Vol.64,No. 12
The
Herpes Simplex
Virus
Type
1UL42
Gene Product:
a
Subunit
of
DNA
Polymerase
That
Functions
To Increase
Processivity
J. GOTTLIEB,' A. I. MARCY,2tD. M. COEN,2 AND M. D. CHALLBERG1*
Laboratory of Viral Diseases, NationalInstitute ofAllergyandInfectiousDiseases, Bethesda, Maryland 20892,' and
Department ofBiological Chemistry andMolecularPharmacology, Harvard MedicalSchool,
Boston,
Massachusetts 021152 Received 2 July 1990/Accepted 12 September 1990Genetic experiments have shown that the products of the herpes simplex virus type 1 (HSV-1) DNA polymerase(UL30)and UL42genesarebothrequiredfor viralDNAreplication,andanumber of studies have suggested that thesetwoproteins specffically interact. We have confirmed and extended these findings. The viral DNA polymerase from HSV-1-infected cells has been purified as a complex containing equimolar
quantities of the UL30 (Pol, the catalytic subunit)and UL42 polypeptides. Sedimentation and gel filtration analyses of this complexareconsistentwith the idea that thecomplexconsistsofaheterodimer of Pol and UL42. A complexwithidentical physicalandfunctional propertieswasalsopurifiedfrom insectcellscoinfectedwith
recombinant baculoviruses expressingthe twopolypeptides. Therefore,theformationofthe Pol-UL42complex does not require the participation of any other HSV-encoded protein. We have compared the catalytic propertiesofthePol-UL42 complexwiththose oftheisolatedsubunits oftheenzymepurified from recombinant
baculovirus-infected insectcells.Thespecificactivityof thecatalyticsubunitalonewasnearly identicaltothat ofthecomplexwhenassayedonactivated DNA.Whenassayedon adefinedtemplatesuchassingly primedM13
DNA, however,the combination of PolandUL42utilized fewerprimers andformed larger products than Pol alone. Template challenge experiments demonstratedthatthePol-UL42 complexwas morehighly processive thanPol alone. Our dataareconsistent withthe ideathat theUL42polypeptideisanaccessorysubunit of the
DNApolymerase that acts to increase the processivity of polymerization.
Thegenome ofherpes simplex virus type 1 (HSV-1) is a
153-kb linear double-stranded DNA molecule containing three cis-acting origins ofreplication (43-45, 49, 53) and at least 72 open reading frames (27). Genetic studies have
shown that seven viralgenes are essentialfor HSV
origin-dependentDNAreplication (4-9, 24, 39, 40, 51, 52, 54, 55),
and biochemical functions have been ascribed to the prod-ucts of several of these genes: UL9 encodes an origin recognition protein (32); ULS, UL8, and UL52 encode the three subunits of a helicase-primase complex (11); UL29 encodes a single-stranded DNA-binding protein (ICP8) (9,
36, 52); UL30 encodes a DNA polymerase (7, 8, 39); and UL42 encodesa double-stranded DNA-binding proteinthat has been reported to stimulate the activity of the DNA
polymerase (13, 14, 35). Although genetic studies strongly
suggest that no other HSV gene products areessential for viral DNA replication, it is possible that one or more as-yet-unidentified host cell proteins might also play an essential role.
The HSV DNApolymerase was first detected as anovel
enzyme activity present in extracts ofHSV-infected cells. Thisactivitywasshowntobe dependent onthe product of the pol gene (UL30) by analysis of temperature-sensitive mutants and viral mutants with altered sensitivities to PP1
and nucleoside analog inhibitors (7, 8, 39). DNA sequence
analyses ofthe polgene predict apolypeptide chain of137 kDa(15, 41), and purified preparations oftheenzymefrom
HSV-infected cells have been shown tocontain a
polypep-tideofapproximately that size (31, 37). Sequence
compari-*Correspondingauthor.
tPresentaddress:DepartmentofBiophysical Chemistry, Merck, Sharpe,andDohmeResearchLaboratories, Rahway, NJ 07065.
sonstudieshavedemonstrated that thepolgenepolypeptide
isamember of the eucaryotic DNA polymerase afamily of
DNApolymerases (29). Thepolgenepolypeptidehas been
showntobecatalyticallyactive intheabsenceof otherviral polypeptides by overexpression in yeast cells (16), by in
vitrotranscription-translation (12), and by overexpression in
insect cellsinfectedwitharecombinant baculovirus(25). In addition to DNA polymerase activity, the Pol polypeptide
also hasanintrinsic 3'-5' exonucleaseproofreading activity
(19, 25, 31)aswellas a5'-3' exonuclease activity capable of
functioning asa RNase H(10, 25).
ItwasreportedsometimeagothatHSVDNApolymerase
activityisassociated witha secondpolypeptideof about50 to60 kDa(37, 48).Thispolypeptidehas since been shown to be the product of the UL42gene (14, 35). The nature and stoichiometry of the association between Pol and UL42 is notknown. There is evidence that the UL42 protein
stimu-latesthe DNA polymerase activity of the Pol polypeptide, although the mechanistic basis for this stimulation has not been explored (13). UL42 is more abundant than Pol in
HSV-infected cells and can bepurified of detectable DNA
polymerase activity (13, 14, 48). This form of the UL42 protein bindsdouble-strandedDNAinanapparently
nonco-operative, sequence-independentmanner(14, 48). The
rela-tionship between the DNA-binding activity of UL42 and DNA replication is not known. UL42 is a phosphoprotein
(26), but again, thefunctional significance of
phosphoryla-tion has notyetbeen studied.
In this report, we have examined both the physical and functional interactions between the Pol polypeptide and UL42protein.Wehaveshown that the HSV DNA
polymer-ase can be purified from virus-infected HeLa cells as a
heterodimer of the Pol and UL42 polypeptides. We have
purifiedtheindividualcomponentsofthisheterodimer from
5976
JOURNALOFVIROLOGY, Dec. 1990, p.5976-5987 0022-538X/90/125976-12$02.00/0
CopyrightC 1990, AmericanSociety forMicrobiology
on November 10, 2019 by guest
http://jvi.asm.org/
insect cells infected with recombinant baculoviruses, and haveadditionally shown that the Pol-UL42 heterodimercan
be purifiedfrom insect cells coinfected with thetwo recom-binant baculoviruses. Finally, by comparing the catalytic properties ofthese purified proteins on singly primed M13 DNA templates, wehave shown that the UL42proteinacts to increase the processivity of the DNApolymerase.
MATERIALS AND METHODS
Cells and viruses. Vero and ICP8-transformed U35 Vero cells (34) were propagated in Eagle's medium containing 10%fetal bovine serum. HeLa suspension cells were main-tained in minimal essential medium containing 5% horse
serum. Virusstocks of the KOS strain ofHSV-1weregrown
and assayed as described previously (6). Spodoptera
fru-giperda (Sf9) cells were maintained in TMNFH medium (GIBCO) containing 10% fetal bovine serum, and wild-type Autographica californica nuclear polyhedrosis virus (Ac-NPV) and recombinant baculoviruses were propagated as
previously described (46).
Constructionof the AcNPV/UL42 baculovirusrecombinant.
The baculovirus recombinant AcNPVUL42 contains the HSV-1 UL42-coding region inserted downstream of the baculovirus polyhedrin promoter and expresses the 51-kDa UL42 gene product. The plasmid pNN4, which was the
source of the UL42-coding sequences, contains the entire UL42 gene and approximately 300 bp of 5' and 3' flanking
sequencesinserted into the SmaI site ofpBluescribe (54). A
unique BglII site wasengineered 8bp upstreamof theATG
start site of the UL42 open reading frame in pNN4 by oligonucleotide-directed mutagenesis (20).TheUL42-coding regionwas removed by digestionwithBglII and EcoRIand
ligated into the BamHI and EcoRI sites ofpVL1392 (22)to
producetheplasmid pVLUL42.Sf9 cellswerecotransfected with AcNPV DNA and pVLUL42 plasmid DNA, and sev-eral viral plaques with altered morphology were isolatedas
previously described (46). Lysates of infected cells were screened for UL42 sequencesbyDNAhybridizationand for
the expression of UL42 protein by Western immunoblot
analysis of sodiumdodecyl sulfate (SDS) protein gels using
rabbit antiseraas describedpreviously (33).
Buffers. BufferAcontained20 mM Trishydrochloride (pH 8.0), 0.5 mM dithiothreitol (DTT), 0.5 mM EDTA, 0.5 mM
phenylmethylsulfonyl fluoride, and 10%glycerol. Buffer B
contained 20 mM HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid) (pH 7.6), 0.5 mM DTT, 0.5 mM
MgCl2, 10 mM NaHSO3, 0.5 mM phenylmethylsulfonyl fluoride,and2 ,ug (each)ofleupeptinandpepstatinAperml.
Buffer Cwas20 mMHEPES(pH 7.6),0.5mMDTT,0.5 mM
EDTA, 0.5 mM phenylmethylsulfonyl fluoride, and 10%
glycerol. Buffer Dwas20 mM Trishydrochloride (pH 7.5),3
mM MgCl2, 4% glycerol, 0.5 mMDTT, 0.1 mM EDTA, 50
,ug of bovine serum albumin (BSA) per ml, 150 mM
(NH4)2SO4. Buffer E contained 20 mM HEPES (pH 7.6), 1
mMMgCl2,4%glycerol,0.1mMEDTA, 1 mMDTT,and 75
mMNaCl.
