site III
region.
Therepli-cation
capacity
andOBP-binding capacity
of thesemutantswere
analyzed (Fig.
8 and9).
In this context,bs-14, bs-15,
and 16 del all had very low levels of
replication.
These mutantsreside inaregion
thatwould, by
sequencesimilar-ity,
represent the criticalbinding
sequence as seenin site I(3,
10, 11,
18,
24).
Mutantbs-14resulted inasignificant
lossof
complex
Iformation. The centralquestion
remains,
sinceno
binding
has yet been demonstratedtositeIII,
what is theroleof theseresidues in
mediating
efficient DNAreplication
and overall OBP
binding
to theorigin?
We find it veryinteresting
thatspecific
substitutionsinaregion
welloutsideof defined
OBP-binding
sites would have such a negative effect onorigin
function. There are severalpossible
expla-nations for
this,
all of which arespeculative
at this time and willrequire
furtheranalysis.
First,
it ispossible
thatOBP
actually recognizes
the site III sequence inan in vivosetting.
This may be due to the presence of otherfactorsthat
change
the overall structure of theorigin, thereby
making
the site moreaccessible,
or to factors thatthrough
protein-protein
interactions allow OBP to sit at this site.However,
our studies show that strong OBPbinding,
asevidenced
by
studies withmutantsiteI-A,
is detrimentaltoorigin
function.Thus,
strong DNAbinding
alone cannotexplain
the functional role of these sequences. It is clearfrom
previous
studies that the site III sequence alone is notsufficient for
sequence-specific
OBPbinding
(9, 28). Thus,
any interaction of OBP with the
origin
at these sites mustbe aided
by
the in vivo environment and is not seen in thecurrent conditions used for in vitroDNA-binding
stud-ies.
Second,
it is alsopossible
that OBP does not interactwith site III
directly.
Thus,
one can propose several func-tions that may beprovided
by
thisregion.
Itispossible
that these residues areimportant
in order toprovide
sites for other factors to act upon as thereplicon
is formed. Onecan envision OBP
binding
and thegeneration
ofaspecial-ized
nucleoprotein
structure whoseintegrity
would becru-cial in order to
provide
for theappropriate
interaction of other factors involved inreplication.
Any
mutations in these residues woulddrastically
reduce theefficiency
of the reaction. Within the same context, residues in site III may beimportant
in thesecondary
structure of the origin.It is
interesting
thatchanges
in site III result in an overallloss of
origin binding.
Thus,
thesecondary
structure of theorigin
may bechanged
such that OBP cannot interact atwild-type
levels. This would then be manifested in the invivo loss of DNA
replication. Third,
thefar-left armoftheOris
region
contains ashort stretch which isA-Trich.This,along
with the central A-T-richregion,
may mediate aconformationonthe
origin
that isrequired
for efficient OBPbinding
and DNAreplication.
If this were the case, themutations should result inan alteration oforigin conforma-tion. Thismay manifest itselfby a loss in OBPbindingand DNAreplication.
The sequenceof the leftarmof the HSV-1
Oris
regionis well conserved among several other herpesvirus origins of DNA replication (Fig. 11). A comparison between HSV-1Oris
andOriL
(27, 29), HSV-2Oris (30)
andOriL
(19),
varicella-zoster virus Ori (26), and the equine herpesvirus
Ori (1) demonstrates a region of high conservation that corresponds to the critical sequences required for OBP binding at site I. This 9-bp region, 5'-CGTTCGCAC-3', is conserved in alloriginscompared. This conservation
corre-lates well withourresults, since these residueswerecritical fororigin function. Other residues throughout the leftarm
arealso well conserved. A total of 10 other residues
through-out were present in four or more of the origins analyzed.
Thus, it is expected that these residues represent crucial functionalelements in theseregions. Mutagenesis of site III would disrupt the integrity of these residues and result in diminishedorigin function. Thiswasthecaseinourstudies. The far-left arm containsa stretch in which 7 of 8 residues are A orT. These residues may mediate the adoption ofa conformation important for origin function. This general
motif is also maintained in these virus origins. In the varicella-zoster virus origin, although there is not aperfect sequence match, the general A-T-rich character is main-tained at this site. This may represent a common element necessary for efficientoriginfunction.
