Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Unusual Distribution of Mutations Associated with Serial Bottleneck
Passages of Human Immunodeficiency Virus Type 1
ELOISA YUSTE,
1CECILIO LÓPEZ-GALÍNDEZ,
2ANDESTEBAN DOMINGO
1*
Centro de Biología Molecular “Severo Ochoa,” Universidad Autónoma de Madrid, Cantoblanco,
28049 Madrid,
1and Centro Nacional de Biología Fundamental, Instituto de
Salud Carlos III, Majadahonda, 28220 Madrid,
2Spain
Received 8 May 2000/Accepted 18 July 2000
Repeated bottleneck passages result in fitness losses of RNA viruses. In the case of human immunodeficiency
virus type 1 (HIV-1), decreases in fitness after a limited number of plaque-to-plaque transfers in MT-4 cells
were very drastic. Here we report an analysis of entire genomic nucleotide sequences of four HIV-1 clones
derived from the same HIV-1 isolate and their low-fitness progeny following 7 to 15 plaque-to-plaque passages.
Clones accumulated 4 to 28 mutations per genome, with dominance of A
¡
G and G
¡
A transitions (57% of
all mutations) and 49% nonsynonymous replacements. One clone—but not three sibling clones—showed an
overabundance of G
¡
A transitions, evidencing the highly stochastic nature of some types of mutational bias.
The distribution of mutations along the genome was very unusual in that mutation frequencies in
gag
were
threefold higher than in
env
. Particularly striking was the complete absence of replacements in the V3 loop of
gp120, confirmed with partial nucleotide sequences of additional HIV-1 clones subjected to repeated bottleneck
passages. The analyses revealed several amino acid replacements that have not been previously recorded
among natural HIV-1 isolates and illustrate how evolution of an RNA virus genome, with regard to constant
and variable regions, can be profoundly modified by alterations in population dynamics.
Retroviruses and in particular human immunodeficiency
vi-rus type 1 (HIV-1) mutate and recombine at high rates (14, 15,
39, 55, 60, 61, 70, 74). Rapid genetic variation, together with
the short replication times of HIV-1 (8), generates complex
and highly dynamic mutant swarms termed viral quasispecies
(10, 18–22, 42, 66, 69). The mutant spectra of viral quasispecies
constitute reservoirs of phenotypically relevant variants, as
ev-idenced by the nonsyncytial-to-syncytial switch in infected
in-dividuals or the rapid selection of antibody-, cytotoxic-T-cell-,
or inhibitor-resistant mutants in viral populations in vivo (2–4,
32, 33, 36, 37, 41, 46, 47, 56, 59, 65). Although most individual
mutations in mutant swarms of RNA viruses may not be of
immediate or even long-term selective value for the virus (63),
evolution of viral quasispecies can be adaptive and may exert
an influence in viral pathogenesis (26, 34, 76; reviews in 10, 28,
29). The adaptive potential of viral quasispecies is manifested
by quantitatively important fitness variations as viruses evolve
in constant or changing environments (for recent examples and
reviews, see 10, 11, 13, 31, 48, 50–52, 75).
Large-population passages under a constant environment
tend to produce fitness gains in viral populations (10, 11, 13,
25, 51, 52). In contrast, bottleneck events—experimentally
re-alized in an extreme case by serial plaque-to-plaque transfers
of virus on cell monolayers—often lead either to average
fit-ness losses (6, 17, 24, 78) or to limitations of fitfit-ness gains (23,
51, 52). The decrease in fitness mediated by repeated
bottle-necks has been interpreted as the result of an accentuation of
Muller’s ratchet effect (45). According to this model, asexual
populations of organisms with a small population size will tend
to incorporate deleterious mutations unless compensatory
mechanisms such as recombination can restore mutation-free
genomes (45). For RNA virus quasispecies, accumulation of
deleterious mutations is expected from successive rounds of
random sampling of genomes from the mutant spectrum
(re-viewed in 10, 11).
In retroviruses, decreases in fitness as a result of serial
bot-tleneck passages were first documented with HIV-1 following
plaque-to-plaque passages on MT-4 cells (78). In this virus,
fitness losses were unexpectedly drastic when compared with
the fitness losses experienced by other RNA viruses, such
as bacteriophage
6, vesicular stomatitis virus, or
foot-and-mouth disease virus (FMDV) subjected to similar passage
reg-imens (6, 17, 24). Only 4 out of 10 HIV-1 clones could produce
viable progeny after 15 plaque-to-plaque transfers, and 3 of the
4 survivors displayed important decreases in fitness (78). Very
little is known about the numbers and types of mutations which
accompany fitness decreases of RNA viruses when they are
subjected to sequential bottleneck passages. In the case of
the animal picornavirus pathogen FMDV, debilitated clones
showed some unusual genetic lesions (infrequent or absent in
populations evolved without intervening bottlenecks; 24, 25).
Such lesions included mutations that resulted in amino acid
replacements at internal sites of the viral capsid and a unique
elongation of five adenylate residues which resulted in an
in-ternal polyadenylate tract of variable length preceding the
sec-ond functional AUG initiation codon of the FMDV genome
(24, 25). No information on genetic lesions associated with
fitness losses in retroviral genomes is available. Here we report
complete HIV-1 genomic sequences of HIV-1 clones subjected
to plaque transfers that led to severe fitness losses. The results
reveal a broad spectrum of mutations associated with fitness
decrease and an unexpected distribution of mutations along
the HIV-1 genome.
MATERIALS AND METHODS
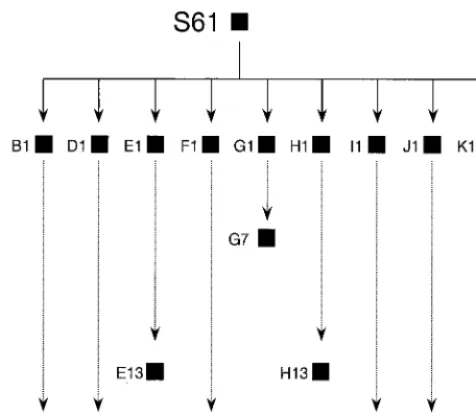
HIV-1 clones.The origin and passage history of the biological clones of HIV-1
used in the present study have been previously described (78). Briefly, virus clones were isolated by plating a natural isolate of HIV-1, termed S61, on MT-4 cells. Virus populations D1, G1, I1, and K1 are from randomly chosen, individual plaques. After the first plating, viruses from individual plaques (in the range of
* Corresponding author. Mailing address: Centro de Biología
Mo-lecular “Severo Ochoa,” Universidad Autónoma de Madrid,
Canto-blanco, 28049 Madrid, Spain. Phone: 34-91-397 8485. Fax: 34-91-397
4799. E-mail: [email protected].
9546
on November 9, 2019 by guest
http://jvi.asm.org/
102to 105PFU) were diluted in 300l of culture medium and plated on fresh
MT-4 cells, and this process was repeated a number of times (Fig. 1). Plaques appeared 7 to 10 days after infection. Fitness of the clonal populations of HIV-1 was determined by growth-competition experiments in MT-4 cells, and popula-tions were analyzed by the heteroduplex tracking assay as previously described (78).