Proteinpurification. (i)HSVDNApolymerase. Six liters of
HeLasuspension cellswas infected with HSV-1 ata
multi-plicity of infection of20. At 16 h after infection, the cells
were harvested by centrifugation, washed with 200 ml of
coldphosphate-buffered saline,andresuspendedin 40 ml of
hypotonicbuffer(buffer B). The cellswereallowed toswell
on ice for 15 min and were lysed with a Dounce
homoge-nizer. Nuclei were pelleted by low-speed centrifugation
(2,000 x g)andwereresuspendedin 20 ml of bufferB,lysed
with the addition ofan
equal
volume ofbufferBcontaining
3.4 MNaCl,
and incubated on ice for 1 h. The nuclearextractwasclarified
by
centrifugation
at100,000
x gfor1h,
and the
supernatant
fraction was removed anddialyzed
extensively against
buffer Ccontaining
50 mM NaCl. Insol-ubleprotein
was removed from the nuclear extractby
centrifugation,
and the solubleprotein
fractionwasapplied
to a 25-mlphosphocellulose
column(5.1
by
2.5cm)
previ-ously
equilibrated
in the same buffer. Proteinswere eluted from the column with a 250-ml lineargradient
containing
0.05 to0.8 M NaClin buffer C. Fractions
containing
HSV DNApolymerase (assayed
asdescribedbelow)
werepooled
and
dialyzed
against
buffer Ccontaining
0.1 M NaCl andapplied
to a 10-mlsingle-stranded
DNA agarose column(Bethesda
ResearchLaboratories)
equilibrated
inthe samebuffer.Proteinswereelutedwitha100-mllinearsalt
gradient
containing
0.1to0.8 MNaClinbufferC. Fractionscontain-ing
DNApolymerase activity
werepooled
anddialyzed
against
buffer A. Thepartially purified polymerase
was further fractionated on a MonoQ
HR5/5
anionexchange
column
(Pharmacia)
at 0.5ml/min
with a 40-ml(80 min)
linear saltgradient
from0to 1MNaClinbufferA. Fractionscontaining
DNApolymerase
activity
were concentratedto 0.6 ml in Centricon 30concentrators(Amicon)
and loadedonto two12.4-ml15to35%
glycerol gradients
inbufferC-0.1
MNaCl-0.01%
Nonidet P-40. Aftercentrifugation
in anSW40
Tirotor for 56 hat40,000
rpm, 0.3-ml fractionswere collected from the bottoms ofthetubes,
assayed
for DNApolymerase activity,
andanalyzed by
SDS-polyacrylamide
gel
electrophoresis
(PAGE).
For the
purification
of theisolatedcatalytic
subunit(Pol),
40
225-cm2
flasksofSf9 cellswereinfected with thebaculo-virus BP58
(25)
atamultiplicity
of10 PFUpercell;
forthepurification
of recombinant Pol-UL42complex,
40225-cm2
flasks of Sf9 cells were coinfectedwith BP58 and AcNPV/UL42at
multiplicity
of10PFUofeach viruspercell. In both cases, the infected cells were shaken offthe flasks at60 hafter
infection,
washed in serum-freeGrace'smedium,
andresuspended
in40 mlof bufferB.Theisolation of nucleiand thesubsequent
purification
oftheseproteins
were identicaltothe
procedure
described abovefor the Pol-UL42complex
from HSV-1-infectedcells.
(ii)
UL42protein.
Approximately
90%
ofthe UL42protein
was
physically
separated
from the Pol-UL42complex during
early
stages
ofPol-UL42purification.
Fractionscontaining
UL42protein
were identifiedby
Western
blotanalysis
ofSDS
protein
gels using
rabbit antisera to the UL42 geneproduct
(33).
Themajority
of free UL42 eluted before thePol-UL42
complex during
fractionationonphosphocellulose
(see
above).
Fractionscontaining
UL42protein
werepooled
and
dialyzed against
0.05 MNaCl inbufferC andreapplied
to a 5-mlphosphocellulose
column. Boundproteins
wereeluted from the column with a 50-ml linear
gradient,
as described above. Fractionscontaining
UL42 werepooled
and
dialyzed
against
0.05 M NaCl inbufferC andapplied
toa 3-ml double-stranded DNA cellulose column
(Sigma).
Proteins were eluted from the column with a 30-ml linear
gradient containing
0.05 to 0.70 M NaCl in bufferC. Frac-tionscontaining
UL42protein
werepooled
anddialyzed
against
buffer A andapplied
to a MonoQ
HR5/5
column(Pharmacia).
Thecolumnwaselutedasdescribed above forthePol-UL42
complex by using
alinear saltgradient
from 0to0.5MNaCl in bufferA. Toremoveresidualpolymerase
activity, UL42-containing
fractionswerepooled
andconcen-trated to 0.6ml with Centricon 30concentrators
(Amicon)
on November 10, 2019 by guest
http://jvi.asm.org/
5978 GOTTLIEB ET AL.
and sedimented through 12.4-ml glycerol gradients as de-scribed above forPol-UL42.
UL42 protein from recombinant baculovirus-infected in-sect cells was purified from 40 225-cm2 flasks of Sf9 cells infected withAcNPV/UL42 atamultiplicityof 10 PFU/cell. Nuclear extracts were prepared 60 h after infection as described aboveandroutinely contained 15mg ofUL42per
109 cells. The procedurefor the purificationofthe recombi-nantUL42 protein wasidenticalto thatdescribed for UL42 from HSV-infected cells except that the second phosphocel-lulose step wasomitted.
(iii)ICP8. ICP8waspurifiedfrom U35 Verocells(34) bya modification of the method ofO'Donnell etal. (31). Nuclei wereprepared from40 rollerbottles ofU35 Vero cells 18 h afterinfection withHSV-1 at amultiplicity of infectionof5 andextracted in buffer B and high salt as described for the Pol-UL42 complex. The clarified nuclear extract was
dia-lyzed against 0.1 M NaCl in buffer C and fractionated
through a 15-ml single-stranded DNA agarose column. The columnwaselutedwitha150-ml linear saltgradient
contain-ing 0.1 to 1.5 M NaCl in buffer C. Column fractions were
analyzed by immunoblotting of protein gels with rabbit antisera to ICP8 (33). Fractions containing ICP8 were
dia-lyzed againstbufferC andapplied toa4-mlheparin agarose
column (Sigma) equilibrated in the same buffer. Proteins
wereelutedfrom thecolumnwitha40-mllinearsaltgradient
containing0to0.75 MNaCl inbufferC.Fractionscontaining ICP8 were pooled and concentrated to a volume of0.3 ml andlayeredontoa15to35%glycerolgradient, asdescribed forthepurification of thePol-UL42complex.
Determination ofhydrodynamic properties. The sedimen-tation coefficient (S20,,,) ofeachpurified protein was deter-minedfromthe rate ofsedimentation oftheprotein during
theglycerol gradient sedimentationstep ofpurification (see above). The proteinswithknownsedimentationcoefficients
thatwere employed asstandards onparallel gradientswere asfollows: BSA, 4.4S;alcoholdehydrogenase, 7.6S;
p-amy-lase, 8.9S. The Stokes radius of each purified protein was determinedby gelfiltrationanalysis on aSuperose 12 10/30
column(Pharmacia) accordingtothe methodof Ackers(1). The standards of known Stokes radii used to calibrate the columns were as follows: BSA, 3.64 nm; alcohol
dehydro-genase, 4.52nm;
P-amylase,
5.14nm.DNAtemplates. Singly primed single-stranded DNA sub-strateswere produced by hybridizing 50pmol of synthetic
oligonucleotide primer to 5 pmol of the appropriate phage
DNA (M13mpl9 or XX174) in a reaction volume of100 ,ul containing50 mMTrishydrochloride (pH 7.5),5 mMMgCl2,
and100mMNaCl.Thehybridizationmixturewasincubated
at 90°C for 5 min and allowed to cool slowly to room
temperature for 1 h. Excess primer was removed by gel filtration on a 10-ml Bio-Gel AiSm column (26 by 0.7 cm;
Bio-Rad) equilibrated in a solution containing 10 mM Tris hydrochloride (pH 7.5), 1mMEDTA,and50 mMNaCl. The oligonucleotides used were 5'-GGCGCATAACGATACCA
CTGACC, which is complementary to nucleotides 2067 to 2045 of 4~X174 DNA, and 5'-CAGTCACGACGTTGTAA AACGACGGCCAGT, which is complementary to
nucleo-tides 6300 to6271 ofM13mpl9DNA.
DNA polymerase assays. DNA polymerase activity was
assayed during purification in 50-,u reaction mixtures
con-taining5 p.gofactivated calfthymusDNA(Sigma); 0.5mM
ATP; 50 puM (each) ofdGTP, dATP, anddTTP; and 5 ,uM [oc-32P]dCTP (5 x 103 to 15 x 103 cpm/pmol) in buffer D.
