DNase I-Hypersensitive Sites Are Associated with Both
Long
Terminal
Repeats
and
with the Intragenic Enhancer of
Integrated Human Immunodeficiency Virus
Type
1ERIC VERDIN
Laboratory of Viral and Molecular Pathogenesis, NationalInstitute of Neurological Disorders and Stroke, Building 36,Room5C22, Bethesda, Maryland 20892
Received 12July 1991/Accepted 16 September 1991
Afterreversetranscription and integration ofthegenomeofhumanimmunodeficiency virus (HIV)inatarget
cell, the viral DNA becomes packaged into chromatin. Regions of chromatin associated with regulatory
functions ineukaryotescangenerallybedistinguishedfrom the bulk of chromatinbyanincreasedaccessibility
of the DNA to nucleases(nuclease-hypersensitive sites).In this report, the chromatinstructureof thecomplete
HIV-1genomehas beenanalyzedin three chronicallyinfectedcell lines ofmonocyte/macrophageandlymphoid origins. Digestionofpurifiednuclei from these cells with DNase I followedbyrestrictiondigestionand Southern
blotting identifiedseveralDNaseI-hypersensitive regions throughoutthe viralgenome. Two constitutive sites
were associated with the U3 region ofthe 5' long terminal repeat (LTR) in which the viral promoter and enhancerarelocated. Anadditionalsite in the Rregion ofthe 5' LTRwaspresentonlyafter activation of viral
transcription by phorbol ester or tumor necrosis factor alpha. A fourth site was identified in all cell lines downstream ofthe 5' LTR(nucleotides [nt]656to720),and the bandcorrespondingtothissite decreased in
intensityuponactivation of transcription.Inthe3' LTR,aconstitutivehypersensitivesitewasidentified in all cell lines(nt 9322to9489).Amajorsite(nt4534to4733)waspresentonlyinacell lineofmacrophage/monocyte origininaregion ofthegenomein whichanintragenicenhancerwasrecentlyidentified(E. Verdin,N.Becker, F. Bex, L. Droogmans, and A.Burny, Proc. Natl. Acad. Sci. USA 87:4874-4878, 1990). Thisstudy defines
regions of the HIV genome associated with an open chromatin configuration and points to the potential regulatory role of these elements in the HIV lifecycle.
After entering a host cell, the RNA genome ofhuman immunodeficiency virus type 1 (HIV-1) is transcribed into double-stranded DNAby the viralreversetranscriptase and then becomes integrated into the cell genome. Once inte-grated, the expression of the HIV genome is under the
combined influence of trans-acting cellularandviral regula-toryfactors and the localchromatin environmentatthesite
ofintegration.These factorsexerttheirregulatoryfunctions
by interacting with viral cis-acting elements, both at the
DNA and RNAlevels (for reviews, see references 5, 6, 17, 21, 30, 36, 38, and 49). At the transcriptional level, viral long
terminalrepeats(LTRs), whicharepresentatboththe 5'and
3' extremities of the viral genome, contain all cis-acting
elements necessaryfor transcription initiation and
termina-tion. Transcription is initiated in the 5' LTR, and several transcription factors have been shown to bind in vitro to
each of thefour functionally defined regions in this element. From the 5' endtothe 3'end, the four regionsare asfollows: first, thenegative regulatory element, a silencer(16, 37, 41, 58) which contains binding sites for at least four distinct
factors(15, 16, 29, 40, 42); second, the enhancer (16, 37, 41, 58) has been shown to interact with at least three distinct
proteins:NF-KB (27), HIVEN86A (14), and EBP-1 (56,57);
third, thepromoter contains three Spl binding sites (22), a
TATAbox, and additional sites closetothe site of initiation
of transcription (21); fourth, downstream of the site of initiation oftranscription is the Tat-responsive element (37) andbindingsitesfor threefactors: LBP-1orUBP-1, TCF-1,
and CTF (21, 23, 56, 57). In additionto these elements, we haverecently identifieda newelement inthepolgeneof HIV presenting the characteristics ofa transcriptional enhancer (47). This element is tetradecanoylphorbol acetate (TPA)
inducible in HeLa cells and composed oftwo distinct
sub-domains (47).
Despite rapid growthinourknowledgeof viral and cellular
factors involved in HIV gene regulation, little is known on
the relevance in vivo of the interactions between
transcrip-tion factors and viral elements observed in vitro. Indeed,
since the DNA ofeukaryotesisorganizedinnucleosomes,in
which DNA iswrapped around histone octamercores, it is
probablethat theaccessibilityof DNAto solubleregulatory
factors is restricted. Consequently, factors identified by
virtue of their ability to bind in vitro to viral cis-acting
elements might never gain access to their DNA target in
vivo. Forthisreason, it has been suggested that cis-acting
DNA elements ofeukaryotes might either be nucleosome-freeorthat theDNA-nucleosomecomplexis inamoreopen
configuration. As a consequence, DNA present in those
regionsshould becomemoresensitiveto avarietyof
chem-ical or enzymatic probes. Pioneering studies demonstrated
that discreteregions of the simian virus 40 andDrosophila
genome were hypersensitive to digestion with DNase I in
vivo (39, 46, 55). Several studies, encompassing numerous viralandcellulargenes,have sincegeneralizedthese obser-vations: DNase I-hypersensitive sites have been found in
association withalarge varietyofcis-actingelements includ-ing promoters, enhancers, upstream activating sequences,
silencers, terminators, recombination loci, telomeres, and
centromeres (for a review, see reference 19). Moreover,
variation in thesensitivity ofaparticular hypersensitivesite
has been foundto precedeortoaccompany geneactivation
orsilencingin a numberof systems, thus strengtheningthe
correlation between these sites andgene regulation (19).
We have initiated studies aimed atdefining in molecular 6790
Copyright © 1991, American Society for Microbiology
on November 10, 2019 by guest
http://jvi.asm.org/
terms theinteractions taking place in vivo between cellular
and viral regulatoryfactors and theviralDNAelements. Our
first step consisted in defining chromatin boundaries and examining the HIVgenome for possible
nuclease-hypersen-sitive sites. We have used three well-characterized chroni-cally infected celllines, ACH2 and 8E5 (both derived from
the
CEM
cell line,aCD4+
lymphoid cell line [3, 13]) and Ul(derived from the U937 cell line, a monocyte/macrophage
cell line [12]). Two of these cell lines (ACH2 and Ul) produce very low amounts of virus under basal conditions and canbe induced to produce virus when stimulated with specific cytokines, phorbol esters, and other agents (3, 10-12, 18, 24, 31-34, 43, 44). The 8E5 cell lineconstitutively produces virus (13). These cells were used to examine the virus chromatin in a variety of cellular environments and
under different levels of transcription rates. These experi-mentsrevealed thepresence ofatleastfive major hypersen-sitive sites in the Ul cell line and four major sites in the
ACH2 and 8E5 cell lines. These sites were associated with
previously recognized cis-acting elements located in the 5'
and 3' LTRs and in the intragenic enhancer(47). Inaddition, other regions of the HIVgenome werealso identified
down-stream of the 5' LTR and in the 5' part of the pol gene,
pointing to the presence of potential new regulatory
ele-ments in these regions.
