JOURNAL OFVIROLOGY, Oct. 1977, p. 22-27 Copyright©1977 American Society for Microbiology
Vol.24, No. 1
Printed in U.S.A.
Characterization
of the Autoregulation of Simian Virus 40
Gene A
JAMES C. ALWINE,* STEVEN I. REED,t AND GEORGE R. STARK
Department of Biochemistry, Stanford University School of Medicine,Stanford, California 94305
Received for publication21April1977
Cells infected by tsA mutants of simian virus 40 (SV40) overproduce early RNA. Overproduction results from failure of the temperature-sensitive A
protein (T antigen)to inhibitearly transcription. Theamountofearly RNA in
the cytoplasm, determined quantitatively from the kinetics ofhybridization to
labeled complementary SV40 DNA, was elevated at both permissive (320C) and
nonpermissive (410C) temperatures in all theearly mutants tested (tsA7, -30,
-58, and -209), but not in the late mutant tsB4. The amountofearly RNA ina
culture maintainedat320Cfor72hand thenshiftedto410Cwasmaximumwhen
each cellwasinfected initially withatleastoneplaque-formingunitof tsA58.
Azidocytidine (2'-deoxy-2'-azidocytidine), which inhibits initiation of DNA
syn-thesis, didnotcauseoverproduction of early RNAincellsinfected with wild-type
SV40, showing that the effectseenwithtsA mutantsis notduetointerference
with initiation of DNA synthesis per se. In parallel infections at 41'C, the
amountofearlyRNAper copyof viral DNA was as much as2,000timesgreater
withtsA58 than with wild-typeSV40, eventhough therewas no replicationof
the tsA58 DNA. Synthesis of late RNA couldnotbedetectedduringthefirst20h
ofaninfectionbyeither virusat320C, indicatingthatlateandearly
transcrip-tion areunder different control. Inthree cell lines transformedby tsA mutants,
the amount of early RNA increased moderately after a shift from 32 to410C,
whereas withhomologous cells transformed by wild-type virus, the amount of
early RNA decreased, indicating that the A protein may be able to repress
transcription ofintegrated SV40 DNA. All the observations are consistent with
a simple model inwhich the bindingof A protein atthe origin of replication
blocks eitherbindingof RNApolymerase tothe early promoterorits progress
through theearlygene(s).
In a lyticinfection by simian virus40 (SV40),
gene A istranscribed before theonsetofviral
DNA synthesis ("early"), leadingtothe
appear-anceofearly mRNAinthecytoplasmandtoits
translation intoTantigen,theAprotein (8, 14,
17, 18). Temperature-sensitive A mutants fail
toinitiateviral DNA synthesisat a
nonpermis-sivetemperature (21)andproducea more
ther-molabileA protein (1, 26), indicating that this
protein is directly involved in initiation.
Con-sistent withthis ideaisthat theAproteinbinds
preferentiallytothe originofreplicationin
vi-tro(15).
The A protein is synthesized continuously
duringthe course ofinfection andaccumulates
inthe nucleus (1, 25). Tegtmeyer et al. (22, 25)
found that, although the steady-state amount
ofthe A protein is very low in cells infected
with tsA mutants at anonpermissive
tempera-t Presentempera-t address: Department ofGenetics, University
ofWashington, Seattle, WA98195.
ture, the rate of synthesis of this protein is
more rapid than in a corresponding infection
withwild-type virus. They proposed that theA
proteinregulatesits ownsynthesis, since faster
synthesismight reflect the loss ofaninhibitory
control. Wepreviously confirmedand extended
this initial idea by demonstrating that cells
infected with tsA mutants overproduce early
RNA, that they synthesize it faster, that they
do so even at a permissive temperature, and
thatthiseffectisamplified greatly afterashift
to a nonpermissive temperature late in
infec-tion when many DNA templates are present
(16). Therefore, autoregulation appearsto
oper-ate through negative feedback control ofthe
transcription of gene A. Since the 5' end of
RNA molecules complementary to the early
strand ofSV40 DNA map near the origin of
replication (10; S. I. Reed and J. C. Alwine,
Cell,inpress),control oftranscription may well
be achieved by the bindingof A protein ator
nearthe early promoter, whichmayin turnbe
22
on November 10, 2019 by guest
http://jvi.asm.org/
nearthe origin of replication (6). Wenow
pre-sentadditional informationonthe
autoregula-tionofgeneA and examinemore critically its
relationshipto initiationof viral DNA
synthe-sis.