DNAsynthesiswasinitiatedwith theaddition of1 pu1 ofthe
columnfractions. The reactions were terminated with the
addition of2 mlof cold 10% trichloroacetic acid
containing
400 ,ug of BSA after incubation at 37°C for 30min,
and the amount of incorporation of acid-insoluble radiolabelednu-cleotide was measured in aliquid scintillation counter. One unit of polymerase activity was defined as the amount of enzyme required to incorporate 1 pmol of[ot-32P]dCTP into acid-insoluble material in 30 minat 37°C.
The specific details of DNA synthesis on singly primed
M13 or 4X single-stranded circular DNA are given in the figurelegends. Ingeneral, standardreactionmixtures(50
RIl)
contained 20 to 50 fmol of substrate DNA (ascircles);
0.5 mM ATP; 60,uM (each) of dGTP, dATP, anddTTP;
20,uM [cx-32P]dCTP (3,000 to 8,000 cpm/pmol); 50 pLg of BSA per ml; and 10 to 2,000fmol of DNApolymerase. The reactions were incubated at 37°C and terminated with the addition of an equal volume of 1% SDS-40 mM EDTA-60 p.g of sonicated calf thymus DNA per ml. After precipitation in ethanol, the reactionproducts wereanalyzed bygel electro-phoresis in 1% alkaline or neutral agarose gels or in 6%acrylamide-urea gelsfollowed byautoradiography.
RESULTS
HSV DNA polymerase is a heterodimer of Pol and UL42. It has been reported that the HSV DNA polymeraseis
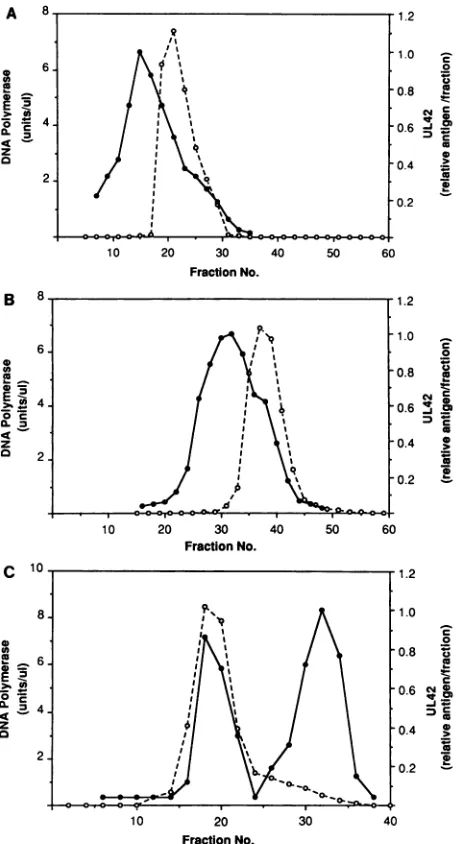
associ-ated with UL42 polypeptide (14, 37, 48). To gain further insight into the nature of this association, we purified the virus-specific DNA polymerase from HSV-infected HeLa cells. During the courseof purification, we assayedfor DNA polymerase activity by using activated DNA as a primer-template and for Pol and UL42 antigen by immunoblot analysiswith monospecific antisera (33) raised againsteither a bacterial fusion protein (Pol) or apeptide predicted from the deduced amino acid sequence (UL42) (Fig. 1; Table 1). At all steps inpurification, DNA polymerase activity copu-rified with Pol antigen, as expected (data not shown). During the early stages of purification the majority of the UL42 protein was separated fromDNA polymerase activity (Fig. 1A and B). A fraction of UL42, however, eluted with the polymerase and was observed as a small shoulder ofthe major UL42 peak during the second chromatographic puri-fication step (heparin agarose chromatography; Fig. 1B).
Glycerol gradient sedimentation of the peak polymerase fractions following Mono Q chromatographyproduced two
distinct peaks of UL42 containing approximately equal amounts of antigen(Fig. 1C). The faster-sedimenting formof UL42 protein cosedimented with the DNA polymerase ac-tivity. These results suggest that a fraction of the UL42 protein forms a stable complex with the polymerase catalytic subunit. The sedimentation rate of the more slowly sedi-menting form of UL42 corresponded to the molecular mass expected for monomeric UL42 protein (51 kDa [28]). We estimate that approximately 5 to 10% of the infected-cell UL42protein copurifies with the DNA polymerase.
The polypeptide composition of the purified DNA poly-merase was examined by SDS-PAGE followed by staining with Coomassie blue (Fig. 2A, lane 4) and by immunoblot analysis (Fig. 2B, lane 1). In the stained gel, three major polypeptides, as well as several minor species, are apparent. The larger major polypeptide, with an observed molecular weight(MW) of 140,000, corresponds to the Pol polypeptide; the smallest major polypeptide, with a MW of 60,000, corresponds to UL42; the third major polypeptide, with a MW of-70,000, does not precisely cosediment with DNA polymerase activity(data not shown) and is present in other preparations of the polymerase in different amounts relative J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
A
0 In 0
0
B
a C
. la
CL,
>4
z
C
0
0-_ _n
)075
<t -a
z
0
10 20 30 40 50 60
FractionNo.
0)
so~
U.
Ui
10 20 30 40 50 60
FractionNo.
10 20
FractionNo.
*1.2
1.0 c
0
*0.8 0
c
*0.6 ,
0.4 0
0.2
30 40
FIG. 1. Copurification of UL42 and DNA polymerase activity. DNA polymerase was purified from HSV-infected HeLacells, as
describedinMaterialsandMethods.DNApolymeraseactivity(0)
was assayed by using activated DNA as a template, and UL42
antigen (0) wasquantitated by densitometric analysis of Western
blots of each fraction; the values plotted refer to relative UL42 antigenperfraction, normalizedtothepeakfraction in eachpanel. (A) Phosphocellulose chromatography; (B) single-stranded DNA
agarosechromatography; (C) glycerol gradientsedimentation.
totheothertwopolypeptides. Webelievethat this
polypep-tide isnotacomponentof the DNApolymerase. The minor polypeptidesintherangefrom 75to130 kDawereshownby
immunoblot analysis to be related to the Pol polypeptide (Fig. 2B) andprobablyrepresent products of limited prote-olysis. Densitometric scanning of the stained gel indicated thatPol andUL42 arepresentinanapparentmolar ratio of 1:1.1. The MW ofthe purified enzyme (Table 2) was esti-mated from its sedimentation coefficient as determined by
glycerol gradientsedimentationanalysisand fromits Stokes radiusasdeterminedby gelfiltrationanalysis.Theobserved value of 181 kDa is in close agreement with the value
[image:4.612.68.296.80.502.2]predictedfrom the deduced amino acidsequencesof Pol(137
TABLE 1. DNA polymerase purification
Total Spat Rliv
Fraction activity % Srat Rliv
(units,
10)
Recovery(units/,u
) purificationPol-UL42
Nuclear extract 660 100 2.3
Phosphocellulose 458 69 10.6 4.6
Single-strandedDNA 337 51 166 72
agarose
Mono Q 280 42 170 73
Glycerol gradient 192 29 571 248
Pol
Nuclear extract 2,600a
Phosphocellulose 341 100 12.7
Single-strandedDNA 186 54 75.5 5.9
agarose
MonoQ 164 48 214 16.8
Glycerolgradient 107 31 348 27
Pol-UL42(bv)b
Nuclear extract 1,800a
Phosphocellulose 810 100 26.1
Single-strandedDNA 378 46 87.5 3.3
agarose
MonoQ 134 16 37.2 1.4
Glycerol gradient 107 13 238 9.1
aNuclearextractalso containsbaculovirusDNApolymeraseactivity (25); yieldandrelativepurificationarecalculated relativetophosphocellulose.
b bv,Purified fromrecombinantbaculovirus-infected Sf9cells.
kDa) and UL42(51 kDa) assuming that the structureofthe
nativeenzymeis aheterodimer ofPol and UL42. We cannot
at present rule out the possibility that one or more of the small, minorpolypeptide speciespresentinthe mostpurified preparations is also a subunit of the enzyme. We think it unlikely, however, since a complex of Pol and UL42 with
identical physical and enzymatic properties, but with dif-ferent contaminating polypeptides, has been purified from
insect cells infected with recombinant baculoviruses ex-pressingPol and UL42 (see below).
Construction of recombinantbaculovirusexpressing UL42. To obtain a source of UL42 protein free of other HSV-encoded proteins, weconstructed a recombinant
baculovi-rusin which theUL42-codingsequenceswere expressed at highlevels under thecontrol ofthepolyhedrin promoter. A
restrictionfragment fromtheplasmidpNN4(54)wascloned
into
M13mpl9,
and oligonucleotide-directed mutagenesiswasused to create a sitefortherestriction enzymeBglII8bp
upstreamfromtheaminoterminusoftheUL42 openreading frame. ABglIIto EcoRIfragment containingthe complete
UL42 open reading frame was then inserted between the
BglII and EcoRI sites of the plasmid vectorpVL1392 (22).