MATERIALS ANDMETHODS
Cell lines. All cell lines (ACH2, 8E5, and Ul) were
obtained from the AIDS Research and Reference Reagent
Program (National Institute of Allergy and Infectious
Dis-eases, Bethesda, Md.). Cells were grownin RPMI medium
(GIBCO/Bethesda Research Laboratories) containing 10% fetal calf serum (HyClone) supplemented with 50 U of
penicillin per ml, 50
,g
ofstreptomycin per ml, and 2 mMglutamine at
37°C
in a 95% air-5% CO2 atmosphere. Cells wereroutinely maintained at adensity of 0.25 x 10'to 1 x106 cellsper ml by dilutingthem with fresh medium. When indicated, cells atadensity of less than5 x 105 cells perml were treated with 10 nM TPAdissolved in dimethyl
sulfox-ide (final concentration, 0.01%) or with dimethyl sulfoxide
alone (final concentration, 0.01%) as a control. In one
experiment, the ACH2 cell line was treated with tumor
necrosis factor alpha (TNF-oa) (Amgen)ataconcentrationof 100U/mlfor 12 h.
Antigen p24 assays. Supernatants from treated and
un-treated cells were collected after low-speed centrifugation
(1,000 rpm in a GPR tabletop centrifuge [Beckman]) of the
cell cultures and kept frozen at -70°C until processed. Antigen p24 content was measured by SRA Technologies
(Rockville, Md.)usingtheCoulterHIV p24antigenassaykit
(Coulter Immunology, Hialeah, Fla.).
DNase I treatment of nuclei. Our experimental protocol
was amodification of the method described by Bushelet al.
(2). Exponentially growing cell were harvested by
centrifu-gationat 1,000rpmfor10min at4°Cand washed twicewith
ice-cold phosphate-buffered saline. All subsequent
opera-tions were performed on ice with precooled buffers. Cells were counted and resuspended at 25 x 106 cells per ml in
RSB (10 mM Tris [pH 7.4], 10 mMNaCl, 3 mM MgCl2)and allowed to swell for 5 min. An equal volume ofRSB-0.2% Nonidet P-40 was added, and the cells were incubated for
another 5 min with intermittent mixing. Nucleiwere
centri-fuged at 2,000 rpm for 10 min, resuspended, washed in 50
volumes of RSB, and centrifuged again at 2,000 rpmfor 10
min. The nuclear pelletwasthoroughly resuspended in RSB
at 25 x 106 nuclei per ml by 15 strokes in a Dounce homogenizer(pestle B). Nuclei were examined and counted after staining with trypan blue, and aliquots were put into cooled 15-ml polypropylene tubes (5 x
107
to 7.5 x 107 nuclei per tube). Variousconcentrations of DNase I(Sigma)(as indicated) were added for 10 min on ice. The digestion
reaction was stopped by adding 3 volumesofproteinase K buffer (50 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA,
0.5% sodium dodecyl sulfate[SDS]) and mixingvigorously.
Samples were solubilized for 1 h at55°C and treated for 1 h at37°C with 50 ,ug of DNase-free RNase A per ml. Protein-ase K was added at 200,ug/ml, and thedigestionwasallowed tocontinueovernight at 55°C. Samples were extracted three times with phenol andthree timeswith
chloroform-isoamyl
alcohol (24:1) and precipitated with ethanol. DNA was resuspended in sterile water, and the DNA concentration
was estimated by measuring the
A26.
Three independentDNase I digestions of nuclei were performed for each cell line and found togenerate similar results.
DNase Itreatment of naked DNA. DNAfrom
exponentially
growing untreated cells was purified after an overnight
digestion with 200 ,ug ofproteinaseKperml inproteinase K buffer. After three phenolextractions and three chloroform-isoamyl alcohol extractions, DNA was ethanol
precipitated
and resuspended in sterile water. DNA was digested for 10 min on ice withincreasingconcentrations of DNase I in RSB buffer at a DNA concentration of 0.3 mg/ml.
Preliminary
experiments wereperformed todetermine theconcentration
range of DNase I necessary to generate a similar level of digestion, asobserved after digestion of intactnuclei.
Reac-tions were stopped by the addition of a fourfold volume excessof proteinase Kbuffer andprocessed asdescribed for the nuclei after DNase I treatment.
Southernblotting. Purified DNA (30 ,ug) wasdigested with restriction enzymes, and thefragments generated were sep-arated by electrophoresis in 0.8 or 1.5% agarose
gels
in Tris-acetate buffer at 1.5 V/cm. Size markers were electro-phoresed along with the samples. Each size marker was generated by digesting HIV-1 DNA (cloned inplasmid
pBru2, a giftfromSimonWain-Hobson)withtworestriction enzymes: the same enzyme used to digest the sample and another enzyme chosen to generate a fragment ofdefined size andlocation in the region under study. Several ofthese markers were mixed together, added to 30 ,ug of cellular DNA,andcoelectrophoresedwith thesamples. Agarose
gels
were incubated twice for 20 min (each time) in
denaturing
solution (1.5 M NaCl, 0.5 M NaOH) and twice for 20 min (eachtime) in neutralizing solution (1.5 M NaCl, 0.5 MTris [pH 7.2], 1 mM EDTA) and transferred overnight
by
capil-larity in 20x SSPE (20x SSPE is 3 M NaCl, 0.2 M
NaH2PO4, 20 mM EDTA, pH 7.4) to nylon membranes (N-Hybond; Amersham). DNA was cross-linked to
nylon
membranes by exposure to UV light (UV Stratalinker
1800;
Stratagene), washed for 20 min in 2x SSPE and
prehybrid-ized for 1 to 2 h at 42°C in hybridization buffer
(50%
formamide, 3.6x SSPE, 1% SDS, 10% dextran
sulfate,
Sx Denhardt's solution [0.1% Ficoll, 0.1%polyvinylpyrroli-done, 0.1% bovine serum albumin], 0.1 mg of sonicated herring sperm DNA per ml). Denatured DNA probes were added to theprehybridization buffer and allowed to
hybrid-ize for atleast 16 h at42°C. Membranes werewashed twice for 20 min (each time) in 2x SSPE-0.1%SDS,
twice for20min (each time) in 0.2x SSPE-0.1% SDS at room tempera-ture, and once for 30 min at 65°C in0.2x SSPE-0.1% SDS.
Autoradiographic exposures with two
intensifying
screenswere carried out for 1 to 5 days at -70°C.
on November 10, 2019 by guest
http://jvi.asm.org/
Earl
BamHI
Lc
LTn
_^
PROBE A - -- PROBE C
--TPA +TPA -TPA + TPA
DNasel ~ 1 __ 2
-f~~~~~~~6m,__.. _
.,_
rR s%3'
4 6 8 10 Kb
I I I I I
[image:3.612.55.294.70.155.2]FIG. 1. Probes used to map hypersensitive sites in the HIV genome.Three sets of probes and restriction enzymes were used to mapthecomplete genomeofHIV. Probe Aspannednt 643 to 1415
andwasusedtoexamine the 5'LTRwithPstI in allcelllines. Probe
B spanned nt 1420 to 2272andwasused to examinethe intragenic regionwithPstI in ACH2 and8E5cells and withSphI in Ulcells.
Probe C containednt8523 to 9113andwasusedtoexaminethe3'
LTRwithBamHIinACH2and8E5cells andwithEarl inUl cells.
DNA probes. Three probes, identified as A, B, and C in
Fig. 1, were used in these studies. Each probe was
synthe-sized by 15cycles(95°C for 2 min, 55°C for2min, and72°C
for 3 min) ofpolymerase chain reaction, using 1 ng ofa
plasmid containing a complete clone of HIV-1 (pBru2) as a template and primers EV1 and EV2 for probe A, primers
EV5 and EV6for probe B, and primers EV3 and EV4for probe C.Polymerasechainreactions were conducted by the protocols provided with the AmpliTaq DNA polymerase
(Perkin-Elmer Cetus), using aPerkin-Elmer Cetus Thermal
Cycler. Probe A spans nucleotides (nt) 643 to 1415 (where +1isthefirst nucleotide in the 5' LTR U3 region), probe B spans nt 1420 to 2272, and probe C spans nt 8523 to 9113. The sequencesof the oligonucleotides were as follows: EV1, 5' (nt 1415)GCTTCCTCATTGATGGTCTC 3'; EV2, 5' (nt
643)CGAACAGGGACTTGAAAGCG 3';EV3, 5' (nt 8523)
GATCCTTAGCACTTATCTGGG 3'; EV4, 5' (nt 9113)
AAAGTGGCTAAGATCTACAGC 3'; EV5, 5' (nt 1420)
GAATGGGATAGAGTGCATCC 3'; and EV6, 5' (nt 2272)
GTTCCTTGTCTATCGGCTCC 3'. The amplified products
wereseparated from thetemplate on a1.5%agarosegel and purified by using the Geneclean procedure (Bio 101, La
Jolla, Calif.). DNAfragments were labeledby therandom primerreaction (8) and purified on a G-50 Sephadex column.