MATERIALS AND METHODS
Cells. The CV-1 line of African green monkey
kidney cells, obtained from the laboratory of Paul Berg, wasgrowninaCO2incubator in Lux plastic
plates (100 by15 mm)inEagle mediumasmodified
by Dulbecco (GIBCO) with 10% fetal bovineserum
(Microbiological Associates).
The cell lines 596HEWT and 596HEA58 (23), hamster embryo cells transformed by SV40
wild-type strain VA45-54or by themutanttsA58,were
thegift of Peter Tegtmeyer. The lines CHLWT15, CHLA209L1, and CHLA239L1 (12), Chinese
ham-sterlung cells transformedby wild-type strain SV-S
orby themutanttsA209ortsA239,werethegift of RobertMartin.
Viruses and nucleic acids. The virus strains used included the wildtype VA45-54 (24) and
tempera-ture-sensitivemutants derived from it:tsA7,tsA30, tsA58, and tsB4 (21, 22, 24) and also tsA209 (4), derived from the SV-Swild-type strain. Nondefec-tivestockswerepreparedasdescribedby Estesetal. (7).SV40 (I) DNA (supercoiled circles)wasprepared
from CV-1 cellsinfected for 11daysat370Cwith 0.01 plaque-forming unit (PFU) of VA45-54 percell as
described previously (15). Cytoplasmic RNA was
preparedasdescribedpreviously (16).
Analysis of hybridization kinetics. SV40 DNA
was labeled in vitro to high specific radioactivity accordingtothe"nicktranslation"procedure ofP.
W. J. Rigby, M. Dieckmann, C. Rhodes,and P. Berg (J. Mol.Biol., inpress),using[3H]TTP (45 Ci/mmol, New England Nuclear). The DNA (1.6 x 107cpm/
gg)wasfragmentedat40Cby sonically treating it for 6to8periodsof10seach atsetting7 ofaSonifier
cell disrupter. The strands were separated with
cRNAaccordingtoSambrooketal. (19).
To determine the fraction ofcytoplasmic RNA that wasviral, we employed the hybridization
ki-neticsanalysis of J. C. Leong, D. E. Garfin, H. S. Smith, P. W. J. Rigby, and H. M. Goodman (manu-script in preparation) as described previously by
Reed etal. (16),inwhichtheinitial rate of hybridi-zation of viral RNA to highly labeled, separated
strands ofSV40DNAis determined.Thefraction(f) ofviralsequencesinthesample iscalculated from theslope ofaplot of the linear equationCo/C =1 +
K~f(Crt), where CO is the initial concentration of
complementary single-stranded DNA, C is the
con-centrationofcomplementary single-stranded DNA atanytime(t),C,is thetotalconcentrationof RNA
(viralandcellular), andKhisthe second-orderrate
constantdetermined withknown concentrations of SV40 cRNA. Viral RNA complementary to the E (minus) DNA strand is referred to as E-RNA and
viral RNA complementary totheL (plus)strandis
L-RNA. Since thecoding region for cytoplasmic
E-RNAorL-RNAisequaltoabouthalf the E-DNAor
L-DNAstrand(6, 10),CO istakentobehalfof the
inputconcentrationoflabeled E-DNAorL-DNA.
The amount ofviral DNA ininfected cells was
determined according to P. W. J. Rigby and P.Berg (manuscript in preparation)by using the hybridiza-tionof"nick-translated" SV40 DNA to determine Cot.
Enzymesandreagents.Escherichia coli DNA
po-lymerase I and E. coli RNA popo-lymeraseholoenzyme
weregifts ofLeroy Bertsch and Michael
Chamber-lin, respectively. TheS,nuclease ofAspergillus
ory-zae waspurchased fromSigma Chemical Co., bovine
pancreatic DNase I (RNase-free) was from
Worth-ington Biochemicals Corp., and proteinase K was
from Merck and Co. 2'-Deoxy-2'-azidocytidine (20)
was agift of PeterReichard.