The resulting plasmidwas cotransfected into Sf9 cells with
infectious AcNPV DNA, and recombinant viruses were
selected on the basis of alteredplaque morphology and the presenceof UL42 DNA sequences. One such recombinant,
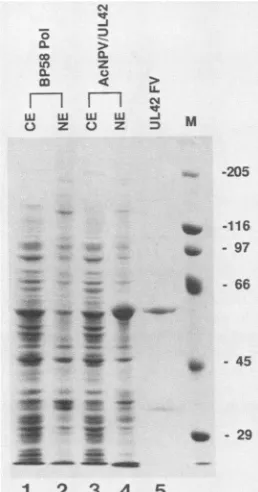
namedAcNPV/UL42,wasselected forfurtherstudy.Figure
3 shows the results from SDS-PAGE analysis of extracts prepared from Sf9 cells infected with AcNPV/UL42. An
abundantpolypeptidewithanelectrophoretic
mobility
iden-tical tothatof UL42purifiedfromHSV-infected HeLa cells (apparent MW, 60,000) was observed in an extract of the nuclei of such cells(Fig. 3,lane4). Thispolypeptide
wasnot observed in a parallel control extract of Sf9 cells infected with therecombinant baculovirus BP58(Fig. 3,lane2)(25);
moreover, it reacted strongly withmonospecific
anti-UL42on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.318.559.93.338.2]5980 GOTTLIEB ET AL.
(N ect
-D
-J 0 0
=D
n.
a.
41 0 -Pol
E_* -ICP8
TABLE 2. Hydrodynamic propertiesa ofpurifiedproteins
ProteinProtein
GO
(105 s)s) Stokes radius~(nm)
MWm flfbPol-UL42 8.0 5.16 181,000 1.18
Pol 6.8 4.49 133,000 1.14
UL42 3.0 4.25 56,000 1.44
a
S20,w
and Stokes radiusweredeterminedasdescribedin Materials and Methods.Molecularweightwascalculated fromthosevaluesaccordingtothe equation: MW=S20,6Rrrqr/k(1 -vp), whereRis theideal gas constant,inis theviscosity of water,ristheStokesradius,k is the Boltzmann constant, vis thepartialspecificvolume of theprotein,assumedtobe 0.74cm3/g,and pis thedensity of water.
b The frictionalcoefficientf/foequalsr/{[3v(MW)/4rrN]033},whereN=6.02
x 1023mol-1.
66
-VI.
4U_
--UL42
45
-1
2
3
4sera on immunoblot analysis (data not shown), confirming the identity of this polypeptide as UL42. We estimatethat
approximately 15 mg of UL42 polypeptidewasproducedin
109 AcNPV/UL42-infected cells, and at least 95% of this
protein was recovered in soluble form in the high-salt nuclear extract. This representsapproximately 10-to15-fold overproduction relative to HSV-infected HeLa cells.
Formation of the Pol-UL42 heterodimerin Sf9cells
express-ing Pol and UL42. The availability of recombinant baculo-viruses expressing both Pol (25) and UL42 allowed us to ask
.0
qTqT a
0 0 -J -1
.00.(L C D:
Pol
, 205
66- UL42
45
29
-1 2 3 4 5
FIG. 2. SDS-PAGEanalysis ofpurified proteins. Electrophoresis
wasperformedaspreviously described (33) by usingaresolving gelof
7.5%acrylamide. The numbers attheleft of each panel indicate the electrophoreticmobility and size (in kilodaltons) of marker proteinsrun
onthesamegel: myosin, 205;P-galactosidase, 116;phosphorylase B, 97; BSA, 66;eggalbumin, 45; carbonic anhydrase, 29. (A) Each lane
contained 10 ,ug of themosthighlypurified fraction of the indicated protein. Followingelectrophoresis, the gelwasstained with Coomassie
brilliantblue. Thearrowindicatesapolypeptide of -70 kDa whichwe
consider acontaminant of theenzyme (see Resultsfor details). (B)
Each lanecontained 1 Fg of themosthighly purified fraction of the indicated protein. Following electrophoresis, the resolved polypep-tidesweretransferredtoapolyvinylidinedifluoride membrane (Immo-bilon;Millipore) and probed withamixtureof antiseraagainst Pol and
UL42,aspreviously described (33).
*,4te ,:.: .. R: :_ :: .: ._
_
4:..;
::'w*._S.
*23sG_:.r:
w '*sF Y wF
_ :_ _ w _W
*W -.s4.
_ _ *'
]X StS.^ * ...
7_ '
., '
_D - -_
lb -116
#* - 97
f
- 66- 45
ft - 29
1 2 3 4 5
FIG. 3. SDS-PAGEanalysis of Sf9 cells infected with AcNPV/ UL42. Sf9 cells were infected with the recombinant baculovirus BP58(expressing Pol)orAcNPV/UL42atamultiplicity of infection
of 10. At60hpostinfection, cells wereharvested andfractionated
into cytoplasmic (CE) or high-salt nuclear (NE) extracts, as de-scribed in Materials and Methods. An aliquot of each extract
correspondingtosolubleproteinfrom5 x 105 cellswasfractionated on anSDS-polyacrylamide gel containing 7.5% acrylamide.
Follow-ing electrophoresis, the gel was stained with Coomassie brilliant
blue. Sixmicrogramsofglycerolgradient-purified UL42 (seeTable
3)wereloaded ontothelast lane onthe right. The numbersatthe rightindicate theelectrophoretic mobilities and sizes (in kilodaltons)
of markerproteins (M)run onthesamegel.
A
116-97
-B
205
-116
-97
--J
IL > co IL
Z 0
m 4:
wLJ Ul w Lw
0) z 0 z U.
c%J
M
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.96.259.80.591.2] [image:5.612.315.554.90.149.2] [image:5.612.372.501.361.607.2]TABLE 3. Purification of UL42
UL42 Total Relative Fraction antigen protein
purification
(mg)a (mg)
Nuclear extract 3.2 411
PhosphocelluloseI 2.7 42.0 8.2
PhosphocelluloseII 1.7 13.2 16.5
Double-stranded DNA cellulose 1.1 1.1 128
Glycerol gradient 0.9 0.9 128
a The values forUL42antigen were determined by densitometric analysis ofWestern blots of glycerolgradient-purified protein, which is essentially
homogeneous (Fig.2),as areference.
whether the formation of the complex between these
poly-peptides can occur in the absence of other HSV-encoded
polypeptides. Sf9 cells were coinfected with BP58 and AcNPV/UL42, and HSV DNA polymerase was purified
from a nuclear extract of these cells. During the course of
purification,DNA polymerase activity was assayed by using activated DNA as primer-template; Pol and UL42 antigen were monitored by immunoblot analysis. As in the case of the DNA polymerase from HSV-infected HeLa cells, Pol
polypeptide copurified with DNA polymerase activity and with a small fraction of the UL42 polypeptide, which was
expressedat amuchhigher level than the Pol polypeptide in coinfected cells. SDS-PAGE and immunoblot analysis ofthe
purified proteinrevealed that Pol and UL42 were present in approximately the same ratioasobserved with the enzyme
purified from HSV-infected cells (Fig. 2B). Moreover, the
sedimentation coefficient and Stokes radius ofthe purified
DNApolymerase(Table 2) and allcatalytic propertiestested (Table 1; see below) were alsoidenticalto thatobserved with thepolymerase fromHSV-infected cells. We conclude that no additional HSV-encoded proteins are required for the
formation of functional Pol-UL42heterodimer.
Purification oftheisolated subunitsof HSVDNA polymer-ase.Todeterminethecontribution ofthe twosubunits of the DNApolymerasetoitsoverallfunction,wehavepurifiedthe
individualsubunits. In the caseofthelargesubunit,this was
accomplished as previously described (25) by using the
baculovirus expression system. The results ofthe purifica-tionare summarized in Table1, and SDS-PAGE analysis of
the purified protein is shown in Fig. 2. The MW of the
purified Pol polypeptide was determined by sedimentation andgel filtration analyses (Table 2). Theobserved value of
133,000 is in excellent agreement with the size of the
polypeptide predicted from the deduced amino acid se-quence (137 kDa) and suggests that the isolatedlargesubunit exists as a monomer in solution. The fact that the purified nativePolsubunithas a smaller observed MW than the DNA
polymerasepurifiedfromHSV-infected cells(or from insect
cells also expressing UL42) lends further support to the conclusion that the HSV DNA polymerase contains more
than one subunit.