We have createdspecific mutationsacrossthe left armof the HSV-1On5 regionin ordertoclearly define theelements requiredfor efficientinitiation of DNA replication andOBP binding. Our results demonstrate the need for residues in site I and site III to mediate efficient DNA replication. Residues in thecritical OBP-binding domainarerequired for bothDNAreplication and OBP binding. However, residues outside this domain, although notcrucial for OBP binding,
are necessary for DNA replication. This suggests that the binding of the origin sequence by OBP is not the only determining factor for the initiation of DNA replication. Single-base-pair mutagenesisis requiredto more finely dis-sect the intricacies oforigin function. Our results demon-stratethe crucialnatureofseveral groups of residues within the origin and begin to implyafunction mediated by these sites.
ACKNOWLEDGMENTS
This workwas supported by grants from the American Cancer Society (MV-420), the Elsa U. Pardee Foundation, andthe Basil O'Conner Starter Scholar Research Award from the March of DimestoSumitraDeb. This work is also supported by anAmerican Cancer Society Institutional Grantto SumitraDeb and an Institu-tional Research Grant to Swati Palit Deb. David W. Martin is supported in part by a NIH predoctoral fellowship in microbial pathogenesis.
Wethank Jane Tatefor excellent typing assistance.
REFERENCES
1. Baumann, R. P., V. Ramana, R. Yalamanchili, and D. J.
O'Callaghan. 1989. Functionalmapping and DNAsequence of
anequine herpesvirus1originofreplication.J. Virol. 63:1275-1283.
2. Bramhill, D.,and A. Kornberg. 1988. A model for initiationat
originsof DNAreplication. Cell 54:915-918. 2a.Challberg, M.Personalcommunication.
3. Deb, S., and S. P. Deb. 1989. Analysis ofOris sequence of J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
HSV-1: identification ofonefunctional DNA binding domain.
Nucleic Acids Res. 17:2733-2752.
3a.Deb, S.,andS. P. Deb. 1991. A269-amino-acidsegmentwitha
pseudo-leucine zipper andahelix-turn-helix motif codes for the
sequence-specific DNA-bindingdomain of herpes simplex virus
type 1origin-binding protein. J.Virol. 65:2829-2838. 3b.Deb, S.,andS. P. Deb.Unpublished data.
4. Deb,S., A.L.DeLucia, C.P. Baur,A. Koff, and P.Tegtmeyer. 1986. Domain structure ofthe simian virus 40 core origin of
replication. Mol. Cell. Biol. 6:1663-1670.
5. Deb, S., A. L. DeLucia, A. Koff, S. Tsui, and P. Tegtmeyer. 1986. Theadenine-thymine domain of the simian virus 40coreorigin
directs DNAbending and coordinately regulates DNA replica-tion. Mol. Cell. Biol. 6:4578-4584.
6. Deb, S., and M. Doelberg. 1988. A 67-base-pair segmentfrom the Orisregion of herpes simplex virus type1 encodesorigin function. J.Virol. 62:2516-2519.
7. Deb, S., S. Tsui, A. Koff, A. L. DeLucia, R. Parsons, and P.
Tegtmeyer. 1987. The T-antigen-binding domain of the simian virus 40coreorigin of replication. J. Virol.61:2143-2149.
8. DeLucia, A. L., S. Deb, K. Partin, and P. Tegtmeyer. 1986. Functional interactions of the simian virus 40 core origin of replication with flanking regulatorysequences.J.Virol. 57:138-144.
9. Elias, P., C. M. Gustafsson, and 0. Hammarsten. 1990. The origin binding protein of herpes simplex virus 1 binds
coopera-tively to the viral origin of replication Oris. J. Biol. Chem. 265:17167-17173.
10. Elias, P., and I. R. Lehman. 1988. Interaction of originbinding protein withanorigin of replication ofherpes simplex virustype
1.Proc. Natl. Acad. Sci. USA85:2959-2963.
11. Elias, P., M. E. O'Donnell, E. S. Mocarski,andI.R.Lehman. 1986. A DNAbinding protein specific foranorigin of replication of herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 83:6322-6326.
12. Frenkel, N.1975. Defective interfering herpesviruses,p.91-120.
In A.J.Nahmias, W. R. Dowdle, and R. S. Schinazi (ed.), The human herpes viruses: an interdisciplinary perspective. Elsevier/North-Holland PublishingCo., New York.