RESULTS AND DISCUSSION Effect of the multiplicity of infection. In
previousexperiments, CV-1 cellswereinfected
withtsA58 virusathighmultiplicity (20to100
PFUpercell) for72hat32°C and then shifted
to41°C for5or10h(16).Therewasa17-fold
in-creaseincytoplasmic E-RNA after10 h at41°C.
To examine the possible effects of different
multiplicities of infection, CV-1 cells were
in-fected with0.5to50PFU oftsA58percellwith
or without a 10-h shift to nonpermissive
tem-perature, and the fraction of E-RNA in the
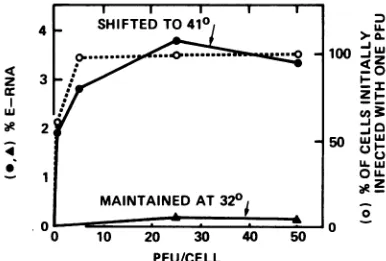
cytoplasmic RNA poolwasdetermined (Fig. 1).
In the experiments with a temperature shift
where the overproduction was greatest, the
percentageofE-RNA didnotincrease
apprecia-bly when multiplicities greater than about 5
PFU per cell were used. The dashed line in
Fig. 1 shows the percentage ofhost cells
ini-tially infectedwithatleast1PFUasafunction
ofmultiplicity, calculated accordingtoa
Pois-son distribution. The agreementbetweenthis
curveandthatofshiftedsamples demonstrates
thatmaximumoverproduction of E-RNAinthe
4
z
*u
li
UA.
>CL
100 JZj
-z
-0
50 "> _
0E
MAINTAINED AT320 v-_
0 0
0 10 20 30 40 50
PFU/CELL
FIG. 1. Effect of multiplicity of infectionon
over-production ofE-RNA by tsA58. The amountof E-RNA inthecytoplasmic fraction wasdeterminedin
CV-1 cells thathadbeeninfected for72hwith(-)or
without (A)ashiftto 41°C foranadditional 10 h.
The dashed line is the calculatedpercentageofhost cellsinfected initiallywithatleast1 PFU.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.504.261.454.475.606.2]24 ALWINE, REED, AND STARK
cultureas awholeisobtained when each cellis
infected initially with atleast 1 PFU; i.e.,
in-fection with more thanone virusper cell does
notfurtherincreasethe amountofE-RNA.
Levels of E-RNA in cytoplasmic samples
from cells infected with different
tempera-ture-sensitivemutants.Wepreviously observed
overproduction of E-RNA with both tsA58
and tsA7 (16). Laiand Nathans (11)showed by
the markerrescuetechniquethat tsA mutants
map in one of three adjacentHindII+III
re-striction endonuclease fragments, all derived
from the early region of the SV40genome. Fora
detailed comparison of mutants, we chose a
representative from each group: tsA7 (B
frag-ment), tsA30 (H fragment), and tsA58 (I
frag-ment).Thesemutants wereallderived from the
VA45-54 strain, which was used as the
wild-typecontrolinthe experiments. Wealsouseda
mutant in the late region, tsB4 (J fragment)
and one additional A mutant tsA209 (I
frag-ment), derived from the SV-S wild-typestrain.
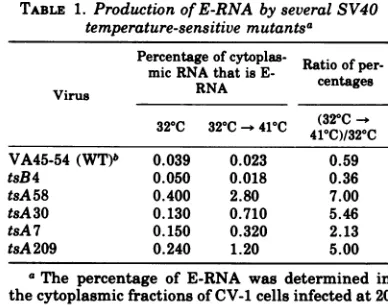
The percentage of E-RNA was determined in
cytoplasmic RNA prepared from CV-1 cells
in-fected at20PFU per cell for 72 h at320Cwith or
withouta 10-h shift to 410C (Table 1). Allthe
tsA mutants overproduced E-RNA, even at a
permissive temperature, and the
overproduc-tion was amplified upon a shift to restrictive
temperature. The late mutant tsB4 had the
same level of early RNA as did thewild-type
controlbothat320Cand afterashiftto410C.