The UL42polypeptide was
purified
fromtwo sourcesby
using immunoblot analysis as an assay duringpurification. Since, as previously mentioned, UL42 is present in HSV-infected cells in large excess relative to the amount of
Pol-UL42 complex, it was possible topurify UL42 free of detectable DNApolymerase fromthatsource; the results of thepurification aresummarized in Table 3,andSDS-PAGE
analysis of the purified protein is shown in Fig. 2. Froma practical perspective, themajordifficultyin purifyingUL42 from HSV-infected cells was contamination by the HSV-encoded alkaline nuclease. The peak of nuclease
activity
substantially overlapped the peak ofUL42antigen in most of thepurification procedures tested.The greatest separation of
thetwoproteinswasachieved byphosphocellulose
chroma-tography; the peak of UL42 elution from the column
oc-curred at 0.3 M NaCI, and the peak of nuclease activity
occurred at0.2 MNaCl. Twosuccessivepassesover phos-phocellulose were required to obtain acceptable levels of separation. UL42 wasalso purified from Sf9 cells infected
with the baculovirus AcNPV/UL42 by a nearly identical
procedure; the major differences between the two
proce-duresare aconsequenceofthefactsthatrecombinantUL42
accountsforahigherpercentageof thestarting materialand
thereforerequiredlesspurificationand thatnuclearextracts of baculovirus-infected Sf9 cells do not contain a nuclease
with chromatographic properties similar to UL42. We did
notobserveanyphysicalorbiochemical differences between
the two
preparations
ofUL42. As shown in Table 2, the observed MW ofpurified
UL42was approximately 56,000, suggesting that isolated UL42 polypeptide is a monomeric protein like thePol subunit.Pol-UL42 utilizes a long single-stranded template more efficiently than Pol alone. As we have previously reported (25),the
specific activity
ofthe Pol-UL42 complex didnotdiffer
significantly
fromthatof thepurified catalytic subunitwhen assayed with activatedDNA asprimer-template
(Ta-ble 1). On the basis of this resultandby analogy with other
DNA polymerase systems, it seemed possible that UL42
mightact as anaccessorysubunittoincreasethe efficiency of
polymerization
onlong
single-stranded templates.
Thispossibility
wastestedby
comparing
theactivity
of Pol-UL42 withthatofPolonsingly primedM13 single-strandedDNAin the presence and absence ofexogenous UL42
protein.
Figure 4 showsan
analysis
byalkaline agarosegelelectro-phoresis ofthe products formed at various ratios of
poly-merase to
primer-template
in such anexperiment.
At allratios of enzyme to template tested, the products
synthe-sized by Pol-UL42 consisted almostexclusively
offull-lengthM13 strands. Identicalresultswereobtained
by using
Pol-UL42
purified
from HSV-infected HeLa cells andPol-UL42
purified
from baculovirus-infected Sf9 cells. Ineithercase,thesize distributionof
products
wasnotalteredby
theaddition of
purified
UL42protein (data
notshown).
In contrast,atlow ratiosoftheisolated Pol subunittotemplate,
thepredominant
products were much smaller in size thanfull-length M13
strands,
with themajority
ofproduct
con-sisting
ofspecific
bandsrepresenting
pause sites on thetemplate.We presumethat these pausesitesrepresentsites
of
template
secondary
structure,butwehavenot character-ized these sites in detail.Nearly
10-fold greateramounts of Pol than of Pol-UL42 wererequired
toproduce
approxi-mately equal
amountsofunit-length product
(compare Pol-UL42 at25 and 50fmol with Pol at200and 400fmol).
Theaddition ofa constant amount
(100
fmol)
ofpurified
UL42to reactionscontaining
Pol resulted inamarkedchange
in the sizedistribution oftheproducts synthesized.
Inthosereac-tions
containing
less than 100fmol of Pol inparticular,
the size distribution ofproducts
wascomparable
with thatobservedforan
equal
amountof Pol-UL42.Again,
identical resultswereobtained with UL42purified
fromHSV-infected HeLa cells and UL42purified
from recombinantbaculovi-rus-infected Sf9 cells. Titrationofthe reaction with
increas-ing amounts of UL42
protein
(Fig. 5)
revealed that theamount of UL42
required
toachieve maximum stimulation ofsynthesis
offull-length
strands wasdependent
on andapproximately
equal
(within
afactoroftwo)
totheamountof Pol in the reaction. UL42protein
purified
fromon November 10, 2019 by guest
http://jvi.asm.org/
5982 GOTTLIEB ET AL.
Pol/UL42
Pol
Pol
UL42
Pol
Pol/UL42 +(bv)
UL42 (bv)
fmol
9.4 6.6 4.4
2.3 2.0
000 8 Me8000 00 c0 00 0
n L .0 0
T-CM ,LvCIU
.56
w
FIG. 4. DNA synthesis on singly primed M13 single-stranded circles. Reactions (50 ,ul) contained 25fmol (as circles)ofprimedM13 single-strandedDNA; 0.5 mMATP;2.5,ug ofBSA;60p.M(each) of dATP,dGTP,anddTTP;20p.M[a-32P]dCTP(4,400cpm/pmol);and25 to400fmol oftheindicatedpolymeraseinbufferE.DNAsynthesiswasinitiatedbytheadditionofpolymerase.InreactionsthatcontainUL42 protein, 100fmol ofUL42 wasaddedpriortotheaddition ofDNApolymerase. Incubationwasfor30minat37°C, and thereactionswere
terminated withthe addition of anequalvolumeof1% SDS-40mM EDTA-60pgof carrierDNAperml. Afterprecipitationwithethanol,
the productsweredissolvedin 15 ,ulof0.1 NNaOH-5% glycerol-1mMEDTA-0.025%bromcresol green andfractionatedon a1% alkaline agarosegel. The radioactivityinthegelwasvisualizedbyautoradiography. Pol-UL42andUL42, ProteinspurifiedfromHSV-infected HeLa cells; Pol, Pol-UL42 (bv), andUL42(bv),proteins purified from recombinant baculovirus-infected Sf9cells. LaneM, Phage lambda DNA digested with HindIIIandend labeledwith32p.Thesizes oftheHindIll restrictionfragments (in kilobases)areindicatedby the numbersat the left.
fected cells and UL42purified from recombinant
baculovi-rus-infected Sf9 cells were equally effective in stimulating thesynthesisoffull-length strandsby Pol (datanotshown). Theeffectof UL42 on the size of the productsynthesizedby Polwas notdependent on the presence of any combination of ribonucleoside triphosphates, nor was it inhibited by
-y-thio-ATP(data not shown).
The effect of the HSV single-stranded DNA-binding pro-tein(ICP8)onthesereactionswas examined (Fig. 6). Addi-tion of ICP8 to levels sufficient to completely coat the template in the reaction had no significant effect on the
activity ofthePol-UL42complexonprimedsingle-stranded M13substrates; theaddition of ICP8toreactions containing Pol stimulated DNA synthesis approximately twofold, but did not affect the overall distribution of product length. In agreementwith thepublished results of Ruyechan and Weir (42), we observed a twofold stimulation of the activity of either Pol or Pol-UL42 on the addition of ICP8 to assays utilizing activated DNA as template (data not shown). In
contrasttothe resultsof O'Donnell et al. (30), we conclude that ICP8-coated single-stranded DNA is an effective
tem-plate for the HSV DNA polymerase.
UL42increasestheprocessivity of the HSVDNA
polymer-ase. Togain some insight into the mechanistic basis for the increase in the size of the products synthesized by Pol in the presence of UL42, we examined the efficiency with which the enzyme utilizes primers. A molar excess of substrate
m
C 60- 40
25 fmolPal 20
20 40 60 80 100
UL42(fmol)
FIG. 5. Titration of Pol with UL42. Reactionscontained25fmol of primed M13 DNA, 25 or 100 fmol of Pol, and the indicated amountof UL42protein(purified from HSV-infectedcells).Other reactionconditionswereidentical to those described in thelegendto Fig. 4.Following incubation, thereactions products were fraction-atedon a1%alkaline agarosegel,and theradioactivityinabandof productthatmigratedasunit-lengthM13 DNAstrandswas quanti-tated with aBetagenBetaScope600.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.139.492.77.353.2] [image:7.612.330.560.487.655.2]Pol/UL42 Pol
I+
CP8 +ICP8000)a0 0 I00 00 0
oC
a aO
a oao
Oa a ao8o
o° C° $
M CM u, Lo Nl ,, in t C inN_In N Ln
10 20 30
time
(min)
B
'.4.
*:'
,:..56 ,,
aC -o
0.
=
=
FIG. 6. ICP8-coated DNA isaneffective template for HSVDNA
polymerase. The reaction conditions and the treatment of the samples were as described in the legend to Fig. 4. Reactions contained20 fmol of singly primed M13single-stranded DNA, 20to
500fmol of Pol-UL42orPol, and 10 pmol of ICP8. Reactionswere
preincubated at37°C for 3 min in thepresence orabsence of ICP8 priortothe addition of DNA polymerase.
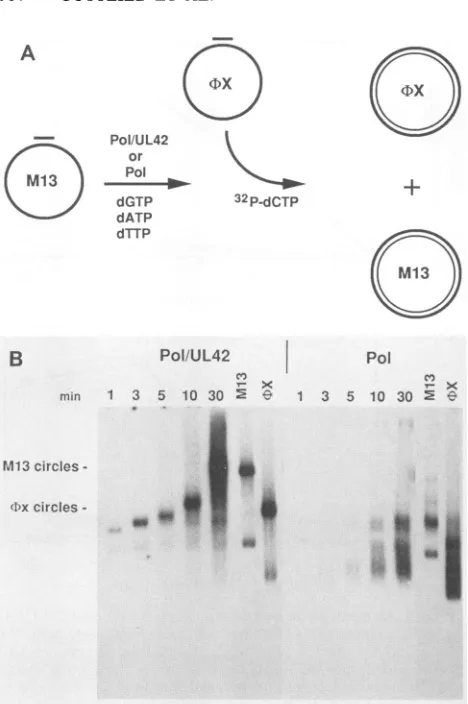
consisting of 5'-32P-labeled oligonucleotide primer annealed toM13single-stranded circleswasincubated with either Pol orwithPol-UL42,and thelabeled productswereanalyzed (i)
on an 8% acrylamide-urea gel to determine the fraction of primer that was elongated (Fig. 7A) and (ii) on analkaline
agarose gel to quantitate the synthesis of full-length M13 DNA strands (Fig. 7B). The results of thisexperiment show quite clearly thatthePol-UL42complex utilizes significantly
fewer primers than an equal amount of Pol alone but elongates them into full-length strands much more
effi-ciently. These results suggestthatUL42acts toincrease the processivity of the HSV DNA polymerase.