RESULTS
Preliminary studies. Since the cell lines used in these studies were generated by infection with a viral strain
(LAVBRU)composedof several distinct quasispecies, it was necessary to establish detailed restriction maps of the vi-rusesintegratedin these cell lines prior to Southern blotting. DNAfromthese infectedcells was extracted, digestedwith
anumberof restrictionenzymes onthe basisofthe sequence
ofBrupreviously published by Wain-Hobsonetal.(50),and examined by Southern blotting. Preliminary experiments
demonstratedthatasinglecopyof HIV hadintegratedinthe ACH2 and 8E5 cell lines and that theUlcellline contained
two integrated copies. None of the cell lines contained detectable amounts of unintegrated HIV.
Unique restriction sites were identified in the 5' and 3'
portionsof theHIVgenome (Fig. 1). The PstI site at nt 1415
was aunique site in 8E5 and ACH2 and was used to study thecompleteHIV genomewith either probe A or B (Fig. 1).
ThisPstIsitewasalso used to examine the 5' LTR inUlbut couldnotbe used to examine theintragenic region, since an
additional PstIsite was present downstream. To examine the
intragenicregion inUl,auniqueSphIsite (nt 1443) was used
withprobeB. Tostudy the 3' LTR, a unique BamHI site (nt
ii,Xi|:
I-
g. 9 bt ~~~~~~~~~e-
.1--FIG. 2. Digestion of ACH2 nuclei with increasing doses of DNase I. Purified nuclei from TPA-treated and untreated ACH2 cells were incubatedwith thefollowing doses ofDNase I:0,0.5, 1, 2, 4, 6, 8, 10, 12, 14, 16, and 20 U/ml. After extraction and purification,DNA waseither digested withPstIandhybridizedwith
probe A to examine the 5' LTR or digested with BamHI and
hybridizedwithprobe Ctoexamine the 3' LTR.Markers aredouble digests ofpBru2 with PstI (nt1415) and HindlIl (nt 1085 [marker a]),
AccI (nt 959 [marker b]), HgaI (nt 792 [marker c]), Sacl (nt 678
[markerd]),AflII (nt517[marker e]),andHpaII(nt309[markerfl). Hypersensitive sitesareindicatedby circles and correspondtosites
II, III, andIVinFig. 3.
8522)wasused inACH2and 8E5 and theunique Earlsite(nt 8564) was used inUl withprobeC.
ExponentiallygrowingACH2 cells(density rangingfrom 2
x
10'
to5x105
cellsperml)wereeither treatedfor 12 h with 10 nM TPA to induce viral expression or not treated and usedas acontrol. The cells were harvested, and the nuclei werepurifiedbycentrifugation afterlysiswithNonidetP-40. Purified nuclei were treated with increasing concentrations of DNase I(from 0to20U/ml) for 10 minat4°Ctomaintain the chromatin architecture ina state as closeaspossible tothe state in vivo. After phenolextraction and purification, genomic DNA was cut with either PstI (for hybridization with probe A) or BamHI (with probe C) and analyzed by
Southernblotting,usingthe indirect endlabellingtechnique
(28,54).Thistechniqueuses asmall labelledprobe abutting
the restriction site andconsequentlyallows the direct
map-pingof thehypersensitive sitebydeterminingthesize of the
fragment generated by the double digestion (DNase I and
restrictionenzyme). Digestionwithaslittleas6 UofDNase
I per ml resulted in the appearance of three new smaller bands(indicatedbycircles inFig. 2)mappingtothe 5' LTR and theregion downstreamof the 5' LTR(probe A). When the 3' LTR was examined with probe C, two new smaller bandsmappingtothe 3' LTRwerevisualized in the absence and presence ofTPA with increasing DNase I
concentra-tions (probe C). These new bands were dependent on a
double digestion by both DNase I and the restriction
en-zyme, since they were absent when DNase I was absent
(Fig. 2) or when the DNA was not cut by a restriction enzyme (PstI orBamHI in this case) (not shown). As the concentration of DNaseI wasfurtherincreased, progressive digestionof the bulk ofgenomicDNAwasobserved, result-ing in thedisappearance of theprimaryband of viral DNA
(Fig. 2). Preliminary titrations of DNase I digestion were
performed for the other cell lines and probes (data not
shown),andonlytheappropriateconcentrations of DNase I will be shown hereafter(indicated in thefigure
legends).
[image:3.612.309.551.79.213.2]Mapping of hypersensitive sites in the 5' region of the
genome. By usingthe approach outlined above, the
hyper-SphI
PstI
A B
LTR 5'_
0 2
L
-N
a-
b-
c-
d-
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 1. Induction ofHIVexpression in chronically
infected cells
Amt(pg/ml) ofp24antigen
Cell line Induction(fold)
WithoutTPA WithTPA
ACH2 132 7,976 60
Ul 64 950 15
8E5 14,564 NDa
a ND,notdetermined.
sensitive sites inACH2, Ul, and 8E5 cell lines were com-pared. ACH2 and Ul were examined both with and without TPA treatment, since this agent was previously shown to induce viral expression in these cells (31). The induction of
viral expression was confirmed by measuring the release of the viral antigen p24 in the culture medium in the absence and presence of TPA (Table 1). Small but detectable
amounts of p24 antigen wasfound in untreated ACH2 and
Ul, indicatingthat thevirusesinthese celllinesare nottruly latent but that these cells are better defined as chronic low producers. In contrast, the 8E5 cell constitutively secreted a
largeamountof p24antigen(Table 1). TPA treatment of Ul andACH2for 12 h resulted ina 15-and60-fold increase in p24 release in the medium, respectively (Table 1). Three
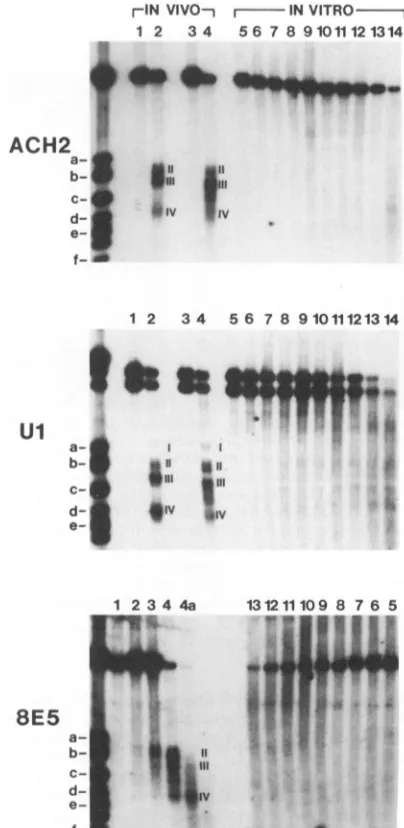
major hypersensitive sites were observed in all three cell lines(sitesII, III, and IV;Fig. 3, lanes 2for ACH2 andUl
cells and lane 4 for 8E5 cells) in the absence ofTPA. To determine theirexactposition in the viral genome,adouble
digestion ofamolecular cloneofHIV(pBru2)byPstIandby several other enzymes scattered throughout the region
ex-aminedwas run along the samples. By using these markers
asreferences, siteIImapped to nt 223 to 325,site III mapped to nt390 to 449, andsite IV mapped to nt 656 to 720 (values averaged fromthree independent experiments, with a stan-darddeviation of12bp). Twochangeswere noted in ACH2 and Ul afterTPA induction: first, siteIIIbecame larger at
theexpenseof its3' boundary,which moved fromnt449 to nt583,its 5' boundarybeing unchanged at nt390 (compare the space betweensitesIIIand IVonlanes 2 and 4 for ACH2 and Ul); second, the intensity of the site IV band was
decreased byatleast 50% (this is mostvisible onFig. 3 for
Ulcells[comparelanes 2 and4]).InUl cells,TPAinduction
also resulted in the appearance ofanewminor site, site Iat
nt56 to 114 (Fig. 3, Ulcells [comparelanes 2and 4]). The
pattern ofhypersensitive sites in untreated 8E5 cells was
similarto thatobserved after TPA induction inACH2 and
Ulcells (Fig. 3, 8E5).