Wereportedpreviously that there wasa
17-fold increaseinE-RNA whencells infected with
tsA58 wereshifted from 32 to 41'C for 10 h late
ininfection (16), yet in Table 1 there isonly a
sevenfold increase. Theamountof E-RNAwas
thesame incells that had been shiftedto410C
[image:3.504.58.252.495.648.2]inthetwostudies, buttherewas more in cells
TABLE 1. Production of E-RNA by several SV40 temperature-sensitive mutantSa
Percentage ofcytoplas- R
micRNA thatisE- Ratioofper.
Virus RNA centages
32°C 32°C-- 41°C (320C
410C)/320C VA45-54 (WT)b 0.039 0.023 0.59
tsB4 0.050 0.018 0.36
tsA58 0.400 2.80 7.00
tsA30 0.130 0.710 5.46
tsA 7 0.150 0.320 2.13
tsA209 0.240 1.20 5.00
aThe percentage of E-RNA was determined in
thecytoplasmicfractions of CV-1 cells infectedat 20
PFUpercell for72h at32°C,withorwithouta10-h
shiftto41°C.
bWT, Wildtype.
maintainedat apermissive temperatureinthe
experiments ofTable 1 than previously. This
discrepancy appearedtobedue to asmall
dif-ference in the permissive temperatures inthe
two cases: ashift from 31 to 330C caused more
than a twofold increase in the amount of
E-RNA. Inprevious workwe foundthat mutant
A proteinswere morelabilethan thewild-type
A protein in vitro at all temperatures (1). If
theywerealsomore labileinvivo, anincrease
in temperature even in the permissive range
wouldincrease the rate atwhichthemutantA
proteins decay, which in turn would be
re-flected in increasedsynthesisofE-RNA.
Phenotypically, tsA mutants are unable to
initiateviral DNA replication and do not
pro-duce virus at restrictive temperatures.
Differ-ent tsA mutants vary in the extent to which
theyaredefective: tsA7 is themostleaky
mu-tant,tsA58 is the least leaky, and tsA30seems
tobe intermediate (21, 22). This relative order
is maintained when the amounts of E-RNA
overproduced by the tsA mutants are
com-pared, both at permissive and at restrictive
temperatures (Table 1). The similarity of the
relative effects oninitiation of viral DNA
syn-thesis and on control of the transcription of
geneA within this set ofthree tsA mutants
indicates that theA protein isinvolvedinboth
processes.
Effectsofinhibitors of DNA replicationon
synthesisofE-RNAininfections bywild-type
SV40. Although the A proteinappears to have
coupled functions ininitiatingviral DNA
syn-thesis andincontrollinggeneA transcription,
overproduction of E-RNA might occur
indi-rectlyas aconsequenceof the cessationof viral
DNAsynthesis. In thiscase,inhibitors of DNA
synthesis should cause overproduction of
E-RNA inaninfectionby thewild-type virus. We
reported previously that cytosine arabinoside
causes nooverproduction (16). Another
inhibi-torof DNA synthesis, 2-azidocytidine (20),
ap-pears to inhibit synthesis ofpolyoma DNA at
initiation (G. L. Bjursell, L. Skoog, L.
Thelan-der, and G. Soderman, Proc. Natl. Acad. Sci.