We directly compared the processivity of the Pol-UL42 complexwith the isolated Pol subunitbymeansofatemplate
challenge assay (Fig. 8). Singly primed M13 was preincu-bated with polymerase and three deoxyribonucleoside triphosphates to allow binding of the polymerase to the template-primer and limited (a single nucleotide)
polymer-ization. Anequivalentamountofasecond substrate, primed
XX174 DNA,wasaddedtothereaction,and DNAsynthesis
was initiated by the addition of [a-32P]dCTP. Aliquotswere removed atvarious times, and the products ofthereaction
were fractionated on a neutral agarose gel. Under these conditions, a highly processive DNA polymerase would be
expected to synthesize predominantly M13 DNA, while a
completely nonprocessive polymerase would incorporate
label equally into both substrates. The products of DNA
synthesisinreactionscontainingPol-UL42 consisted almost
entirelyofpartially duplex M13 circles at early time points
andfully replicated duplex M13 by 30 min (Fig. 8B). Very little, ifany, fully replicated duplex 4X174 DNA was ob-served after 30 min of incubation. In contrast, Pol alone
synthesized approximately equimolar amountsof both M13
10 20 30
[image:8.612.55.293.78.311.2]time(min)
FIG. 7. Comparison of primer utilization by Pol and Pol-UL42. Thereplication reactions contained 300 fmol of single-stranded M13 circular DNA annealed to a single 5'-end-labeled primer (4,100
cpm/fmol), 0.5 mM ATP, 50 ,g of BSA per ml, 60 ,uM each
deoxyribonucleoside triphosphate, and 300 fmol of either Pol or
Pol-UL42 in buffer E in a volume of 600 ,ul. Reactions were
preincubated at 37°C for 5 min prior to the addition of DNA polymerase. Aliquots (100
RI)
wereremovedat0, 2, 5, 10, 20,and 30min and addedtoanequal volume of 1% SDS-40 mM EDTA-60 ,ug
of DNA carrierpermltostopthereaction. Following precipitation with ethanol, the products of the reaction were fractionated by
electrophoresis on a6% acrylamide-urea sequencing gelto
deter-mine the fraction ofprimer that was elongated (A) and on a 1%
alkalineagarosegeltodeterminetherelativeamountofprimerthat
waselongated to produce unit-length M13 DNA strands (B). The quantitation of radioactivity on thegels was accomplished with a
Betagen BetaScope 600.
and 4XX174and produced few fully duplex circles of either substrate.
DISCUSSION
Structure of HSV DNApolymerase.Two lines of evidence have previously suggested the existence ofa specific asso-ciation between Pol and UL42. First, purified preparations
of the HSV-2-induced DNA polymerase were reported to containtwo polypeptides (37, 48). The larger of these two polypeptideswas showntobe theproduct of the Pol gene;
the smallerpolypeptide, withanestimated MW of-55,000,
was shown to be immunologically distinct from the Pol
polypeptideandwasidentifiedasinfected-cell-specific
poly-peptide (ICSP) 34/35. ICSP 34/35 wassubsequently demon-stratedby hybrid-selectedtranslationtobeequivalenttothe product of the HSV-1 gene UL42 (35). Second, immunoaf-finity purification of UL42 with an anti-UL42 monoclonal antibody resulted in the specific enrichment of HSV DNA polymerase activity (14). The results reportedin this paper
9.4 6.6 4.4
2.3 2.0
fmol
A
0 0
o
c
o2Z
Q--o
0.6
0.5
0.4
0.3
0.2
0.1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.316.551.79.360.2]5984 GOTTLIEB ET AL.
A
,
Pol/UL42or
Pol dGTP
dATP dTTP
B
min 1 3 5
32P-dCTF
Po.I!UL42 13 x
10 30 a
M13 circles
(lx
[image:9.612.70.304.60.412.2]FIG. 8. UL42increases the processivit
merase.(A)Diagrammatic representation egy. See Results fordetails. (B) Reactior
fmol ofsingly primed M13 single-strandec ,ugofBSAperml; 60,uM (each) dATP,dG
80fmolofPol-UL42or200fmol of Pol.T
experiment was purifiedfrom HSV-infect were obtained with Pol-UL42 purified fro rus-infected Sf9cells (not shown). Afterpi
min,400fmolofsingly primed
qX
single-sandDNAsynthesiswasinitiatedby thead [ct-32P]dCTP(7,100cpm/pmol). Aliquots(1
3, 5, 10, and 30 min, and following el
productsofthereaction werefractionated gel.TheM13and 4Xmarker lanescontai
tionusing50fmolofeither Pol-UL42orPo
or(X substratealone.
both confirm and extend these previ
shownherethattheHSV-1DNA poly as acomplexcontaining equimolaran
UL42.Determination ofthe sizeof thi
ofitshydrodynamicpropertiesyieldei
is consistent with the idea that the s
enzymeisa heterodimerofPol and I
Althoughthe evidence thatboth P
ponents of the native enzyme is ur
highlypurified preparationscontaino
so we cannot rule out the
possibilit3
thesepolypeptides mightalsobeaspe
DNA polymerase. We view this pos
tworeasons. First,wehavealsopurif
Pol and UL42 from insect cells in w
were expressed by recombinant baculoviruses; the
physical
and enzymaticproperties
ofthe recombinant enzyme were(D
c>X
] identicaltothose ofthe enzymeobtained fromHSV-infected cells.Second,
anapparently
fully
functional enzyme could be reconstituted bymixing equimolar
amounts of the two isolated, highly purified subunits. It remains possible, of course,thatanyputative additional subunits affectanaspect+
ofpolymerase
function thatwas notrequired
in the assaysthatwehave employedtodate. Final resolution of this issue will probably depend on the development ofmore complex invitro assaysystemsthatmore
closely
resemble viral DNA l M13 replication in vivo.ED
i,Function
of UL42. The demonstration ofaspecific
com-plex of Pol and UL42 of defined stoichiometry strongly
implies
thatthe associationbetween these twopolypeptides
is functionally significant. It has been reported previouslyPol that UL42 stimulates the activity of Pol on activated DNA
>,x (13). We addressed this question bypurifying Poland UL42
1 3 5 10
E3
e free of each otherby using recombinantbaculovirus-infectedinsect cells as a source of each subunit and comparing the activity of the isolated subunits,alone and incombination,to the activity of the purified native enzyme. In contrast to a
previous report (13), in our experiments UL42had noeffect
on the activity of Pol when activated DNA was used as W* *
primer-template.
Wedidfind, however,
that the presence of £ * UL42 had aprofound
effect on theability
of the DNAI
|"|polymerase to copy long single-stranded DNA templates,
such as singly primed M13 DNA. Inthe absence ofUL42,
U the isolated Pol subunit synthesized primarily short DNA chains; in the presence of UL42, the DNA polymerase
synthesized long DNA chains equal in size tothe template.
Substrate challenge experiments clearly showed that the increase in the size of the products in the presence of UL42 was due, at least in part, to an increase in processivity of the -y of the HSV DNA poly- enzyme.
Preliminary experiments
havesuggested
that this of the experimental strat- increase in processivity is accompanied by an increase in theas (500
,ul)
contained 400 affinity of the enzyme for the ends of duplex DNA (J. DNA; 0.5 mM ATP; 50 Gottlieb and M. Challberg, unpublished data). While differ-TP, and dTTP; and either ences in processivity per se would not be expected to havee celLs; useiln
rsis
aneffect on the activity ofaDNA polymerase onthe shortFed
cells;
identical resultsm recombinant baculovi-
single-stranded
DNAregions
that arethought
to be thereincubationat37°C
for
5 predominanttemplate
in activatedDNA,
it ispossible
that stranded DNA was added underrelatively
dilute assay conditions an increase in theiditionof50 p.l of 0.2 mM affinity for primer termini brought about by UL42 would
L00
,ul)
were removed at 1, have led to the stimulation of activity observed by other thanol precipitation, the investigators. We have not systematically tried to reproduce on a 1% neutral agarose such conditions. It has also been reported (31), in contrast tonthe
products
ofreplica-
the results reported here, that the purified catalytic subunit l and 100 fmol of the M13 ofHSV DNApolymerasecarries out highlyprocessive DNA synthesis in the absence of other subunits. The reason for this discrepancy is not known with certainty, although theious reports. We have most likely explanation is that the preparation of HSVDNA
(merasecan be purified polymerase used by O'Donnell et al. (31) contained at least nounts of both Pol and some UL42. In that study, the DNA polymerase was purified is complex on the basis from HSV-infected cells. Since the UL42 polypeptide was da value (181,000) that not a recognized component of the viral DNA polymerase at structure of the native that time, no specific immunological assays were used to test
UL42. for its presence.