Toprove that the hypersensitive sites observed werethe consequenceofchromatinorganizationandnotsecondaryto
sequence-dependent cleavage preference by DNase I, the
following experiment was performed: DNA was extracted and purified from each cell line and submitted in vitro to
digestion with increasing dosesofDNase I, and DNAwas
thendigested with PstI andexaminedby indirect end
label-ling as described above. The range of DNase I
concentra-tionswaschosentogeneratesimilar levels ofdigestionunder in vitro and in vivo conditions. This experiment indicated thatalthough DNase Iexhibited some sequence preference forcutting(Fig. 3, lanes5 to 14), nocomparable
hypersen-sitive sites were noted (compare lanes 2 or 4 and 14 for ACH2 andUl cells and lanes4 and 13for 8E5).
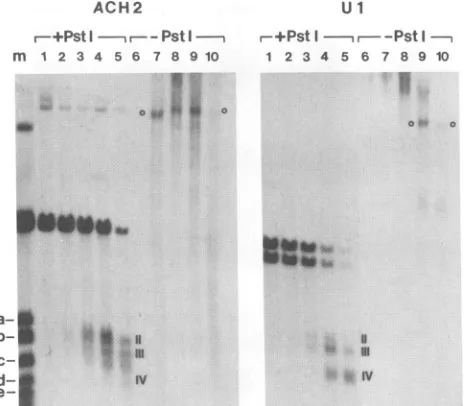
Tounequivocallyprove thateach band observed
(hereaf-ter referred to as a hypersensitive site) was dependent on
digestion by both DNase I on isolated nuclei and PstI
r-IN VIVO-1 IN VITRO--1 2 34 5 6 7 8 91011121314
lb
*a
mu
1 2 3 4 5 6 7 8 91011121314
.
B#
U1
c-
w1"
a11d- |v
I
le
-8E5
FIG. 3. Mapping ofDNase I-hypersensitive sites inthe 5'
por-tionofthe HIV genome. Thepattern ofdigestionby DNase Iof purifiednucleifromACH2 andUlcells (withoutTPA[lanes1 and
2]andwithTPA[lanes3and4])and8E5cells(withoutTPA[lanes
1 to4a]) is comparedwiththatbyDNase Iofnaked DNA in vitro
(lanes5 to14). ForACH2 and Ulcells,eithernoDNase I(lanes 1,
3, and5)or15 UofDNase I(lanes2and4)wasused.For8E5cells,
lanes 1, 2, 3,4, and 4a showdigestion by 0, 3.75,7.5, 15and 30 U
ofDNase Iperml,respectively.Forall celllines,lanes5, 6,7, 8,9, 10, 11, 12, 13, and 14 showdigestion by 0, 0.17, 0.24, 0.33, 0.47, 0.65, 0.91, 1.28, 1.79,and 2.5 U ofDNase Iperml, respectively. Markers aredescribedin thelegendtoFig.2. Hypersensitivesites areindicatedbyroman numbers.
digestion of purified DNA, DNA samples
purified
after DNase I digestionofnuclei were examinedbyindirectendlabelling with and without PstI
digestion (Fig.
4).Samples
digested solelywithDNaseI showedhigh-molecular-weight
bands(indicated byopencircleson
Fig.
4)andnobands with sizes similarto those observed after doubledigestion
withDNase I and PstI (Fig. 4, sites II, III, and IV). This
experiment indicates that the fragments visualized (II, III, and IV) depend on both DNase I andPstI digestions. The
-F11W
F
r
"M-W"
0
4.
i
..4
t ilt
4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.68.310.101.164.2]ACH2
+Pst I --I'-PstI-_
m 1 2 3 4 5 6 7 8 9 10
*0~~~~~
u 1
r-+Pst lI -l Pst I 1 2 3 4 5 6 7 8 9 10
o*1ma
0
r-Control- r-ITpa r-Tnf-X I m 1 23 45 1 2 3 45 1 2 3 45m
i""
-,W_.I
Im
C-.
.0
HId-
f*
gv IVe-l
FIG. 4. Hypersensitive site visualization depends on digestion by bothDNase IandPstI. DNA samples extracted afterDNase I
digestion of purified nuclei from ACH2 (treated with TPA) orUl cells (untreated) were digestedor not withPstI andexamined by using indirect end labelling withprobe A.Size markers(lane m) are
described in the legend to Fig. 2. For ACH2 cells, DNase I concentrations (in units per milliliter) were 0(lanes1and6), 9 (lanes 2and 7), 12(lanes 3 and 8), 15 (lanes 4 and 9), and 18(lanes5and
10). For Ul cells, DNase I concentrations (in unitsper milliliter)
were 0 (lanes 1 and 6), 7.5 (lanes 2 and7), 10 (lanes 3 and 8), 15
(lanes4and 9), and 20 (lanes5 and 10). Hypersensitive sitesare
indicated byromannumerals.
high-molecular-weight band observed in the absence of PstI
digestion probably represents the complete HIV genome
resulting from DNase I digestion in hypersensitive sites located inboth the 5' and 3' LTRs,as willbe seenlater.
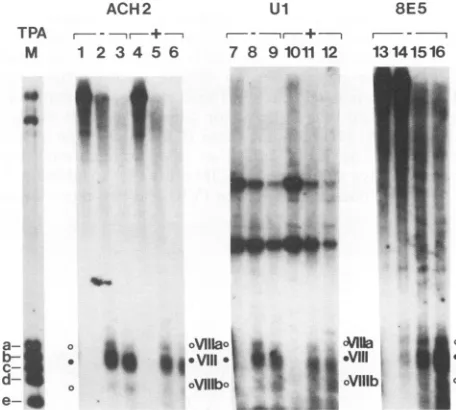
SinceTNF-othas been showntoinduce viralexpressionin ACH2 cells and to mediate in part the induction observed after TPA treatment through an autocrine loop (32), the
chromatin of the 5' LTRwas examined after TNF-ot treat-ment(100U/mlfor 12 h) andcompared with TPAtreatment
in these cells(Fig. 5).
TNF-c.
treatmentwasassociatedwithchanges in the 5' LTR indistinguishable, at this level of
resolution, from those observed after TPA treatment (Fig.
5).
In Fig. 6, the hypersensitive sites described above have beenalignedwithanillustration of the 5'portionof the HIV genome (the LTR and the beginning of the gag gene).