DNA extraction, PCR amplification, and nucleotide sequencing.DNA was
extracted using an Instagene purification matrix (Bio-Rad) according to the man-ufacturer’s instructions. To determine the consensus nucleotide sequence of the entire HIV-1 genome in individual virus clones, a collection of overlapping sets of oligonucleotide primers was used. They either have been previously described (46, 47) or were designed for the present experiments (Table 1). HIV-1 DNA was amplified using nested PCR. The first amplifications (external primers) were carried out using the GeneAmp PCR kit (Perkin-Elmer) and resulted in the copying of 1,235-, 4,253-, and 6,686-bp fragments, comprising residues 1 through 1235, 546 through 4799, and 2975 through 9661, respectively; residue numbers correspond to those of the genome of HIV-1 isolate HXB2 (35). Internal am-plifications yielded fragments of 500 to 1,500 residues, which were used for nu-cleotide sequence determination. For amplification of short regions ofgag
(positions 1337 to 1598) andenv(positions 7071 to 7333), a single PCR ampli-fication was carried out. Both external and internal ampliampli-fications involved 35 cycles with temperatures chosen according to the composition of the oligonu-cleotide primers (78). Before sequencing, the PCR mixture was digested with exonuclease I and shrimp alkaline phosphatase (Amersham Life Sciences). Nu-cleotide sequences were determined on the two cDNA strands, with an ABI 373 automatic sequencer. Multiple sequence alignments were obtained using the CLUSTAL W program (71).
The newly determined nucleotide sequences have been deposited in the EMBL sequence database with accession numbers AF256204, AF256205, AF256206, AF256207, AF256208, AF256209, AF256210, and AF256211.
RESULTS
Mutation accumulation as a result of bottleneck transfers.
HIV-1 clones underwent severe fitness losses as a result of
serial plaque-to-plaque transfers in MT-4 cells (78). To
deter-mine the types and numbers of mutations accumulated during
bottleneck transfers, the entire genomic nucleotide sequence
of four HIV-1 clones (D1, G1, I1, and K1) and their derivatives
after 15, 7, 15, and 15 plaque-to-plaque passages, respectively
(termed D15, G7, I15, and K15, respectively [Fig. 1]), were
obtained. The comparison of genomic nucleotide sequences of
[image:2.612.55.293.71.278.2]each passaged clone relative to the corresponding initial clone
showed that transitions were 2.8-fold more frequent than
transversions and that A
¡
G and G
¡
A accounted for 20%
and 36%, respectively, of all mutation types; nonsynonymous
mutations represented 49% of the total; an insertion of one
nucleotide was present in clone I15 (Table 2). Mutation
fre-quencies varied up to sevenfold among lineages (range, 4.4
⫻
FIG. 1. Scheme of passages of HIV-1 clones subjected to plaque-to-plaque transfers in MT-4 cells. Clonal populations (HIV-1 isolated from individual plaques) are depicted as filled squares. The experimental procedures and the origins of natural HIV-1 isolate S61 and clones B1 to K1 are given in reference 78 and in Materials and Methods. HIV-1 clones are indicated by letters followed by a number which gives the total number of plaque-to-plaque transfers under-gone by the clone. Infectious virus could not be rescued from viral populations B15, E13, G7, H13, and J15 as described in reference 78.
TABLE 1. Synthetic oligonucleotide primers employed for
nucleotide sequence determination of the HIV-1 genome
aNucleotide sequence
GCTTCAAGTAGTGTGTGCCCGTCTG (563, S) AGAGTCACACAACAGACGGG (582, A) TGACTAAAAGGGTCTGAGGG (614, A) CTCTGGTAACTAGAGATCCC (615, S)
TCTCTAGCAGTGGCGCCCGAACAGGGAC (626, S) AGACAGGATCAGAAGAA (995, S)
GGTGATATGGCCTGATGTACCATTTGCCCCTG (1204, A) CCAGGCCAGATGAGAGAACCAAGGG (1462, S) TGTCCAGAATGCTGGTAGGG (1642, A) CTCTCAGAAGCAGGAGCCG (2205, S) GTATTTAGTAGGACCTACACCT (2475, S) TCTTCTGTCAATGGCCATTGTTTAAC (2635, A) GGATTAGATATCAGTACAATGTGCTT (2971, S) CATGGATCCGATATCTAATCCCTGG (2991, A) GCTGGTGACCTTTCCATCC (3022, A)
TAGATATCAGTACAATGTGCTTCCAC (2975, S) TATTGCTGGTGATCCTTTCC (3026, A) AAACATCAGAAAGAACCTCC (3207, S) GTTCATAACCCATCCAAAGG (3249, A)
GCGGAATCTGTATGTCATTGACAGTCCAGCT (3300, A) CATGGAGTGTATTATGACCC (3492, S)
CTTTCCCCATATTACTATGC (3704, A) AGTTTGTCAATACCCCTCCC (3793, S) AATCATTCAAGCACAACCAG (4061, S) TTAGATGGAATAGATAAGGC (4233, S)
CTTGAAGCTTATCTATTCCATCTAAAAATAGT (4257, A) GGCGAATTCACTAGCCATTGCTCTCCA (4284, A) AGTGATTTTAACC (4299, S)
GCTTCTATATATCCACTGGC (4486, A) AAGTATGCTGTTTCTTGCCC (4528, A)
AAGGGGGGATTGGGGGGTACAGTGCAGGG (4792, S) TAGCCCTTCCAGTCCCCCCTTTTCTTTTA (4799, A) CTTTCCCCTGCACTGTACCC (4825, A)
TACTAATCTAGCCTCCCCTAGTGGGATGTG (5235, A) GCCTCTGTGGCCCTTGGTCTTCTGGGG (5595, A) CAGAAAAGCTTGTCGACATAGCAGAATAGG (5576, S) TTAGGCATCTCCTATGGCAGGAAGAAGCGG (5957, S) CCCATAATAGACTGTGACCC (6347, A)
TGTGGGTTGGGGTCTGTGGG (6469, A) ATGGGATCAAAGCCTAAAGCCATGTG (6557, S) AGGATACCTTTGGACAGGCC (6852, A) TCAGCACAGTACAATGTACACATGG (6949, S) ATAAGCTTGCAGTCTAGCAGAAGAAGA (7004, S) CAATCCTCAGGAGGGGAC (7311, S)
TGCATCTCAATTTCTGGGCTCCCCTCCTGAG (7345, A) AGGAGTCCTCCCCTGGGTCTTAAGTA (7648, A) AGTGCTTCCTGCTGCTCCCAAGAACCCAAG (7811, A) TCTTGCCTGGAGCTGTTTGATGCCCCAGAC (7961, A) TTGGAATTGGATAAGTGGGC (8205, S)
GTGAATAGAGTTAGGCAGGG (8337, S)
GAAATGACAATGGTGAGTATCCCTGCC (8376, A) CGATTCCTTCGGGCCTGTCGGGTCCCC (8424, A) TGTGGAACTTCTGGGACGCAGGGGGTGGG (8567, S) CAAGGAGGAGGAGGAGGTGGGTTTTCC (8977, S) TGGAAGGGCTAATTTGGTCCCAGA (9086, S) CTGGGACCAAATTAGCCCTTCCAGTCC (9108, A)
aSequences are written from 5⬘to 3⬘; data in parentheses are the position of the 5⬘nucleotide of each primer—according to the genomic nucleotide number-ing of isolate HXB2 (35)—and primer orientation (S, sense [same orientation as genomic RNA]; A, antisense [complementary to genomic RNA]). Oligonucleo-tide primers were purchased from Isogen (Maarssen).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.314.546.188.680.2]10
⫺4to 3.1
⫻
10
⫺3substitutions per nucleotide [Table 2]).