'ol and UL42 are com- The UL42 protein was originally identified as an abundant,
iambiguous, our most infected-cell-specific protein with high affinity for
double-otherpolypeptides, and stranded DNA. The relationship between the in vitro DNA-y that one or more of
binding
activity
and thefunction of UL42 has notyet been -cific component of the explored. As mentioned, we have shown that the presence of,sibility
as unlikely for UL42 in the native DNA polymerase is associated with an ied a complex between increase in the affinity of the enzyme for the ends ofihich the two subunits double-stranded DNA (J. Gottlieb and M. Challberg, unpub-J. VIROL.
(1)
0
$a 0 .::.
:,:, ft
on November 10, 2019 by guest
http://jvi.asm.org/
lished data). It is tempting to speculate that this increased affinity for termini is related to the DNA-bindingactivity of free UL42 and that the role of UL42 in theDNA polymerase complex is to act as a "clamp," decreasing the probability that the polymerase dissociates from the template after each cycle of catalysis. An analogous role has beenproposed for other DNA polymerase accessory subunits, such as the T4 gene 45 and 44/62 proteins (23) and the thioredoxin subunit of T7 DNA polymerase (17, 18). Since we have shown that it is possible to reconstitute processive DNA synthesis by using isolated subunitsproduced independently in a heterol-ogous expression system, it should now be possible to test this model in more detail.
HSV DNA polymerase and DNA polymerase B. Sequence comparison studies have shown that the Pol subunit of HSV DNA polymerase is a member of the eucaryotic DNA polymerase a family (29). More recent studies have indi-cated, however, that HSV Pol is more closely related to DNA polymerase 8 than to Pol a itself (3). The studies reported here suggestthat thesimilarities between Pol 8 and the HSV DNA polymerase extend beyond primary amino acid sequence alone. Structurally, the two enzymes are at least superficially alike. Both comprise two tightly associ-ated subunits: a large, catalytic subunit and a smaller subunit of about 50 kDa (2, 21). It will be of some interest to determine whether UL42 shares any amino acid sequences with the small subunit of Pol 8 when the geneencoding that polypeptide is cloned and sequenced. The catalytic proper-ties of the HSV DNA polymerase also appear similar to those of Pol8. In contrast to Pol a, both enzymes have an intrinsic 3' to 5' exonuclease (21) and both carry out highly processive DNA synthesis (2, 47). In the case of Pol 8, however, processive synthesis requires the participation of another protein, PCNA (2, 47). Inpreliminary experiments, we have not found any effect of purified PCNA on the activity of the HSV Pol subunit, either in the presence or absence of UL42 (J. Gottlieb, unpublished data), nor have we detected any amino acid sequence homology between UL42 and PCNA. More-detailed structural studies of the two enzymes will be required to establish the basis for the similarities and differences between these two enzymes.
Pol-UL42 and thereplication fork.There is adichotomous requirement for processivity during the synthesis of the two daughter strands at a replication fork. Synthesis of the leading strand is expected to be highly processive, whereas synthesis of the lagging strand isexpected to be less proces-sive, since DNApolymerase must dissociate from the 3' end of the growing chain at thecompletion of synthesis of each Okazakifragment. These two different levels ofprocessivity are met in a number of ways in various DNA replication systems that have been studied to date. For example, the eucaryotic chromosomal replication apparatus uses two dif-ferent DNApolymerases: leading-strand synthesis is carried out by DNA polymerase 8, and lagging-strand synthesis is accomplished by DNA polymerase a (38, 50). In other systems, the same DNApolymerase is used, but its proper-ties are modulated byinteraction with other protein factors which differ on the two sides of the fork. The finding that the processivityofthe HSV DNApolymerase catalytic subunit isaltered by thepresence of UL42 protein suggests a simple model for the HSV DNA replication fork in which the Pol-UL42 complex carries out leading-strand synthesis and the Pol subunit alone is responsible for lagging-strand syn-thesis. Although our data do not bear directly on this question, we have shown that there is a molar excess of UL42relative to Pol in infected cells, and thatvirtually all of
the HSV DNA
polymerase
present in extracts ofinfectedcellscanbe
purified
in the formofaPol-UL42complex. Wetherefore favor a morecomplex model in which UL42 is a
component of the DNA polymerase on both sides of the
fork,
andprocessivityis modulatedby interactionwith otherreplication proteins. Alternatively, ourdata do not rule out
the possibility that there are two different forms of the Pol-UL42complex which differ in the degree ofprocessivity.
Thefactthat UL42 has been showntobe aphosphoprotein
(26) suggests onepossiblemeansby which the properties of
the polymerase could be altered. The data reported inthis paper representa
starting point
for developing strategiestoaddress these issues.
ACKNOWLEDGMENTS
Public Health Service grant RO1 A11838 from the National Institutes of Health to D.M.C. andapostdoctoral fellowship from theAmerican Cancer SocietytoA.I.M. areacknowledged.
LITERATURE CITED
1. Ackers,G. K. 1964.Molecular exclusion and restricted diffusion processes in molecular-sieve chromatography. Biochemistry 3:723-730.
2. Bauer, G. A., and P. M. J. Burgers. 1988. The yeastanalogof mammalian cyclin/proliferating-cell nuclear antigen interacts with mammalian DNA polymerase delta. Proc. Natl. Acad. Sci. USA 83:7506-7510.
3. Boulet, A., M. Simon, G. Faye, G. A. Bauer, and P. M. J. Burgers. 1989. Structure and function ofthe Saccharomyces cervisiae CDC2gene encoding the large subunit of DNA
poly-meraseIII. EMBO J. 8:1849-1854.
4. Carmichael, E. P., M. J. Kosovsky, and S. K. Weller. 1988. Isolation and characterization of herpes simplex virus type 1 host range mutants defective in viral DNA synthesis. J. Virol. 62:91-99.
5. Carmichael,E.P.,andS. K. Weller.1989. Herpessimplexvirus type 1 DNA synthesis requires the product of the UL8 gene: isolation and characterization of an ICP6::lacZ insertion
muta-tion. J. Virol. 62:2970-2977.
6. Challberg,M. D. 1986. A methodforidentifyingthe viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA 83:9094-9098.
7. Chartrand, P., C. S. Crumpacker, P. A. Schaffer, and N. M. Wilkie. 1980. Physical and genetic analysis of the herpes sim-plex virus DNApolymerase locus. Virology 103:311-326. 8. Coen,D. M., D.P. Aschman, P. T.Gelep, M. J. Retondo, S. K.
Weller, and P. A. Schaffer. 1984. Fine mapping and molecular cloning of mutations intheherpes simplex virus DNA
polymer-ase locus. J. Virol.49:236-247.
9. Conley, A. J., D. M. Knipe, P. Jones, and B. Roizman. 1981. Moleculargenetics ofherpes simplex virus. VII. Characteriza-tion of a temperature-sensitive mutant produced by in vitro mutagenesis anddefective in DNA synthesisandaccumulation ofgammapolypeptides. J. Virol. 37:191-206.
10. Crute, J. J., and I. R. Lehman. 1989. Herpes simplex-1 DNA polymerase. Identification of an intrinsic 5' to 3' exonu-clease with ribonuclease Hactivity. J. Biol. Chem. 264:19266-19270.
11. Crute, J. J., T. Tsurumi, L. Zhu, S. K. Weller, P. D. Olivo, M. D. Challberg, E. S. Mocarski, and I. R. Lehman. 1989. Herpes simplex virus 1 helicase-primase: a complex ofthree herpes-encoded gene products. Proc. Natl. Acad. Sci. USA 86:2186-2189.
12. Dorsky, D. I., and C. S. Crumpacker. 1988. Expression of herpes simplexvirus type 1 DNA polymerase gene by in vitro translation and effects ofgene deletions on activity. J. Virol. 62:3224-3232.
13. Gallo, M.L., D.I.Dorsky, C. S. Crumpacker,and D. S. Parris.
on November 10, 2019 by guest
http://jvi.asm.org/
5986 GOTTLIEB ET AL.
1989. The essential 65-kilodalton DNA-binding proteinof
her-pessimplexvirus stimulates the virus-encoded DNA
polymer-ase.J. Virol. 63:5023-5029.
14. Gallo,M.L.,D. H.Jackwood,M.Murphy,H. S.Marsden, and
D. S. Parris.1988. Purification of theherpes simplexvirustype 1 65-kilodalton DNA-binding protein: propertiesof the protein
and evidence of its association with the virus-encoded DNA
polymerases.J. Virol. 62:2874-2883.
15. Gibbs, J. S., H. C. Chiou, J. D. Hall, D. W. Mount, M.J. Retondo, S. K. Weller, and D. M. Coen. 1985. Sequence and
mapping analysesof theherpes simplexviruspolymerasegene predict a C-terminal binding domain. Proc. Natl. Acad. Sci. USA82:7969-7973.
16. Haffey, M. L., J.T. Stevens, B.J. Terry, D. I. Dorsky, C. S. Crumpacker,S. M.Wietstock,W. T.Ruyechan,and A. K. Field. 1988.Expressionofherpes simplexvirustype 1 DNA
polymer-aseinSaccharomycescerevisiae and detection ofvirus-specific
enzymeactivity incell-freelysates. J. Virol. 62:4493-4498.