U.S.A., in press), and, therefore, its action is
more similar to the defectin a nonpermissive
infection by atsA mutant at 410C than is the
action of cytosine arabinoside, which inhibits
primarily elongation. CV-1 cells wereinfected
withwild-typevirus at 20PFUpercellfor 72h
at320C, and some ofthe cellswere shifted to
41°C. Atthetime of theshift, samples ateach
temperature were treated withinhibitory
con-centrations of 2'-azidocytidine, and the cells
wereharvested6hlater. AsshowninTable 2,
blocking viral DNAsynthesiswiththis
inhibi-tormadenodifferenceintheamountofearlyor
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 2. Effectof2'-azidocytidineonSV40RNA in infections by wild-type virus
E-RNA(9) L-RNA (%) Temp(MC)
-AzidoC +AzidoC -AzidoC +AzidoC
32 0.030 0.030 1.05 1.04
32 -i 41 0.021 0.021 2.70 2.97
aThe percentages ofE-RNA and L-RNA were
determined in the cytoplasmic fractions ofCV-1cells infected with VA45-54 at 20PFU per cell for 72 h at 32TC with or without a shift to 4100. At the time of
theshift, the medium above some of the cells was
made 2 mM in 2'-deoxy-2'-azidocytidine (AzidoC), and incubation at 32 or 41'0 was continued for an additional 6 h. Theinhibitoryeffect of 2'-azidocyti-dine had been previously tested in CV-1 cells in-fected for 24 h at 37'0 with wild-type virus at 50 PFU per cell. Various amounts of inhibitor were added for4h, followedby a 15-min pulse of [3H]thymidine. The amount of label in viral DNA was taken to be the same as the label in supernatants prepared ac-cording to Hirt (9). At 0.3 mM, 2'-azidocytidine in-hibitsSV40 DNA synthesis by at least 95%, an effect similar to the onereportedwithpolyoma (G. Bjur-sell et al. Proc. Natl. Acad. Sci.U.S.A., inpress).
lateviralRNAinthecytoplasmateither
tem-perature. Therefore, blocking viral DNA
syn-thesis at initiationdoes notresultin
overpro-ductionof E-RNA in thepresenceof normalA
protein. This resultsupports amodelinwhich
failure to initiate DNAsynthesis and
overpro-duction of E-RNAarebothrelatedtorapidloss
ofa morethermosensitiveA protein.
Viral DNA synthesis and control of
tran-scription in infections with no temperature
shift. To examine further the linked functions
of the A proteinincontrolling viral DNA
syn-thesis and transcription ofE-RNA, CV-1 cells
wereinfectedwithwild-typeortsA58 virusat a
multiplicity of20PFU percell for times
rang-ingfrom 0 to 98h at
320C
orfrom 0 to 48h at410C.
Theamountsof E-RNAandL-RNAinthecytoplasmic fractionsareshowninFig.2and3.
After0and72hat
320C,
and after0and48hat410C,
the number of SV40genomes percellwasdetermined by
Cot
analysis (Table 3). Asex-pected, lessviral DNAwasmade inthe tsA58
infection than in the wild-type infection at
320C,
and there seemedtobesomelossof inputtsA58 DNA after 48 h at 410C. The impaired
ability of tsA58 to grow even at 320C can be
attributed to instability of the mutant A
pro-tein. Overproduction of E-RNA by tsA58
oc-curred even at
410C,
where theCot
data showthat therewas no netsynthesis of DNA (Fig. 2).
It can be estimated that tsA58 produced
ap-proximately 180 times more cytoplasmic E-RNA per genome than did wild-type virus at
320C,
whereas at410C
tsA58 produced approxi-mately 2,000 times more.Initiation of L-RNA synthesis has been
re-portedto dependonthe initiation, butnotthe
continuation, ofviral DNA synthesis (5). As
shown in Fig. 3, accumulation of L-RNA did
not start until 24 hafterinfectionat3200,after
z
w
cc
0.3
0.2
0.11
V
-0 20 40 60
HOURS
80 100
FIG. 2. Amount ofcytoplasmic E-RNA in cells
infectedwithtsA58orwild-typevirusas afunctionof
time at 32 or 41°C. CV-1 cells were infected with
tsA58 (0)orVA45-54 (@)virusata multiplicity of
20PFUpercellateither32or41°C.
z
INFECTION AT 320 INFECTION AT 410
0 20 40 60 80 100 0 HOURS
FIG. 3. Amountof cytoplasmicL-RNA in cells in-fected by tsA58 or wild-type virus as a function of
time. Theexperimentisthesameasdescribed in the
legendtoFig.2.
I I I
INFECTION AT410
tsA58
WT
I_ I I
I
n _
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.504.57.246.74.150.2] [image:4.504.263.451.144.422.2] [image:4.504.261.453.483.634.2]26 ALWINE, REED, AND STARK
thestartof DNAsynthesis, and the
accumula-tion wasdiminishedinthe case oftsA58,
proba-bly because there were fewer gene copies. At
410C, accumulation ofL-RNA in the infection
by wild-type virus started after about 12 h,
andverylittle L-RNAwasformedinthe tsA58
infection, asexpected, sincetsA58 DNA is not
synthesizedatthistemperature.