Binding sites forsomeofthe transcription factors knownto interact with the LTR are indicated as landmarks. Site I, which is present only in Ul cells and slightly increases in
intensity after TPA treatment, corresponds to aregion of the LTR in which binding sites for transcription factors have been identified including AP-1 (15) (Fig. 6). Site II
partially overlaps a silencer (negative regulatory element) containing a binding site for transcription factor USF (16, 37, 41, 58) and contains a region of the viral promoter in
which several proteins have been shown to bind in vitro (16, 40, 42, 51). Site III is separated from siteII by a 65-nt space which probably indicates the protection of DNA from DNaseI digestion in purified nuclei by a DNA-bound
factor. Interestingly, the protected region maps to the
en-hancer (16, 37, 41, 58), where at least three distinct
pro-a_ _
b-
SI i
II-b
d_so
4.0I
IV Bg-e
e-~~~~~~
f--f
FIG. 5. TPA andTNF-a treatment induce the same modifica-tions in DNase I hypersensitivity in the 5' LTR. Exponentially
growing ACH2 cells were treated for 12 h with TPA (10 nM) or TNF-a (100U/ml). Purified nucleiweretreated withDNaseIatthe following concentrations (in unitspermilliliter):0(lanes1),9(lanes
2),12(lanes 3),15(lanes 4), and18(lanes 5). Markers (lanesm)are described in thelegendtoFig.2. PurifiedDNAweredigestedwith PstIandexamined withprobeA.Hypersensitive sitesareindicated byromannumbers.
teins have been shown to bind, NF-KB (Fig. 4) (27), HIVEN86A (14), and EBP-1 (56, 57). However,we cannot
completely exclude the possibility that this protection is
secondary to a phased nucleosome present in this region. SiteIII, in the absence ofTPA, covers aregion containing three Spl binding sites (22), the TATA box where TFIId
binds, and additional sites close to the site of initiation of
transcription, at the U3-R junction (21). TPA induction
results inanextensionof siteIIIdownstreamtoincludethe
Tat-responsive region (36). Finally, site IV, which is most
apparent in uninduced Ul cells(Fig. 6),maps3' of the LTR
in a region for which a regulatory function has not been
discovered yet.
Mapping of hypersensitivesites inthe3'LTR.The 5' and 3'
LTRs in retroviruses exert different functions despite an
identical nucleotide sequence. The 5' LTR acts as a
pro-moter: transcription is initiated at the U3-Rjunction. The
3' LTR functions as a site of polyadenylation of the
vi-ral transcripts. Different mechanisms have been invoked
toexplain why transcripts are notpolyadenylatedin the 5' LTR at the R-US junction, a region located downstream of the initiation site (20, 52) and why transcription is not or poorly initiated in the 3' LTR (7). It was therefore of interest to determine whether these functional differences
mightbeaccompanied bystructuraldifferencesinchromatin
organization.
DNase Idigestion
ofpurified
nuclei resulted in the appearance ofone major site (VIII) and two minor sites (Vllla and VlIIb) in the 3' LTR in all three cell lines(Fig. 7). Contrarytowhathad beenobserved in the 5' LTR, nochange was observed inACH2 and Ul cells after TPAtreatment. Using markers,asdescribed for the 5'LTR,these siteswere mapped asfollows: VIIIa, nt 9178to 9221
(nt94 to 137 in the 5' LTR); VIII, nt 9322 to9489 (nt 238
to 405 in the 5' LTR); and VlIIb, nt 9535 to 9586 (nt 451
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.320.537.79.286.2] [image:5.612.58.290.81.284.2]_0 U 3 -N. o-R--o U
5_--.N
Ap- I
GA"
USF kB SplTF.ld TAR
:11:
IIII I IV
"N.
..v..
'li\..
[image:6.612.116.534.83.355.2].:
FIG. 6. Alignment ofhypersensitive sites in the 5' LTR with cis-acting regulatory element. An enlargement of hypersensitive sites in Fig. 2from ACH2and Ul cells in thepresence(+)and the absence(-)of TPAarealignedtoscale with the5' LTRand itscis-acting regulatory elements.TAR,Tat-responsive element; kB, NF-KB.
to 502 in the 5' LTR). If one compares the patterns of hypersensitivity in the5'and 3' LTRs, it isapparentthat site VIIIapproximately covers aregion encompassing both site
IIand IIIandthe interveningspace. However, evenshorter
exposure times showed a single site with no intervening
space, indicatingthat the protein(s) that protected thisarea
inthe 5' LTRis notbound in thehomologous region in the
3' LTR.
Theintragenic region containsahypersensitivesitemapping
tothepolgene. Sincewehadpreviously identified an
intra-genic enhancer in the pol gene of HIV-1 (47) and since
enhancer elementsarefrequently associatedwith
hypersen-sitivesites(19), the possibility of DNaseIhypersensitivity in
this region was particularly intriguing. All three cell lines were testedas described above with probeB(Fig. 1). In all
celllines, amajor hypersensitive sitewas observed in both
the presence and absence ofTPA at nt 9400 (Fig. 8). This corresponds approximately, given the uncertainty of size
determination of such alarge fragment, to site VIII, which was described above as the major site in the 3' LTR. No
othermajorsitewasobservedinACH2or8E5 cells (Fig. 8).
InUlcells,however, threeadditional sites(V, VI,andVII)
wereapparentin the absence ofTPA(Fig.8,comparelanes
7 with 8 and 9). Site VII mapped to nt 4534 to 4733. This region is precisely located between the two functional
do-mains of the intragenic enhancer identified in HeLa cells (47).Twoadditional sites (V andVI[Fig. 8])wereidentified
in Ul andmapped to nt2849 to2956 for site V andnt 3073
to 3187 for site VI. Both sites V and VI were also faintly
visible in ACH2uponlonger exposuretimes of the autora-diograms (datanotshown).
ACH2 Ul 8E5
TPA - r + - - +
-M 1 2 3 4 5 6 7 8 91011 12 13141516
a- 0 o*v: 'illaom i
d- o0 oVilibo oVillb;
e-~d- 0 oVli~~~~~~~~~~~~li.! f1tAftb |
FIG. 7. The 3' LTR containsasingle majorDNase
I-hypersen-sitivesite. DNA samplesfrom TPA-treated(+) and untreated(-)
cellsweredigestedwithDNase Iatthefollowingconcentrations(in
unitspermilliliter):0(lanes 1, 4, 7, 10,and13), 12(lanes 2, 5, 8, 11,
and15), and 18(lanes 3, 6, 9, 12, and 16). DNAwas purifiedand
digestedwith BamHI(forACH2 and 8E5cells)orwith Earl(forUl
cells) and analyzed by electrophoresis on a 1.5% agarose gel.
Markers (lane M) are double digests of pBru2 with BamHI (for
ACH2 and 8E5)or EarI (for Ul) andAflII (nt 9649 [marker a]),
BanII(nt9541 [markerb]), HpaII (nt9442 [markerc]),DraIll (nt 9339 [markerd]),andEcoRV(nt9165 [marker e]). Hypersensitive
sitesareindicatedbyromannumbers.
+
+ N u
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.337.565.416.621.2]ACH2
TPA r, +
M 1 2 3 4 5 6
^
a-
aF
' rb-v
I-,
h-@
-_
j_w
Ul
I-I-- +T
7 8 9101112
.vIII
I
VII.
9.
*..Vil
VIl
V°
* 0 - *VI
oV
FIG. 8. Colocalization of ahypersensitive site inthe intragenic region. DNA from nuclei that had DNase I (same concentrations as in the legend
digestedwithPstI(forACH2 and 8E5 cells) or
Sp
analyzedona0.8%agarose gel, andhybridized w
transfertonylon membranes. Markers (lanes M)a of pBru2 withPstI(forACH2and 8E5) orSphI(for
9619[markera]),XhoI(nt 8944[markerb]),BamH c]), HhaI(nt 7863 [markerd]), DraIll (nt 6627 [m;
5821[markerfl),AlwnI(nt5423 [markerg]),EcoR] h]),KpnI(nt 3862 [markeri]),PvuII(nt3335[mark
(nt 2532[markerk]).