There was no obvious correlation between fitness decrease and
mutation types or frequencies; for example, clone G7, which
was debilitated to the point of not allowing a reliable
determi-nation of fitness value (78), showed a mutation frequency that
was sevenfold lower than that of I15 (Table 2).
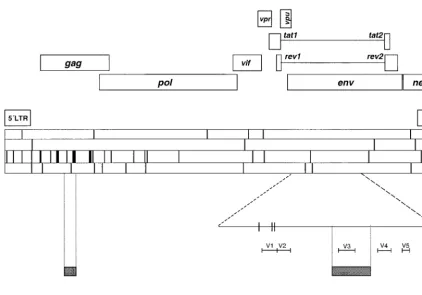
Unusual distribution of mutations.
Mutation frequencies in
env
are generally higher than in
gag
and
pol
when natural
HIV-1 isolates are compared (35, 44, 57, 58). In contrast, the
HIV-1 clones debilitated by serial plaque transfers showed
average mutation frequencies that were threefold higher for
gag
than
env
(Fig. 2). No mutations were found in
tat, vpu
, and
rev
. Most remarkable was the absence of mutations in the
regions encoding variable loops of gp120, particularly the V3
loop. The asymmetric distribution of mutations was visualized
by dividing the HIV-1 genome into three arbitrary regions of
similar length: region 1, residues 1 through 3028; region 2,
residues 3029 through 6065; and region 3, residues 6066
through 9035. Taking into account all mutations scored for
clones D15, G7, I15, and K15 (as quantitated in Table 2), we
found mutation frequencies for regions 1, 2, and 3 of 2.5
⫻
10
⫺3, 9.0
⫻
10
⫺4, and 6.6
⫻
10
⫺4substitutions per nucleotide,
respectively. The results suggest that plaque-to-plaque
trans-fers of HIV-1 lead to accumulation of mutations at multiple
sites of the HIV-1 genome, following a pattern which is quite
different from that observed in the natural evolution of HIV-1
in infected hosts.
Conservation of the V3 loop.
The unusual distribution of
mutations found in D15, G7, I15, and K15 and the absence of
mutations in the region encoding the variable V3 loop of gp120
prompted us to extend nucleotide sequence determinations to
additional HIV-1 clones subjected to serial bottleneck events.
These additional analyses included two clones from each of the
lineages D15, G7, and K15, one clone from I15, and clones
from independent lineages which were previously described
(78), including three clones from F15 and one clone from H13
(Fig. 1). The analysis involved nucleotide sequences of residues
7071 to 7333 (which include the V3-coding region) and
resi-dues 1337 to 1598 (within the p24-coding region). The
com-parison of nucleotide sequences with those of the
correspond-ing initial clones fully confirmed the bias in the distribution of
mutations; in all, 13 replacements were found in the
gag
region
analyzed (which represents a mutation frequency of 4.5
⫻
10
⫺3substitutions per nucleotide), while no replacements were
found in the V3-coding region (mutation frequency,
⬍
3.5
⫻
10
⫺4substitutions per nucleotide). The statistical significance
of the biased distribution of mutations was evaluated by
com-paring the expected versus the actual number of mutations in
gag, pol
, and
env
for regions 1, 2, and 3 into which the genome
was arbitrarily divided (Fig. 2), as well as for the short
gag
and
env
stretches for the additional clones from populations D15,
F15, G7, H13, I15, and K15. These results (Table 3) indicate
high statistical significance of the biased mutant distribution.
The degree of statistical significance of the biased distribution
did not vary when either all G
¡
A mutations or just G
¡
A
mutations found in clone I15 were excluded from the
calcula-tions (in all cases,
P
⬍
0.001 [
2test]). Therefore,
accumula-tion of mutaaccumula-tions that were associated with serial bottleneck
events and with fitness loss of HIV-1 affected genomic regions
that are less variable during the natural evolution of the virus.
DISCUSSION
The results reported here describe the numbers and types of
mutations associated with fitness loss of HIV-1 as a result of
the operation of Muller’s ratchet (Table 2; Fig. 2). For clones
D15, G7, and K15, the average mutation frequency (1.3
⫻
10
⫺4substitutions per nucleotide, measured relative to the genomic
nucleotide sequence of their respective parental clones D1,
G1, and K1) was 12 mutations per genome (range, 10 to 15),
with a predominance of transitions (75% of all mutations) over
transversions. These figures are similar to those of previous
de-terminations of the number of mutations accompanying
Mul-ler’s ratchet in the FMDV genome—an average of six
muta-tions per genome, with 77% of these transition mutamuta-tions (24).