17. Huber, H. E., S. Tabor, and C. C. Richardson. 1987. Esche-richiacoli thioredoxin confers processivity on the DNA
poly-merase activity of the gene 5 protein ofbacteriophage T7. J. Biol. Chem. 262:16212-16223.
18. Huber, H. E., S. Tabor, and C. C. Richardson. 1987. Esche-richiacolithioredoxin stabilizescomplexesofbacteriophageT7 DNA polymerase and primed templates. J. Biol. Chem. 262: 16224-16232.
19. Knopf, K. 1979. Properties of herpes simplex virus DNA
polymerase and characterization of its associatedexonuclease
activity.Eur. J. Biochem. 98:231-244.
20. Kunkel,T.A., J.D.Roberts,and R. A. Zakour.1987.Rapidand efficientsite-specific mutagenesiswithoutphenotypicselection. Methods Enzymol. 155:166-180.
21. Lee, M. Y., C. K. Tan, K. M. Downey, and A. G. So. 1984.
Further studiesoncalfthymusDNApolymerasedeltapurified
to homogeneity by a new procedure. Biochemistry
23:1906-1913.
22. Luckow,V.A.,and M. D. Summers. 1989.Highlevelexpression
of nonfusedforeigngeneswithAutographa californicanuclear
polyhedrosisvirusexpressionvectors.Virology179:31-39. 23. Mace,D. C., and B. M. Alberts. 1984. Characterization of the
stimulatory effect of T4 gene 45 protein and the gene 44/62
protein complexonDNAsynthesis byT4 DNApolymerase. J.
Mol. Biol. 177:313-327.
24. Marchetti, M. E., C. E. Smith, and P. A. Schaffer. 1988. A
temperature-sensitive mutation in herpes simplexvirus type 1
gene required for viral DNA synthesis maps to coordinates 0.609through0.614 in UL.J. Virol. 62:715-721.
25. Marcy,A. I.,P. D. Olivo, M. D. Challberg, andD. M. Coen.
1990. Enzymatic activities of overexpressed herpes simplex
virus DNA polymerase purified from recombinant
baculo-virus-infected insect cells. Nucleic Acids Res. 18:1207-1215.
26. Marsden,H.S.,M. E. M.Campbell,L.Haarr,M.C.Frame,D. S. Parris, M. Murphy, R. G.Hope, M. T. Muller, and C. M. Preston. 1987. The 65,000-Mr DNA-binding and virion trans-inducing proteins of herpes simplex virus type 1. J. Virol. 61:2428-2437.
27. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan,
M.C.Frame,D.McNab,L.J. Perry, J.E.Scott,and P.Taylor.
1988.ThecompleteDNAsequenceof thelong unique regionin
the genome of herpes simplex virus type 1. J. Gen. Virol.
69:1531-1574.
28. McGeoch, D.J., M. A.Dalrymple, A. Dolan, D. McNab, L. J. Perry, P. Taylor, and M. D. Challberg. 1988. Structures of herpes simplex virus type 1 genes required for replication of
virusDNA.J. Virol. 62:444-453.
29. Miller,M.A.,D.Korn,and T.S.Wang. 1988. The evolutionary
conservation of DNA polymerase alpha. Nucleic Acids Res.
16:7961-7973.
30. O'Donnell, M. E., P. Elias, B. E. Funnell, and I. R. Lehman.
1987. Interaction between the DNA polymerase and
single-strandedDNA-binding protein (infected cell protein 8) of herpes simplex.J. Biol. Chem.262:4260-4266.
31. O'Donnell, M. E., P. Elias, and I. R.Lehman. 1987. Processive replication of single-stranded DNA templates by the herpes simplex virus-induced DNA polymerase. J. Biol. Chem. 262: 4252-4259.
32. Olivo, P. D.,N.J.Nelson, and M. D.Challberg. 1988.Herpes simplex virus DNA replication: the UL9 gene encodes an origin binding protein.Proc. Natl.Acad. Sci. USA 85:5414-5418. 33. Olivo,P.D.,N.J. Nelson,and M. D.Challberg. 1989. Herpes
simplex virus type 1 gene products required for DNA repli-cation: identification and overexpression. J. Virol. 63:196-204.
34. Orberg, P.K., and P. A. Schaffer. 1987. Expressionofherpes simplex virus type 1 major DNA-binding protein, ICP8, in transformed cell lines:complementationof deletionmutantsand inhibition ofwild-type virus.J.Virol. 61:1136-1146.
35. Parris, D. S., A. Cross, L. Haarr, A. Orr, M. C. Frame, M. Murphy, D. J. McGeoch, and H. S. Marsden. 1988. Identifica-tion of thegeneencoding the 65-kilodaltonDNA-binding protein ofherpes simplex virus type 1. J. Virol. 62:818-825.
36. Powell, K. L., E. Littler, and D. J. M. Purifoy. 1981. Nonstruc-turalproteinsofherpes simplex virus. II.Major virus-specific DNA-binding protein. J. Virol. 39:894-902.
37. Powell, K. L., and D. J. M. Purifoy. 1977. Nonstructural proteins of herpes simplex virus. l.Purification ofthe induced DNApolymerase. J. Virol.24:618-626.
38. Prelich, G., and B. Stillman. 1988. Coordinated leading and lagging strandsynthesis during SV40DNAreplicationin vitro requiresPCNA. Cell 53:117-126.
39. Purifoy, D. J. M., R. B. Lewis, and K. L. Powell. 1977. Identification of the herpes simplex virus DNA polymerase gene. Nature(London) 269:621-623.
40. Purifoy, D. J. M., and K. L. Powell. 1981. Temperature-sensitive mutants in two distinct complementation groups of herpessimplex virustype 1specify thermolabileDNA polymer-ase.J. Gen. Virol. 54:219-222.
41. Quinn, J. P., and D. J. McGeoch. 1985. DNAsequence ofthe region inthe genomeofherpessimplex virustype 1containing the genes for DNA polymerase and the major DNA binding protein. Nucleic AcidsRes. 13:8143-8163.
42. Ruyechan,W.T.,and A. C. Weir.1984.Interactionwithnucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J. Virol. 52:727-733.
43. Spaete,R.R.,and N. Frenkel. 1982. Theherpes simplex virus amplicon: a new eucaryotic defective-virus cloning-amplifying vector. Cell30:295-304.
44. Stow, N.D. 1982. Localization ofanorigin ofDNArepliation withintheTRs/IRs repeated region of the herpes simplexvirus type 1 genome. EMBOJ. 1:863-867.
45. Stow,N. D., and E. C. McMonagle. 1983. Characterization of theTRs/IRsorigin ofDNAreplication of herpes simplexvirus type 1. Virology 130:427-438.
46. Summers,M.D.,andG. E. Smith. 1987. Amanual of methods forbaculovirus vectorsandinsect cellprocedures. Texas Agri-cultural Experimental Station, bulletin no. 1555. Texas A&M
University, College Station,Tex.
47. Tan, C. K., C. Castillo,A.G.So,and K. M.Downey. 1986. An auxiliary protein for DNA polymerase-delta from fetal calf thymus. J. Biol. Chem. 261:12310-12316.
48. Vaughan, P. J., D. J. M. Purifoy, and K. L. Powell. 1985. DNA-binding protein associated with herpes simplex virus DNApolymerase. J.Virol.53:501-508.
49. Vlasny, D. A., and N. Frenkel. 1982. Replication of herpes simplexvirus DNA:localization ofreplication recognition sig-nals within defective virus genomes. Proc. Natl. Acad. Sci. USA 78:742-746.
50. Weinberg,D.H., and T. J. Kelly.1989.Requirementfor 2 DNA polymerasesin thereplication of simian virus-40DNAin vitro. Proc.Natl. Acad. Sci. USA 86:9742-9746.
51. Weller,S.K.,E. P.Carmichael,D. P.Aschman, D. J. Goldstein, andP. A. Schaffer. 1987.Geneticandphenotypic characteriza-tion ofmutantsinfouressential genes that maptotheleft half of HSV-1UL DNA. Virology161:198-210.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
52. Weller, S. K., K. J. Lee, D. J. Sabourin, and P. A. Schaffer. 1983. Genetic analysis of temperature-sensitive mutantswhich define the gene for the major herpes simplex virus type 1 DNA-binding protein. J. Virol. 45:354-366.
53. Weller,S.K.,A.Spadaro,J. E.Schaffer,A. W.Murray,A. M. Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functional analysis oforiL,aherpessimplexvirustype 1origin of DNAsynthesis. Mol.Cell. Biol. 5:930-942.
54. Wu, C. A., N. J. Nelson, D. J. McGeoch, and M. D. Challberg. 1988. Identification ofherpes simplex virus type 1 genes
re-quired for origin-dependent DNA synthesis. J. Virol. 62:435-443.
55. Zhu, L., and S. K. Weller. 1988. UL5, aprotein required for
HSVDNAsynthesis: genetic analysis, overexpressionin Esch-erichia coli, andgenerationofpolyclonalantibodies. Virology 166:366-378.