DISCUSSION
In this report, I have used three HIV-1
fectedcell lines to study the chromatin
stru4
Byusingthree setsof probes and restriction,, sites and five minor sites for DNase I cut
identified in isolated nuclei. These results,
Fig. 9 indicate that three major sites are pi
portion ofthe HIV genome(sites II, III, and cell lines. The three cell lines also cont hypersensitive site in the 3' LTR (site VIII).
Ulcell linecontainsone major(VII)and tw
8E5 VI) sites located in the pol gene. TPA treatment, which is
- associated with the induction of viral expression in ACH2
131415 M andUlcells, results inadecrease in intensity of three sites, IV, VI,andVII, andmoreremarkably, in a displacement of the 3' boundary of site II into the R region of the 5' LTR. Someofthese sitesareassociatedwithpreviouslyidentified cis-actingregulatory elements, suchasthe HIV promoter in the 5' LTR(sites II and III), or with an intragenic enhancer
(site VII). Other sites (IV, V, and VI), however, cover regions of the HIV genome with noknown
regulatory
role. Thesefindings raiseseveralinterestingquestions whichwillbediscussedbelow.
Our data on the pattern of digestion of the 5' LTR is
consistent with alarge nucleosome-free region spanning nt 223 to449in theabsence of TPA and nt 223 to 583 after TPA treatment. We suspect that the minor hypersensitive site I v observed in Ul represents a region of internucleosomal
linkerDNA, since it islocated138 ntfrom the 5' limit of site b. II, which corresponds approximately tothe length ofDNA andanenhancer included inonenucleosome. Preliminary observations with beentreatedwith micrococcalnuclease, whichpreferentially cleaves chroma-to Fig. 5) were tin DNA in theinternucleosomal linker region, corroborate 7hI (for Ul cells), this hypothesis (47a). Binding sites for AP-1 transcription
vith
probe
Bafter factor have been identified in this region (15), and moreiredouble
digests
recently, a site with homology to steroid or thyroid hor-Ul)andSak
(ntmone-responsive
elements was identified in the same
region
[(nt8522
[marker
(29) andfoundtobindafactor calledCOUP-TF (4). SeveralIk(nt
4684 [marker transcription factors have been shown to bind specifically toIerj]),
and HindII theregion spanning sites II and III (reviewed in Results and shown inFig. 4).Theregion separatingsiteII from III(65 nt)containsanenhancer(16,37, 41,58) and severalfactors have been shown to bind within this short domain (NF-KB, EBP-1, and HIVEN86A). Since this region is too small to
accommodateanucleosome,although thispossibilitycannot
chronically in- completely be excluded, the absence ofdigestion indicates
cture ofHIV-1. that the DNA is protected from DNase I digestion by sites,fivemajor DNA-boundfactor(s).These results could indicate, with the
ting have been resolution afforded by this method (±12 nt), that DNA-summarized in binding proteins are bound to the enhancer even before
resent in the 5' activation by TPA. Since NF-KB activity is dependent on IV)in all three cellularactivation (by TPA, for example), it is probable that tain one major anotherfactor,suchasEBP-1(56, 57),binds to the enhancer Inaddition,the before cellular activation. After TPA or TNF-ot induction,
tominor (V and site III increases markedly in size, largely by moving its 3'
ACH2 + + + + +
Ul + + + + + + + + + +
+ + +
I II III IV V VI VII Villa Vill Villb
IS//X<<
nI
GAG POL
LTR
2
1 1
4 1
ENV
LTR
[image:7.612.53.284.79.267.2]6 8
FIG. 9. Summary of DNase I-hypersensitive sitesin the HIVgenome.All sites observed in the absence ofTPAdescribedpreviouslyare
aligned withadrawing of the HIVgenome.Thickarrowsindicatemajor hypersensitive sites, whereas thinarrowsindicateminor sites. The
presence(+)orabsence (-)of sites in the three cell lines is indicated atthetopof thefigure.
8E5 - + + +
5'
0 10 Kb
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.135.461.535.699.2]boundaryfrom nt 449 to nt583.ThisTPA-dependent
hyper-sensitiveregioncontainsbinding sites for three transcription
factors: LBP-1 or UBP-1,TCF-1, and CTF (21, 23, 56, 57)
and at theRNAlevel,thecis-actingTat-responsive element,
anRNAhairpin. Theincrease insensitivity noted after TPA induction could be secondary to displacement of a
nucleo-somebythe rapidlyinitiatingRNApolymerase typeII or by
aconformationalchange atthe DNA level. Indeed, the DNA inthis regioncontainsalargeinverted repeat which could, in theory, adopt a cruciform secondary structure. Site IV is locateddownstream of the 5' LTR in the region of theprimer binding site. To date, no regulatory function has been
ascribed tothisregion.However, Moloney murine leukemia
virus contains a silencer in asimilar region (primer binding site) thatspecifically represses viral expression in embryonal carcinomacelllines (1,9, 25,26, 45, 53). We haveobserved
inseveralindependentexperiments, especially in the Ul cell line, a marked decrease (>80%) in the intensity of this site
afterinduction, suggesting that this site could alsofunction
as a silencer(Fig. 3 and 6).
The 5' LTR and 3' LTR have the same nucleotide
se-quence but different functional properties as discussed in Results. The molecular mechanism(s) underlying these dif-ferences is still not fully understood. A major functional difference between the two LTRs is the apparent low rateof transcription starting in the 3' LTRin comparison with the 5' LTR. Studies by Cullen et al. (7) have established a modelof promoter occlusion in which transcripts initiating in the 5'
LTR and continuing through the U3 region of the 3' LTR prevent the assembly of a stable preinitiation complex, thereby inhibitingtranscription initiation. The present study demonstrates a significant structural difference between the two LTRs in terms of DNase I sensitivity, specifically, the absence of a footprint in a region of U3 that corresponds to the enhancer in the 3' LTR, whereas the same site is protected from digestion in the 5' LTR. The absence of protein(s) bound to this region could represent the molecular basis of promoter occlusion. These results would suggest that even low levels of transcription, as observed before TPA induction, are able to inhibit binding of proteins to DNA in this region of the promoter.
Amajorhypersensitive site is located in the part of the pol geneencoding the integrase protein, in close proximity to a region of HIV-1 in which we previously identified an en-hancer (47). The enhancer contains two domains in HeLa cells, fragment 5103 (nt 4079 to 4342) andfragment 5105 (nt 4781 to 6026), and site VII maps to nt 4534 to 4733,precisely between the two functional domains. These results provide evidence that the intragenic enhancer is an element partici-pating in the control of HIV transcription in vivo. TPA
induction resulted in athreefold reduction in theintensity of this site (bydensitometry scanning), whereas theactivity of the enhancer was increased by TPA in HeLa cells. The reason for thisdiscrepancy is unclear but could be a resultof cell-type differences between HeLa cells and Ul cells. The fact that thishypersensitive site is observed only in Ul cells, which are of monocytic origin, and not in ACH2 and 8E5 cells, both of lymphoid origin, could point to a cellular specificity associated with this intragenic element. Itis also possible that the nature of chromatin at the site of viral integration in the cellular genome determines the appearance or the nonappearance of this site. Studies in other viral systems have indeed shown that hypersensitive sites can
induce or suppress the appearance of other hypersensitive sites, even over long distances, and retroviruses frequently
become integrated near DNase I-hypersensitive sites in
chromatin (35, 48). Consequently, depending on the site of proviral integration in the cellular genome, the provirus couldencounter anenvironment favorableorunfavorableto
the establishment of a given hypersensitive site. A last
possibility is that the nucleotide sequence of viruses inte-gratedin the different cell lines differs in the region of the intragenic enhancer and that these differences account for the binding or the absence of binding of specific factors
necessary for the establishment of a hypersensitive site. Thesepossibilities arecurrently underinvestigation.