Control of E-RNA transcription in
trans-formed cells. Cells transtrans-formed by tsA
mu-tants lose both phenotypic indicatorsof
trans-formation and detectable T antigen (Aprotein)
upon incubation at nonpermissive
tempera-tures(3, 12, 13, 23).Furthermore, theAprotein
from cells transformed with a tsA mutant is
more thermolabile than the A protein from
cells transformed by wild-type virus (2, 26).
Therefore, the A protein is strongly implicated
inmaintainingthe SV40transformedstate.We
tested several cell lines transformed by
wild-typevirusand tsA mutants to seewhether the
A protein controls transcription of E-RNA in
these cells. Transformed cells were grown to
confluency at 320C, and some were shifted to
39.5 or410Cfor20h. As showninTable4,both
CHL cell lines transformed by tsA mutants
overproduced E-RNAmoderately after the shift
to anonpermissive temperature. The hamster
TABLE 3. Copies ofSV40 DNA per cellafter infectionat32or41'Ca
Conditios.Wildtype tsA58
Conditions (copies/cell) (copies/cell) Input DNA, zero time 250 230
72h at320C 19,400 2,500
48h at410C 43,100 110
aThe number ofSV40 genomes per cellwas
de-terminedby Cotanalysis of total DNA from CV-1 cells infected with VA45-54 or tsA58 at 20 PFU per cell at 32 or41°C. The amount of input DNA was determined after 2 h ofadsorptionat32°C.
TABLE 4. E-RNA inSV40-transformedcellsa
% E-RNAa Ratio
Cellline 32'C 32°C (32°C-o
32°C 410C
39.5°C
41C)/32CCHLWT15 0.035 0.024 0.70
CHLA209L1 0.040 0.120 3.00
CHLA239L1b 0.036 0.084 2.33
596HE(WT)Y 0.051 0.028 0.55
596HE(A58) 0.062 0.063 1.00
aThe perecentage of E-RNA was determinedin
cytoplasmic fractions of cells growntoconfluency at 320C with or without a shift to39.5 or41°C for 20h
before harvest.
bInthe experiment withCHLA239L1, total RNA
wasused instead of thecytoplasmic fraction. cWT, Wild type.
embryo line transformed by tsA58didnot
sur-vive well at nonpermissivetemperatures,
per-hapsaccountingfor itsfailuretoproducemore
E-RNA aftera temperatureshift.However,the
amount of E-RNA didnot decrease, as it did
with both lines transformed by wild-type virus.
These results mayindicate that theA protein
can function to control E-RNA synthesis in
transformed cellsaswellas ininfected cells. In
transformed cells such regulation could
func-tionattheviralpromoter, at acellular
promo-ter, or in conjunction with another factor(s)
involved in transformation.
In summary, a tsA mutant has diminished
ability to synthesize viral DNA at both
per-missive and restrictivetemperatures, and this
property canbe correlatedquantitatively with
theamountof E-RNAoverproduced, indicating
the dual function of the A proteinininitiation
of viral DNA synthesis and in negative
feed-back regulation of E-RNA synthesis. The
coupled effects are consistent with a simple
modelinwhichtheA proteinbindsat or near
the origin of replication and blocks early
transcription from a promoter at or near the
origin. The large amount of E-RNAmade per
genome in infections by tsA58 at 320C, and
especiallyat410C, indicates that the early
pro-moteris veryefficient, givingthevirusthe
ad-vantage of being able to initiate early
tran-scription, and the processes that follow and
depend on it, with high probability. With an
efficient early promoter, efficient control
be-comes necessarylater, to divertgenomesfrom
early transcription to replication, late
tran-scription,andpackaging of complete virions.
ACKNOWLEDGMENTS
Thisinvestigation was supported by Public Health Ser-vicegrantCA17287 from the National CancerInstituteand
byPublicHealth Research FellowshipgrantCA00053 from the National Cancer Institute (to J.C.A.). S.I.R. was sup-ported by apredoctoral fellowship from the H. J. Kaiser
FamilyFoundation.
We are mostgrateful to Peter Reichard and Lionel Craw-ford forhelpful suggestionsand discussions.