However, clone I15 displayed a different pattern in that its
mutation frequency corresponded to 28 mutations per genome
with an overabundance of G
¡
A transitions (43% of all
mutations [Table 1]). G
¡
A is one of the substitutions that
have been associated with hypermutagenesis in HIV-1 (72,
73) and a number of other viruses (reviewed in 5, 43). In clone
I15, G
¡
A transitions were distributed rather uniformly along
the genome. Several possible mechanisms have been proposed
to explain biased hypermutagenesis (5, 43), including
alter-ations in intracellular deoxynucleotide pools. The sequence
context of 67% of the G
¡
A transitions in clone I15 was GpA
or GpG, which suggests a possible influence of low dCTP levels
during minus cDNA synthesis in the origin of this mutation
type (43). Our results with debilitated HIV-1 clones emphasize
the stochastic nature of the G
¡
A mutation bias because it
was observed in only one of four clones derived from the same
viral isolate, and these clones were subjected to identical
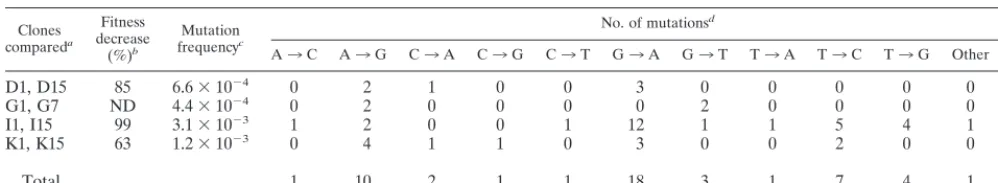
treat-TABLE 2. Number and types of mutations in the genome of HIV-1 clones subjected to serial plaque transfers
Clones compareda
Fitness decrease
(%)b
Mutation frequencyc
No. of mutationsd
A3C A3G C3A C3G C3T G3A G3T T3A T3C T3G Other Total
D1, D15
85
6.6
⫻
10
⫺40
2
1
0
0
3
0
0
0
0
0
6
G1, G7
ND
4.4
⫻
10
⫺40
2
0
0
0
0
2
0
0
0
0
4
I1, I15
99
3.1
⫻
10
⫺31
2
0
0
1
12
1
1
5
4
1
28
K1, K15
63
1.2
⫻
10
⫺30
4
1
1
0
3
0
0
2
0
0
11
Total
1
10
2
1
1
18
3
1
7
4
1
49
aThe entire genomic nucleotide sequence of the indicated clones was determined as described in Materials and Methods.
bFitness decrease refers to the comparison of D15, I15, and K15 with their corresponding initial clones D1, I1, and K1; it is expressed as percent reduction, calculated
as described in reference 78. For G7 a relative fitness value could not be calculated because the virus yield was insufficient to perform competition passages (78).
cMutation frequency is the number of mutations found in the genome of D15, G7, I15, or K15 (when compared with the corresponding initial clones D1, G1, I1,
or K1) divided by the total number of nucleotides sequenced (in this case, the entire HIV-1 genome, or 9.1 kb); therefore, values are expressed in substitutions per nucleotide.
dTransversions A3T and G3C were not found in the genomes of the clones analyzed. Other, nsertion of one nucleotide at genomic residue 584 (according to
the numbering for the genome of HXB2 [35]) in the leader sequence. The location of mutations in the HIV-1 genome is given in Fig. 2 and is discussed in the text.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.53.552.84.175.2]ment during the serial plaque transfers (Fig. 1). If the
mech-anism of nucleotide pool bias was in operation (43), it must
have been triggered either by extremely subtle perturbations in
the intracellular environment or by differences among the
ge-nomes of the four clones, differences that existed initially or
that were generated in the course of passaging. In the latter
case, the mutational biases must be subjected to
indetermina-tions derived from the dynamics of mutant generation within
the quasispecies swarms (reviewed in 10, 20).
A dominance of G
¡
A transitions was also found in an
analysis of mutations in a
lacZ
␣
-based reporter gene, which
was constructed to study a single cycle of HIV-1 replication
(39). However, there are important differences in the mutant
repertoire found following a single cycle of replication and in
our clones subjected to serial plaque transfers. In the former
study, G
¡
A, C
¡
T, and T
¡
C mutations occurred at
frequencies of 1.7
⫻
10
⫺3, 7.1
⫻
10
⫺4, and 1.3
⫻
10
⫺4substi-tutions per nucleotide, respectively, while in our study these
same mutations occurred at frequencies of 4.9
⫻
10
⫺4, 2.7
⫻
10
⫺4, and 1.9
⫻
10
⫺4, respectively (Table 2). After a
single-cycle replication, T
¡
G, T
¡
A, and A
¡
G each occurred at
a frequency of 6.5
⫻
10
⫺5substitutions per nucleotide, while in
our clones the values for these substitutions ranged from 2.7
⫻
[image:4.612.91.514.69.364.2]10
⫺4to 2.7
⫻
10
⫺5substitutions per nucleotide. In addition,
A
¡
C, C
¡
A, C
¡
G, and G
¡
T mutations found in HIV-1
FIG. 2. Location of mutations found in HIV-1 clones D15, G7, I15, and K15, relative to their parental counterparts. The upper part indicates HIV-1 genes and regulatory regions based on the compilation of Korber et al. (35). The four horizontal bars in the center of the figure indicate the positions of mutations along the genome (9.1 kb) in the four clones analyzed (from top to bottom, D15, G7, I15, and K15 [described in Materials and Methods]); vertical lines within these bars indicate one, two, or three mutations, according to thickness. Mutations were found at positions 35, 171, 377, 379, 570, 584, 760, 807, 833, 988, 1128, 1161, 1188, 1351, 1467, 1545, 1578, 1596, 1810, 1863, 1875, 1937, 1961, 1966, 2145, 2174, 2329, 2668, 2804, 3068, 3114, 3129, 3945, 4458, 5239, 5270, 5342, 5422, 5684, 5686, 6588, 6655, 6670, 7962, 8095, 8890, 8900, and 8989 according to the numbering of HIV-1 isolate HXB2 (35). The gp120-coding region ofenvhas been enlarged to depict the positions of variable loops V1 to V5; two mutations affected the V1-coding region. The two shaded rectangles correspond to genome positions 1337 to 1598 (gag[left shaded rectangle]) and 7071 to 7333 (env[right shaded rectangle]), which have been sequenced for a number of additional HIV-1 clones to confirm the asymmetric distribution of mutations (see text). The mutations found in these additional clones are not included in this scheme. The bottom part shows the three arbitrary regions into which the HIV-1 genome was divided to illustrate the bias in the distribution of mutations along the genome. Procedures used for nucleotide sequence determination are described in Materials and Methods, and the oligonucleotide primers are listed in Table 1.