This study has identified several regions of the HIV genome that probably play a role in the control of HIV
transcription by virtue of their accessibility in chromatin. In orderto generalize these observations andtodraw conclu-sions in terms of cell specificity, these studies have to be
extended to a larger number of cell lines, particularly to
primarycultures, and to several sites ofintegration percell
line.Thesestudies alongwith thehigh-resolution mappingof
proteinbinding sites within these regions by genomic foot-printingwill undoubtedly increaseourunderstandingofHIV regulation invivo.
ACKNOWLEDGMENTS
I thank Peter Paras, Jr., for superb technical assistance and
Monique Dubois-Dalcq for support and encouragement. I thank
Tom Folks, Guido Poli, Anthony Fauci, and the AIDS Reagent Reference Program at NIAIDforthe generous gift of the celllines
used in these studies; Simon Wain-Hobson for the gift of pBru2
plasmid; and the membersoftheLaboratoryof Viral andMolecular
Pathogenesis for comments on the manuscript. The initial part of
this project was conducted in the laboratories of Arsene Burny (UniversiteLibre deBruxelles, Brussels, Belgium) and Susie Spre-cher (Institut Pasteur du Brabant, Brussels, Belgium) and I thank them forencouragement andsupport.
REFERENCES
1. Barklis,E., R. C.Mulligan,and R.Jaenish. 1986.Chromosomal
position or virus mutation permits retrovirus expression in
embryonalcarcinoma cells. Cell 47:391-399.
2. Bushel, P., K. Rego, L. Mendelsohn, and M. Allan. 1990.
Correlation between patterns of DNase I-hypersensitive sites
andupstream promoteractivity of the human e-globin geneat
different stages of erythroid development. Mol. Cell. Biol. 10:1199-1208.
3. Clouse, K. A., D. Powell, I. Washington,G. Poli,K. Strebel, W.
Farrar, P. Barstad, J. Kovacs, A. S. Fauci, and T. M. Folks. 1989. Monokine regulation of human immunodeficiency virus type 1 expression in achronically infected human Tcell clone. J.Immunol. 142:431-438.
4. Cooney, A. J., S. Y. Tsai, B. W.O'Malley,andM.-J.Tsai. 1991.
Chicken ovalbumin upstream promoter transcription factor
binds to a negative regulatory region in the human
immunode-ficiency virus type 1 long terminal repeat. J. Virol.
65:2853-2860.
5. Culien, B. R. 1990. The HIV-tat protein: an RNA sequence-specific processivity factor? Cell 63:655-657.
6. Cullen, B. R., and W. C. Greene. 1989. Regulatory pathways
governing HIV-1 replication. Cell 58:423-426.
7. Cullen, B. R., P. T. Lomedico, and G.Ju. 1984. Transcriptional interference in avian retroviruses-Implications for promoter insertion modelofleukaemogenesis. Nature (London) 307:241-245.
8. Feinberg, A. P., and B. Vogelstein. 1983. A technique for
radiolabeling DNA restriction endonuclease fragments to high
specific activity. Anal. Biochem. 132:6-13.
9. Feuer, G., M. Taketo, R. C. Hanecak, andH. Fan. 1989. Two blocks in Moloney murine leukemia virus expression in undif-ferentiated F9 embryonal carcinoma cells as determined by
transientexpression assays. J. Virol. 63:2317-2324.
10. Folks, T. M., K. A. Clouse, J.Justement, A. Rabson, E. Duh,
on November 10, 2019 by guest
http://jvi.asm.org/
J. H. Kehrl, and A. S. Fauci. 1989. Tumor necrosis factor-a induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. USA 86:2365-2368.
11. Folks, T. M., J. S. Justement, A. Kinter, C. A. Dinarello, and A. S. Fauci. 1987. Cytokine-induced expression of HIV-1 expression in a chronically infected prom nocytic cell line. Science 238:800-802.
12. Folks, T. M., J. S. Justement, A. Kinter, S. Schnittman, J. Orenstein, G. Poli, and A. S. Fauci. 1988.Characterization ofa promonocytic clone chronically infected with HIV and induc-ible by 13-phorbol-12-myristate acetate. J. Immunol. 140:1117-1122.
13. Folks, T. M., D. Powell, M. Lightfoote, S. Koenig, A. S. Fauci,S. Benn, A. Rabson, D. Daugherty, H. G. Gendelman, M. D. Hoggan, S.Venkatesan,and M. A.Martin.1986. Biological and biochemical characterization of a cloned leu-3- cell surviving infection with the acquired immune deficiencysyndrome virus. J. Exp. Med. 164:280-290.
14. Franza, B. R., S. F. Josephs, M. Z. Gilman, W. Ryan, andB. Clarkson. 1987. Characterization of cellularproteinsthat recog-nize the human immunodeficiency virus enhancer using a mi-croscale DNA-affinity precipitation assay. Nature (London) 330:391-395.
15. Franza, B. R., F. J. Rauscher, S. F. Josephs, and T. Curran. 1988. The fos complex and fos-related antigens recognize se-quence elements that contain AP-1 binding sites. Science 239: 1150-1153.
16. Garcia,J. A., F.K. Wu,R.Mitsuyasu, and R. B. Gaynor. 1987. Interactions of cellular proteinsinvolved in the transcriptional regulation of the human immunodeficiency virus. EMBO J. 6:3761-3770.
17. Greene, W. C. 1991. The molecularbiology ofhuman immuno-deficiency virus type 1 infection. N. Engl.J. Med. 324:308-317. 18. Griffin, G. E., K. Leung, T. M. Folks, S. Kunkel, and G. J. Nabel. 1989. Activation of HIVgene expression during mono-cyte differentiation by induction of NFKB. Nature (London) 339:70-73.
19. Gross, D.S., andW. T.Garrard. 1988. Nucleasehypersensitive
sites inchromatin. Annu. Rev. Biochem. 57:159-197.
20. Iwasaki, K., and H. M. Temin. 1991. The efficiency of RNA 3'-endformationisdetermined by the distance between the cap site and the poly(A) site in spleen necrosis virus. Genes Dev. 4:2299-2307.
21. Jones, K. A. 1989. HIVtrans-activation and transcription con-trolmechanisms. The New Biol. 1:127-135.
22. Jones, K. A., J.T. Kadonaga, P. A. Luciw, and R. Tjian. 1986. Activation of the AIDS retrovirus promoter by the cellular transcription factorSpl. Science232:755-759.
23. Jones, K. A., P. A. Luciw, andN. Duchange. 1988. Structural arrangements of transcription control domains within the 5'-untranslated leader regions of the HIV-1 and HIV-2 promoters. GenesDev. 2:1101-1114.
24. Kinter, A. L., G. Poli, W. Maury, T. M. Folks, and A. S. Fauci. 1990. Direct andcytokine-mediated activation of protein kinase C induces humanimmunodeficiency virus expression in chron-ically infected promonocyticcells. J. Virol. 64:4306-4312. 25. Loh, T.P., L. L. Silvert, and R. Scott. 1987. Proviral sequences
that restrict retroviral expression in mouse embryonal carci-noma cells. Mol. Cell. Biol. 7:3775-3784.
26. Loh,T.P., L. L. Silvert, and R. Scott. 1988. Negative regulation ofretrovirus expression in embryonal carcinoma cells mediated by an intragenic domain. J. Virol.62:4086-4095.