13. Frenkel, N., R. J. Jacob, R. W. Honess, G. S. Hayward, H. Locker, and B. Roizman. 1975. Anatomy of herpes simplex virus
DNA. III. Characterization ofdefective DNAmolecules and
biological properties of virus populations containing them. J. Virol. 16:153-167.
14. Frenkel, N., H. Locker, W. Batterson, G. S. Hayward, and B. Roizman. 1976. Anatomy of herpes simplex virus DNA. VI. Defective DNA originates from the S component. J. Virol.
20:527-531.
15. Hernandez, T. R., R. E. Dutch, I. R. Lehman, C. Gustafsson,
andP.Elias. 1991. Mutations in aherpes simplex virus type1 origin that inhibit interaction with origin-binding protein also
inhibitDNAreplication. J. Virol. 65:1649-1652.
16. Kaerner, H. C., A. Ott-Hartmann, R.Schatten, C. H. Schroder, and C. P. Gray. 1981. Amplification of a short nucleotide sequencein therepeatunits ofdefectiveherpes simplex virus
type1AngelottiDNA.J. Virol. 39:75-81.
17. Kaerner, H. C., I. B. Maichle, A. Ott, and C. H. Schroeder. 1979. Origin of two different classes of defective HSV-1 An-gelottiDNA. Nucleic Acids Res. 6:1467-1478.
18. Koff, A., and P. Tegtmeyer. 1988. Characterization of major recognition sequences for aherpes simplex virus type 1origin binding protein. J. Virol. 62:4096-4103.
19. Lockshon, D., and D. A. Galloway. 1986. Cloning and charac-terizationof oriL2, a large palindromic DNA replication origin ofherpes simplex virus type 2. J. Virol. 58:513-521.
20. Lockshon, D., and D. A. Galloway. 1988. Sequence and struc-tural requirements ofaherpes simplex viral DNAreplication origin. Mol. Cell. Biol. 8:4018-4027.
21. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C.Frame, D. McNab, L. J. Perry, J. E. Scott, and P.Taylor. 1988. The complete DNA sequence of the long uniqueregion in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.
22. McGeoch, D. J., M. A. Dalrymple, A. Dolan, D. McNab, L.J. Perry, P. Taylor, and M. D. Challberg. 1988. Structures of herpes simplex virus type 1 genes required forreplication of virusDNA. J.Virol. 62:444 453.
23. Mocarski, E. S., and B. Roizman. 1982.Herpesvirus dependent amplification and inversion ofcell-associated viral thymidine kinase gene flankedbyviral sequences and linked toanoriginof replication. Proc. Natl. Acad. Sci. USA 79:5626-5630. 24. Olivo, P. D., N. J. Nelson, and M. D. Challberg. 1988. Herpes
simplex virus DNA replication: the OBP gene encodes an
origin-binding protein. Proc. Natl. Acad. Sci. USA 85:5414-5418.
25. Spaete, R. R., and N. Frenkel. 1982. Theherpes simplex virus amplicon: a neweukaryotic defective-virus cloning-amplifying vector. Cell 30:295-304.
26. Stow, N. D., and A. J. Davison. 1986.Identificationofavaricella zoster virus origin of DNA replication and its activation by herpes simplextype 1 geneproducts. J. Gen. Virol. 67:1613-1623.
27. Stow, N. D., and E. C. McMonagle. 1983. Characterization of theTRs/IR5originof DNA replicationofherpes simplexvirus type 1.Virology 130:427-438.
28. Weir, H. M., and N. D. Stow. 1990. Twobindingsites for the herpes simplex virus type 1 OBP protein are required for efficientactivity of the Oris replication origin. J. Gen. Virol. 71:1379-1385.
29. Weller, S.K., A. Spadaro, J. E.Schaffer,A. W.Murray, A. M. Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functionalanalysisofOriL,aherpessimplexvirus type 1origin ofDNAreplication. Mol. Cell.Biol.5:930-942.
30. Whitton, J.L., and J. B. Clements. 1984.Replication originsand asequenceinvolvedincoordinate induction of the immediate-earlygenefamilyareconserved inanintergenic regionofherpes simplex virus. Nucleic AcidsRes. 12:2061-2079.
31. Wu, C. A., N.J. Nelson, D. J.McGeoch,and M. D.Chaliberg. 1988. Identification ofherpes simplex virus type 1 genes
re-quired for origin-dependent DNA synthesis. J. Virol. 62:435-443.
65,