TABLE 3. Expected versus actual number of mutations in different
genomic regions of HIV-1 clones subjected to
serial plaque transfers
Genomic regions compared
No. of mutations (Pf)
Expected Found
gag, pol, enva 8gag, 15.7pol, 13.2envb 19gag, 10pol, 5env
(⬍0.001) R1, R2, R3a,c 15.8 R1, 15.8 R2,
15.8 R3b 30 R1, 11 R2, 8 R3(⬍0.001)
gag(1337–1598),
env(7071–7333)d 6.8gag, 6.8env
e 13gag, 0env
(⬍0.001)
aComparisons involve mutations found in clones D15, G7, I15, and K15 relative to their corresponding parental clones D1, G1, I1, and K1 (Table 2).
bThe expected numbers of mutations are based on the mutation frequencies given in Table 2, assuming a random distribution of mutations along the genome. cR1, R2, and R3 indicate genomic regions 1, 2, and 3, as depicted in Fig. 2. dComparisons involve mutations at genomic residues 1337 through 1598 and 7071 through 7333 found in three clones from lineage F15, two clones each from lineages D15, G7, and K15, and one clone each from lineages H13 and I15, as described in the text and in Fig. 1.
eThe expected numbers of mutations are based on the average mutation frequencies for genomic residues 1337 through 1598 (gag) and 7071 through 7333 (env) assuming a random distribution of mutations along the two genomic stretches.
fPvalues were calculated by the2test.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.311.551.527.614.2]clones (Table 2) were not represented among the mutations
found after a single infectious cycle (39). These variations in
mutation types and frequencies probably arose not only from
differences in the number of replication cycles but also from
the sequence context in the template being copied in a
differ-ent biological environmdiffer-ent (43).
The most unexpected finding in the analysis of low-fitness
HIV-1 clones was the distribution of mutations along the
ge-nome (Fig. 2), with a statistically significant accumulation of
mutations in
gag
and the first third of the genome, relative to
env
, which appears as the most conserved genomic region in all
clones examined (Fig. 2; Table 3). This is in sharp contrast to
results with natural HIV-1 isolates (35, 44, 57, 58) and with
large-population passages of HIV-1 clones—derived also from
natural isolate S61—in cell culture (64); in all of these
analy-ses,
gag
and
pol
showed more nucleotide and amino acid
sequence conservation than
env
. Several mechanisms could
contribute to this striking difference in the distribution of
mu-tations. In the course of plaque-to-plaque transfers, fitness
gains or purifying selection is probably diminished since
com-petition among genomes from the quasispecies is limited to the
period of plaque development (10, 25). In this view, the higher
variation normally seen in
env
relative to
gag
and
pol
would be
essentially due to selection for immune evasion, for adaptation
to alternative cellular receptors, increased particle stability,
etc. Evidence for selection in vivo has been obtained for HIV-1
and for simian immunodeficiency virus (2, 3, 26, 34, 48, 54, 62,
77). However, even if replicative optimization of the mutant
spectrum was limited as a result of bottlenecking, the deficit in
mutations in
env
remains to be explained. There are at least
two possibilities (which are not mutually exclusive):
env
may
have a lower tolerance for mutations than
gag
and the 5
⬘
third
of the genome under the cell culture environment in which the
passages were carried out, or HIV-1 genome replication may
be more error prone in the process of copying
gag
than when
copying
env
. Constraints to accept replacements in surface
proteins were documented through functional and structural
studies with FMDV (12 and references therein). The mutant
repertoire in viral quasispecies could be strongly influenced by
tolerance to nucleotide and amino acid substitutions,
includ-ing silent replacements in codinclud-ing regions (12, 38, 40, 67). In
HIV-1, constraints in surface proteins could come about from
the need always to use the cellular receptors presented by
MT-4 cells in culture and to enter this same cell type
monot-onously in an invariant cell culture environment. Purifying
selection during plaque growth, which must include many
cy-cles of replication, may have contributed to the observed bias
in the distribution of mutations along the genome.
A possible molecular basis for a difference in accuracy
dur-ing copydur-ing of different genome segments of HIV-1 is not
ob-vious. The lowest number of mutations was seen in the
geno-mic region copied immediately after the first strand transfer in
the synthesis of minus-strand DNA by reverse transcriptase
(RT) (reviewed in reference 68). Since processivity of RT is
limited, it could be proposed that as synthesis proceeds inside
the nucleocapsid, accuracy may decrease as a result of
envi-ronmental alterations (ionic composition, deoxynucleoside
tri-phosphate pools, etc. [43]). It has been suggested that
misalign-ment mutagenesis could be more frequent during dissociation
and reinitiation of RT-catalyzed reactions (reviewed in 1).
However, examination of the sequence context of each
muta-tion suggests that the frequency of mutamuta-tions which may have
occurred as a result of template misalignment (1, 53) is not
significantly different for regions 1, 2, and 3, into which we have
divided the HIV-1 genome (52, 64, and 63%, respectively).
Other effects on fidelity or, more likely, a combination of
factors outlined in previous paragraphs may converge to
pro-duce the unusual distribution of mutations seen in HIV-1
clones as a result of operation of Muller’s ratchet.
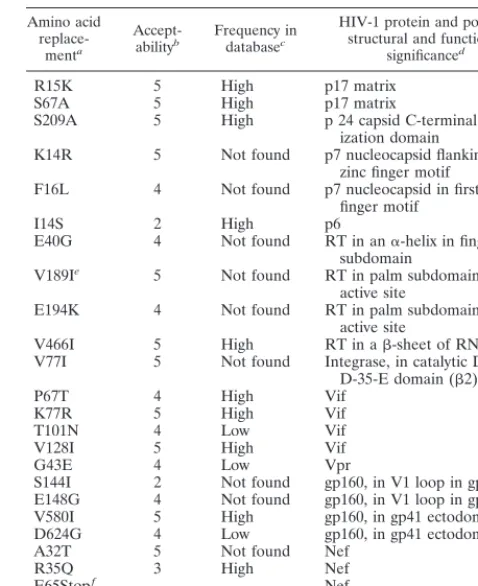
A total of 8 out of 20 nonsynonymous replacements found
among the HIV-1 clones analyzed have not been recorded in
current sequence data banks (35; Table 4). An attractive
pos-sibility is that replacements in
gag
could have multiple effects in
RNA-protein interactions, nucleocapsid assembly, and protein
stability (16, 30) and that such effects could contribute to
fit-ness loss. It must also be considered that Vif, Vpr, and Nef are
dispensable functions for HIV-1 replication in some permissive
cell lines, including MT-4 (7, 49). Therefore, the accumulation
of nonsynonymous replacements in
vif, vpr,
and
nef
genes (Fig.
2) could be of little consequence for plaque formation in MT-4
cells. However, an evaluation of the influence of amino acid
replacements (individually or in combination) in viral fitness
would require analysis of the effects of candidate mutations
when introduced into infectious clones or examination of
pos-sible reversions and fixation of compensatory mutations upon
fitness recovery of the debilitated HIV-1 clones. These studies
are now in progress.