27. Nabel, G., and D. Baltimore. 1987. An inducible transcription
factor activatesexpressionofhumanimmunodeficiencyvirus in T cells. Nature (London)326:711-713.
28. Nedospasov, S. A., and G. P. Georgiev. 1980. Non-random
cleavage ofSV40 DNA in the compact minichromosome and free in solution by micrococcal nuclease. Biochem. Biophys. Res. Commun. 92:532-539.
29. Orchard, K., N. Perkins, C. Chapman, J. Harris, V. Emery, G.
Goodwin, D. Latchman, and M. Collins. 1990. A novel T-cell protein which recognizes a palindromic sequence in the negative
regulatoryelement of the human immunodeficiencyvirus long terminal repeat. J. Virol.64:3234-3239.
30. Pavlakis, G.N.,and B. K. Felber.1990. Regulationof expres-sion of humanimmunodeficiencyvirus. The New Biol.2:20-31. 31. Poli, G., P. Bressler, A. Kinter, E. Duh, W. C. Timmer, A. Rabson, J. S. Justement, S. Stanley, and A. S. Fauci. 1990.
Interleukin 6 induces human immunodeficiency virus expres-sion in infectedpromonocytic cells alone and insynergy with
tumornecrosis factor-a bytranscriptional and
post-transcrip-tional mechanisms. J.Exp. Med. 172:151-158.
32. Poli,G.,A.Kinter, J.S.Justement,J.H.Kehrl,P.Bressler,S.
Stanley,and A. S. Fauci. 1989. Tumor necrosis factor-a( func-tions inanautocrinemannerintheinduction of HIVexpression. Proc. Natl. Acad. Sci. USA 87:782-785.
33. Poli, G., J. M. Orenstein, A. Kinter, T. M. Folks, and A. S. Fauci. 1989.Interferon-abutnotAZTsuppresses HIV expres-sion inchronicallyinfected cell lines. Science 244:575-577. 34. Pomerantz, R.J.,M.B.Feinberg,D.Trono,and D. Baltimore.
1990. Lipopolysaccharide is a potent
monocyte/macrophage-specific stimulator ofhuman immunodeficiency virus type 1
expression. J. Exp. Med. 172:253-261.
35. Rohdewohld, H., H. Weiher, W. Reik, R. Jaenish, and M. Breindl. 1987. Retrovirusintegrationand chromatin structure: Moloney murine leukemia proviral integration sites map near DNaseI-hypersensitivesites. J. Virol. 61:336-343.
36. Rosen, C. A. 1991. Regulation of HIV gene expression by RNA-proteininteractions. Trends Genet. 7:9-14.
37. Rosen, C. A., J. G. Sodroski,and W. A. Haseltine. 1985. The
locationofcis-actingregulatorysequencesin the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 41:813-823.
38. Rosenberg, Z. F., and A. S. Fauci. 1990. Immunopathogenic
mechanisms of HIV infection: cytokine induction of HIV
expression. Immunol.Today 11:176-180.
39. Scott,W.A.,and D.J.Wigmore. 1978.Sitesinsimian virus40 chromatinwhich arepreferentially cleaved byendonucleases. Cell 15:1511-1518.
40. Shaw, J. P.,P.J. Utz,D. B.Durand,J. J.Toole,E.A.Emmel,
and G. R. Crabtree. 1988.Identification ofaputativeregulator
ofearlyTcellactivationgenes. Science 241:202-205.
41. Siekevitz, M., S. F.Josephs,M.Dukovich, N.Peffer,F.
Wong-Staal,and W.C. Greene.1987. Activationofthe HIV-1 LTR
by
T cell mitogens and the trans-activator protein of HTLV-1.
Science238:1575-1578.
42. Smith, M. R., and W. C. Greene. 1989. The same 50-kDa cellularproteinbindstothenegativeregulatoryelementsofthe
interleukin-2receptora-chaingeneand the HIV-1 LTR. Proc. Natl. Acad. Sci. USA 86:8526-8530.
43. Stanley,S.K.,P. B.Bresler,G.Poli,and A.S. Fauci. 1990. Heat shock induction of HIV production from chronically infected promonocyticand Tcelllines. J.Immunol. 145:1120-1126. 44. Stanley,S.K.,T. M.Folks, andA. S. Fauci. 1989.Inductionof
expression of human immunodeficiency virus in a chronically
infectedhumanpromonocyticcelllinebyultraviolet irradiation.
AIDS Res. Hum. Retroviruses5:375-384.
45. Tsukiyama,T., N.Ohtsura,and K. Yokoro.1990. Characteriza-tion of the negative regulatory element of the 5'
noncoding
regionofMoloneymurine leukemia virus in mouseembryonal
carcinomacells. Virology 177:772-776.
46. Varshavsky, A.J., 0. H. Sundin, and M.J. Bohn. 1978. SV40 minichromosome: preferentialexposureof theoriginof
replica-tionasprobedbyrestriction endonucleases. Nucleic Acids Res. 5:3469-3479.
47. Verdin, E., N. Becker, F. Bex, L. Droogmans, and A. Burny. 1990. Identification and characterization ofan enhancer in the
codingregion of the genome of humanimmunodeficiencyvirus type 1. Proc. Natl. Acad. Sci. USA 87:4874-4878.
47a.Verdin, E., and P. Paras. Unpublisheddata.
48. Vijaya, S., D. L. Steffen, and H. L. Robinson. 1986. Acceptor
sites for retroviral integrationmapnearDNaseI-hypersensitive
sites in chromatin. J. Virol. 60:683-692.
49. Virelizier, J. L. 1990. Cellular activation and human immuno-deficiency virus infection. Curr. Opin. Immunol. 2:409-413.
on November 10, 2019 by guest
http://jvi.asm.org/
50. Wain-Hobson, S., P.Sonigo,0.Danos,S. Cole, and M. Alizon.
1985. Nucleotide sequence of the AIDS virus, LAV. Cell 40:9-17.
51. Waterman, M. L., and K. A. Jones. 1990.Purification ofTCF-1 a,aT-cell specific transcription factor that activates the T-cell
receptorCageneenhancer inacontext-dependentmanner.The
NewBiol. 2:621-636.
52. WeichsanderGlon,C., J. Moonks, and N. J. Proudfoot. 1991.
Occlusion of the HIV poly(A) site. Genes Dev. 5:244-253. 53. Weiher, H., E. Barklis, W. Ostertag, and R. Jaenish. 1987. Two
distinct sequence elements mediate retroviralgene expression inembryonal carcinoma cells. J. Virol. 61:2742-2746.
54. Wu, C. 1980. The 5' ends of Drosophila heat shockgenes in chromatin are hypersensitive to DNase I. Nature (London)
286:854-860.
55. Wu, C., P. M. Bingham, K. J. Livak, R. Holmgren, and S. C. R. Elgin. 1979. The chromatin structure of specific genes. 1. Evidence for higher order domains of defined DNAsequence. Cell 16:797-806.
56. Wu, F. K., J. A. Garcia, D. Harrich, and R. B. Gaynor. 1988. Purification of the human immunodeficiency virus type 1 en-hancer and TAR binding proteins EBP-1 and UBP-1. EMBO J. 7:2117-2129.
57. Wu, F., J. Garcia, R. Mitsuyasu, and R. Gaynor. 1988.
Alter-ations inbinding characteristics of the human immunodeficiency virus enhancer factor. J. Virol. 62:218-225.
58. Zeichner, S. L., J. Y. H. Kim, and J. C. Alwine. 1991. Liniker-scanning mutational analysis of the transcriptional activity of the human immunodeficiency virustype1long terminalrepeat.
J. Virol. 65:2436-2444.