[image:5.612.312.551.93.386.2]In conclusion, some mutations associated with the operation
TABLE 4. Amino acid replacements associated with nonsynonymous
mutations during plaque-to-plaque transfers of HIV-1
Amino acid
replace-menta
Accept-abilityb Frequency indatabasec
HIV-1 protein and possible structural and functional
significanced
R15K 5 High p17 matrix
S67A 5 High p17 matrix
S209A 5 High p 24 capsid C-terminal
dimer-ization domain
K14R 5 Not found p7 nucleocapsid flanking first zinc finger motif
F16L 4 Not found p7 nucleocapsid in first zinc finger motif
I14S 2 High p6
E40G 4 Not found RT in an␣-helix in fingers subdomain
V189Ie 5 Not found RT in palm subdomain near
active site
E194K 4 Not found RT in palm subdomain near
active site
V466I 5 High RT in a-sheet of RNase H
V77I 5 Not found Integrase, in catalytic D, D-35-E domain (2)
P67T 4 High Vif
K77R 5 High Vif
T101N 4 Low Vif
V128I 5 High Vif
G43E 4 Low Vpr
S144I 2 Not found gp160, in V1 loop in gp120
E148G 4 Not found gp160, in V1 loop in gp120
V580I 5 High gp160, in gp41 ectodomain
D624G 4 Low gp160, in gp41 ectodomain
A32T 5 Not found Nef
R35Q 3 High Nef
E65Stopf Nef
E154K 5 Low Nef
aAmino acids are numbered for each individual protein, according to the
numbering for isolate HXB2 found in the data banks (35).
bThe degree of acceptability of the amino acid substitution is given according
to reference 27; the acceptability scale is from 0 to 6, with the latter value representing replacement by the same amino acid.
cThe database consulted is the one presented in reference 35, and it was again
retrieved on April 18, 2000, by entering GenBank and EMBL databases.
dLocation of amino acid replacements and their possible significance is based
on current databases (35) and overviews on HIV-1 structure-function relation-ships (references 7, 9, 28, and 30 and references therein).
eThis replacement has been correlated with resistance to nonnucleoside RT
inhibitors (references 11, 28, 33, 35, and 46 and references therein).
fThis mutation leads to an amber termination codon in Nef.
on November 9, 2019 by guest
http://jvi.asm.org/
of Muller’s ratchet in HIV-1 have not been previously reported
among natural isolates evolved over more than two decades
(35; Table 4). Furthermore, the mutations were distributed
along the viral genome, unlike mutations in natural HIV-1
isolates, and
env
was the most conserved genomic region. An
interesting possibility is that, in vivo, HIV-1 is not subjected to
as severe bottleneck events as in experiments designed to
ac-centuate Muller’s ratchet. The observations reported here add
to the complexities inherent in the relationships between
oc-currence of mutations and what can be eventually observed
upon examination of genomic nucleotide sequences. The
HIV-1 mutational pattern could be made to vary with respect to
hundreds of sequences recorded in data banks simply by
changing the passage regimen, without intervening, externally
applied, selective forces.
ACKNOWLEDGMENTS
Work at Centro de Biología Molecular “Severo Ochoa” was
sup-ported by grants FIS98/0054-01 and PM97-0060-C02-01 and that at
Centro Nacional de Biología Fundamental was supported by grants
FIS00/0266 and FIS98/0054-02. E.Y. was supported by a postdoctoral
fellowship from Comunidad Autónoma de Madrid.
REFERENCES
1. Bebenek, K., and T. A. Kunkel. 1993. The fidelity of retroviral reverse tran-scriptases, p. 85–102.InA. M. Skalka and S. P. Goff (ed.), Reverse tran-scriptase. Cold Spring Harbor Laboratory Press, Gold Spring Harbor, N.Y. 2. Borrow, P., H. Lewicki, X. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A.
Nelson, J. E. Gairin, B. H. Hahn, M. B. Oldstone, and G. M. Shaw.1997.
Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211.
3. Borrow, P., and G. M. Shaw. 1998. Cytotoxic T-lymphocyte escape viral variants: how important are they in viral evasion of immune clearance in vivo? Immunol. Rev. 164:37–51.
4. Carrillo, C., J. Plana, R. Mascarella, J. Bergada, and F. Sobrino. 1990. Genetic and phenotypic variability during replication of foot-and-mouth disease virus in swine. Virology 179:890–892.
5. Cattaneo, R., and M. A. Billeter. 1992. Mutations and A/I hypermutations in measles virus persistent infections. Curr. Top. Microbiol. Immunol. 176:63– 74.
6. Chao, L. 1990. Fitness of RNA virus decreased by Muller’s ratchet. Nature
348:454–455.
7. Chen, I. S. Y., H. Koprowski, A. Srinivasan, and P. K. Vogt (ed.). 1995. Transacting functions of human retroviruses. Springer-Verlag KG, Berlin, Germany.
8. Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483–489.
9. Coffin, J. M., S. H. Hughes, and H. E. Varmus. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
10. Domingo, E., C. Biebricher, J. J. Holland, and M. Eigen. 2000. Quasispecies and RNA virus evolution: principles and consequences. Landes Bioscience, Austin, Tex.
11. Domingo, E., C. Escarmís, L. Menéndez-Arias, and J. J. Holland. 1999. Viral quasispecies and fitness variations, p. 141–161.InE. Domingo, R. G. Web-ster, and J. J. Holland (ed.), Origin and evolution of viruses. Academic Press, San Diego, Calif.
12. Domingo, E., M. G. Mateu, C. Escarmís, E. Martínez-Salas, D. Andreu,
E. Giralt, N. Verdaguer, and I. Fita.1996. Molecular evolution of
aphtho-viruses. Virus Genes 11:197–207.
13. Domingo, E., R. G. Webster, and J. J. Holland (ed.). 1999. Origin and evolution of viruses. Academic Press, San Diego, Calif.
14. Dougherty, J. P., and H. M. Temin. 1988. Determination of the rate of base-pair substitution and insertion mutations in retrovirus replication. J. Vi-rol. 62:2817–2822.
15. Drake, J. W. 1993. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 90:4171–4175.
16. Druillennec, S., A. Caneparo, H. de Rocquigny, and B. P. Roques. 1999. Evidence of interactions between the nucleocapsid protein NCp7 and the reverse transcriptase of HIV-1. J. Biol. Chem. 274:11283–11288. 17. Duarte, E., D. Clarke, A. Moya, E. Domingo, and J. Holland. 1992. Rapid
fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc. Natl. Acad. Sci. USA 89:6015–6019.
18. Eigen, M. 1971. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften 58:465–523.
19. Eigen, M. 1996. On the nature of virus quasispecies. Trends Microbiol. 4: 216–218.
20. Eigen, M., and C. K. Biebricher. 1988. Sequence space and quasispecies distribution, p. 211–245.InE. Domingo, P. Ahlquist, and J. J. Holland (ed.), RNA genetics, vol. 3. CRC Press, Inc., Boca Raton, Fla.
21. Eigen, M., J. McCaskill, and P. Schuster. 1988. Molecular quasispecies. J. Phys. Chem. 92:6881–6891.
22. Eigen, M., and P. Schuster. 1979. The hypercycle: a principle of natural self-organization. Springer-Verlag KG, Berlin, Germany.
23. Elena, S. F., F. González-Candelas, I. S. Novella, E. A. Duarte, D. K. Clarke,
E. Domingo, J. J. Holland, and A. Moya.1996. Evolution of fitness in
experimental populations of vesicular stomatitis virus. Genetics 142:673–679. 24. Escarmís, C., M. Dávila, N. Charpentier, A. Bracho, A. Moya, and E.
Do-mingo.1996. Genetic lesions associated with Muller’s ratchet in an RNA
virus. J. Mol. Biol. 264:255–267.
25. Escarmís, C., M. Dávila, and E. Domingo. 1999. Multiple molecular path-ways for fitness recovery of an RNA virus debilitated by operation of Mul-ler’s ratchet. J. Mol. Biol. 285:495–505.
26. Evans, D. T., D. H. O’Connor, P. Jing, J. L. Dzuris, J. Sidney, J. da Silva,
T. M. Allen, H. Horton, J. E. Venham, R. A. Rudersdorf, T. Vogel, C. D. Pauza, R. E. Bontrop, R. DeMars, A. Sette, A. L. Hughes, and D. I. Watkins.
1999. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat. Med. 5:1270– 1276.
27. Feng, D. F., M. S. Johnson, and R. F. Doolittle. 1984. Aligning amino acid sequences: comparison of commonly used methods. J. Mol. Evol. 21:112– 125.
28. Flint, S. J., L. W. Enquist, R. M. Krug, V. R. Racaniello, and A. M. Skalka. 2000. Virology: molecular biology, pathogenesis, and control. ASM Press, Washington, D.C.
29. Forns, X., R. H. Purcell, and J. Bukh. 1999. Quasispecies in viral persistence and pathogenesis of hepatitis C virus. Trends Microbiol. 7:402–410. 30. Freed, E. O. 1998. HIV-1 gag proteins: diverse functions in the virus life
cycle. Virology 251:1–15.
31. Goudsmit, J., A. de Ronde, E. de Rooij, and R. de Boer. 1997. Broad spectrum of in vivo fitness of human immunodeficiency virus type 1 sub-populations differing at reverse transcriptase codons 41 and 215. J. Virol. 71: 4479–4484.
32. Goulder, P., D. Price, M. Nowak, S. Rowland-Jones, R. Phillips, and A.
McMichael.1997. Co-evolution of human immunodeficiency virus and
cy-totoxic T-lymphocyte responses. Immunol. Rev. 159:17–29.
33. Havlir, D. V., S. Eastman, A. Gamst, and D. D. Richman. 1996. Nevirapine-resistant human immunodeficiency virus: kinetics of replication and esti-mated prevalence in untreated patients. J. Virol. 70:7894–7899.
34. Kimata, J. T., L. Kuller, D. B. Anderson, P. Dailey, and J. Overbaugh. 1999. Emerging cytopathic and antigenic simian immunodeficiency virus variants influence AIDS progression. Nat. Med. 5:535–541.
35. Korber, B., C. Kuiken, B. Foley, B. Hahn, F. McCutchan, J. Mellors, and J.
Sodroski.1998. Human retroviruses and AIDS: a compilation and analysis of
nucleic acids and amino acid sequences. Theoretical Biology and Biophysics Group T-10. Los Alamos, N.M.
36. Kurosaki, M., N. Enomoto, T. Nouchi, I. Sakuma, F. Marumo, and C. Sato. 1995. Fraction-specific populations of the hypervariable region of the hep-atitis C virus in a patient with cryoglobulinemia. J. Med. Virol. 46:403–408. 37. Lech, W. J., G. Wang, Y. L. Yang, Y. Chee, K. Dorman, D. McCrae, L. C.
Lazzeroni, J. W. Erickson, J. S. Sinsheimer, and A. H. Kaplan.1996. In vivo
sequence diversity of the protease of human immunodeficiency virus type 1: presence of protease inhibitor-resistant variants in untreated subjects. J. Vi-rol. 70:2038–2043.
38. Lobert, P. E., N. Escriou, J. Ruelle, and T. Michiels. 1999. A coding RNA sequence acts as a replication signal in cardioviruses. Proc. Natl. Acad. Sci. USA 96:11560–11565.
39. Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094.
40. McKnight, K. L., and S. M. Lemon. 1996. Capsid coding sequence is re-quired for efficient replication of human rhinovirus 14 RNA. J. Virol. 70: 1941–1952.
41. McMichael, A. J., and R. E. Phillips. 1997. Escape of human immunodefi-ciency virus from immune control. Annu. Rev. Immunol. 15:271–296. 42. Meyerhans, A., R. Cheynier, J. Albert, M. Seth, S. Kwok, J. Sninsky, L.
Morfeldt-Manson, B. Asjo, and S. Wain-Hobson.1989. Temporal
fluctua-tions in HIV quasispecies in vivo are not reflected by sequential HIV isola-tions. Cell 58:901–910.
43. Meyerhans, A., and J.-P. Vartanian. 1999. The fidelity of cellular and viral polymerases and its manipulation for hypermutagenesis, p. 87–114.InE. Domingo, R. G. Webster, and J. J. Holland (ed.), Origin and evolution of viruses. Academic Press, Inc., San Diego, Calif.
44. Montagnier, L. 1999. Human immunodeficiency virus (Retroviridae).InA. Granoff and R. G. Webster (ed.), Encyclopedia of virology. Academic Press, Inc., San Diego, Calif.
on November 9, 2019 by guest
http://jvi.asm.org/
45. Muller, H. J. 1964. The relation of recombination to mutational advance. Mutat. Res. 1:2–9.
46. Nájera, I., A. Holguín, M. E. Quiñones-Mateu, M. A. Muñoz-Fernández, R.
Nájera, C. López-Galíndez, and E. Domingo.1995. Pol gene quasispecies of
human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J. Virol. 69:23–31. 47. Nájera, I., D. D. Richman, I. Olivares, J. M. Rojas, M. A. Peinado, M.
Perucho, R. Najera, and C. Lopez-Galindez.1994. Natural occurrence of
drug resistance mutations in the reverse transcriptase of human immunode-ficiency virus type 1 isolates. AIDS Res. Hum. Retrovir. 10:1479–1488. 48. Nijhuis, M., R. Schuurman, D. de Jong, J. Erickson, E. Gustchina, J. Albert,
P. Schipper, S. Gulnik, and C. A. Boucher.1999. Increased fitness of drug
resistant HIV-1 protease as a result of acquisition of compensatory muta-tions during suboptimal therapy. AIDS 13:2349–2359.
49. Nishino, Y., M. Kishi, M. Sumiya, K. Ogawa, A. Adachi, K. Maotani-Imai, S.
Kato, K. Hirai, and K. Ikuta.1991. Human immunodeficiency virus type 1
vif, vpr, and vpu mutants can produce persistently infected cells. Arch. Virol.
120:181–192.
50. Novella, I. S., E. A. Duarte, S. F. Elena, A. Moya, E. Domingo, and J. J.
Holland. 1995. Exponential increases of RNA virus fitness during large
population transmissions. Proc. Natl. Acad. Sci. USA 92:5841–5844. 51. Novella, I. S., C. L. Hershey, C. Escarmis, E. Domingo, and J. J. Holland.
1999. Lack of evolutionary stasis during alternating replication of an arbo-virus in insect and mammalian cells. J. Mol. Biol. 287:459–465.
52. Novella, I. S., J. Quer, E. Domingo, and J. J. Holland. 1999. Exponential fitness gains of RNA virus populations are limited by bottleneck effects. J. Virol. 73:1668–1671.
53. Pathak, V. K., and H. M. Temin. 1992. 5-Azacytidine and RNA secondary structure increase the retrovirus mutation rate. J. Virol. 66:3093–3100. 54. Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards,
A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. Bangham, C. R. Rizza, and
A. J. McMichael.1991. Human immunodeficiency virus genetic variation
that can escape cytotoxic T cell recognition. Nature 354:453–459. 55. Preston, B. D., and J. P. Dougherty. 1996. Mechanisms of retroviral
muta-tion. Trends Microbiol. 4:16–21.
56. Quiñones-Mateu, M. E., J. L. Albright, A. Mas, V. Soriano, and E. J. Arts. 1998. Analysis ofpolgene heterogeneity, viral quasispecies, and drug resis-tance in individuals infected with group O strains of human immunodefi-ciency virus type 1. J. Virol. 72:9002–9015.
57. Quiñones-Mateu, M. E., A. Holguín, J. Dopazo, I. Nájera, and E. Domingo. 1996. Point mutant frequencies in the pol gene of human immunodeficiency virus type 1 are two- to three-fold lower than those of env. AIDS Res. Hum. Retrovir. 12:1117–1128.
58. Quiñones-Mateu, M. E., A. Holguín, V. Soriano, and E. Domingo. 1996. env gene diversity of HIV type 1 isolates from Spain. AIDS Res. Hum. Retrovir.
12:955–957.
59. Ribeiro, R. M., S. Bonhoeffer, and M. A. Nowak. 1998. The frequency of resistant mutant virus before antiviral therapy. AIDS 26:461–465. 60. Robertson, D. L., B. H. Hahn, and P. M. Sharp. 1995. Recombination in
AIDS viruses. J. Mol. Evol. 40:249–259.
61. Robertson, D. L., P. M. Sharp, F. E. McCutchan, and B. H. Hahn. 1995. Recombination in HIV-1. Nature 374:124–126.
62. Rodrigo, A. G., and J. I. Mullins. 1996. Human immunodeficiency virus type 1 molecular evolution and the measure of selection. AIDS Res. Hum. Ret-rovir. 12:1681–1685.
63. Sala, M., and S. Wain-Hobson. 1999. Drift and conservation in RNA virus evolution: are they adapting or merely changing? p. 115–140.InE. Domingo, R. G. Webster, and J. J. Holland (ed.), Origin and evolution of viruses. Academic Press, Inc., San Diego, Calif.
64. Sánchez-Palomino, S., J. M. Rojas, M. A. Martínez, E. M. Fenyö, R. Nájera,
E. Domingo, and C. López-Galíndez.1993. Dilute passage promotes
expres-sion of genetic and phenotypic variants of human immunodeficiency virus type 1 in cell culture. J. Virol. 67:2938–2943.
65. Schuitemaker, H., N. A. Kootstra, R. E. de Goede, F. de Wolf, F. Miedema,
and M. Tersmette.1991. Monocytotropic human immunodeficiency virus
type 1 (HIV-1) variants detectable in all stages of HIV-1 infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J. Vi-rol. 65:356–363.
66. Schuster, P., and P. F. Stadler. 1999. Nature and evolution of early replicons, p. 1–24.InE. Domingo, R. G. Webster, and J. J. Holland (ed.), Origin and evolution of viruses. Academic Press, Inc., San Diego, Calif.
67. Simmonds, P., and D. B. Smith. 1999. Structural constraints on RNA virus evolution. J. Virol. 73:5787–5794.
68. Telesnitsky, A., and S. P. Goff. 1993. Strong-stop strand transfer during reverse transcription, p. 49–83.InA. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
69. Temin, H. M. 1989. Is HIV unique or merely different? J. Acquir. Immune Defic. Syndr. 2:1–9.
70. Temin, H. M. 1993. The high rate of retrovirus variation results in rapid evolution, p. 219–225.InS. S. Morse (ed.), Emerging viruses. Oxford Uni-versity Press, Oxford, United Kingdom.
71. Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
72. Vartanian, J. P., A. Meyerhans, B. Asjo, and S. Wain-Hobson. 1991. Selec-tion, recombinaSelec-tion, and G3A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 65:1779–1788.
73. Vartanian, J. P., A. Meyerhans, M. Sala, and S. Wain-Hobson. 1994. G3A hypermutation of the human immunodeficiency virus type 1 genome: evi-dence for dCTP pool imbalance during reverse transcription. Proc. Natl. Acad. Sci. USA 91:3092–3096.
74. Wain-Hobson, S. 1996. Running the gamut of retroviral variation. Trends Microbiol. 4:135–141.
75. Weaver, S. C., A. C. Brault, W. Kang, and J. J. Holland. 1999. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 73:4316–4326.
76. Weiner, A., A. L. Erickson, J. Kansopon, K. Crawford, E. Muchmore, A. L.
Hughes, M. Houghton, and C. M. Walker.1995. Persistent hepatitis C virus
infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. USA 92:2755–2759. 77. Wolinsky, S. M., B. T. Korber, A. U. Neumann, M. Daniels, K. J. Kunstman,
A. J. Whetsell, M. R. Furtado, Y. Cao, D. D. Ho, and J. T. Safrit.1996.
Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272:537–542.
78. Yuste, E., S. Sánchez-Palomino, C. Casado, E. Domingo, and C.
López-Galíndez.1999. Drastic fitness loss in human immunodeficiency virus type 1
upon serial bottleneck events. J. Virol. 73:2745–2751.
on November 9, 2019 by guest
http://jvi.asm.org/