LITERATURE CITED
1. Alwine, J. C., S. I. Reed, J. Ferguson, and G. R.Stark. 1975.Properties of T antigens induced bywild-type
and tsA mutantsinlyticinfection. Cell 6:529-533. 2. Anderson, J. L., C. Chang, P. T. Mora, and R. G.
Martin. 1977. Expression and thermal stability of simian virus40tumor-specifictransplantation anti-gen and tumorantigeninwild type-andtbA mutant-transformed cells. J. Virol. 21:459-467.
3. Brugge, J. S.,and J.5.Butel.1975.Role ofsimian virus 40gene A functioninmaintenanceoftransformation. J. Virol.15:619-635.
4. Chou,J.Y.,and R. G.Martin.1974.Complementation analysisofSV40mutants. J.Virol.13:1101-1109. 5. Cowan, K.,P. Tegtmeyer, and D. D.Anthony. 1973.
Relationshipofreplicationandtranscription of sim-ian virus 40 DNA. Proc. Natl. Acad. Sci. U.S.A. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
70:1927-1930.
6. Dhar, R., K. N.Subramanian, J. Pan, and S. M. Weiss
man.1977.Nucleotidesequenceofafragment ofSV40 DNA thatcontainsthe origin of DNA replication and specifies the5'ends of 'early" and 'late" viral RNA. J.Biol. Chem. 252:368-376.
7. Estes, M. K., E.-S. Huang, and J. S. Pagano. 1971. Structuralpolypeptides of simian virus 40. J. Virol. 7:635-641.
8. Graessmann, M., and A. Graessmann. 1976. 'Early" simian virus 40-specific RNA contains information fortumorantigenformationand chromatin replica-tion.Proc. Natl. Acad. Sci. U.S.A. 73:366-370. 9. Hirt, B. 1967. Selective extraction of polyoma DNA
from infected mouse cell cultures. J. Mol. Biol. 26:365-369.
10. Khoury, G., P. Howley, D. Nathans, and M. Martin. 1975.Posttranscriptionalselection of simianvirus
40-specific RNA. J. Virol. 15:433-437.
11. Lai, C.-J., and D. Nathans. 1975. Amapoftemperature sensitivemutantsof simian virus 40.Virology 66:70-81.
12. Martin, R. G., and J. Y. Chou. 1975. Simian virus40 functionsrequired for the establishment and
mainte-nanceofmalignant transformation. J. Virol. 15:599-612.
13. Osborn,M.,and K. Weber. 1975. Simian virus40gene
A function and maintenance of transformation. J. Virol. 15:636-644.
14. Prives,C., E.Gilboa, M. Revel, and E. Winocour.1977. Cell-free translation of simian virus40early
messen-gerRNAcoding for viral T-antigen. Proc. Natl. Acad. Sci. U.S.A. 74:457-461.
15. Reed,S.I., J. Ferguson,R. W. Davis,and G. R. Stark.
1975.T antigen bindstosimian virus 40DNAatthe origin of replication. Proc. Natl. Acad. Sci. U.S.A. 72:1605-1609.
16. Reed, S. I., G.R. Stark, and J. C. AMwine. 1976. Auto-regulation of simian virus 40geneA by T antigen. Proc.Natl. Acad. Sci. U.S.A. 73:3083-3087. 17. Roberts, B. E., M. Gorecki, R. C. Mulligan, K. J.
Danna, S. Rozenblatt, and A. Rich. 1975. Simian virus40DNA directs the synthesis of authentic viral polypeptides in a linked transcription-translation
cell-free system. Proc. Natl. Acad. Sci. U.S.A. 72:1922-1926.
18. Rundell, K., J. K.Collins, P. Tegtmeyer, H. L. Ozer, C.-J. Lai, and D. Nathans. 1977. Identification of simian virus 40 protein A.J. Virol. 21:636-646. 19. Sambrook, J., P. A. Sharp, and W. Keller. 1972.
Tran-scription of simian virus 40. J. Mol.Biol. 70:57-71. 20. Skoog, L.,G. Ijursell, L. Thelander, T. Hagerstrom, J.
Hobbs, and F. Eckstein.1977. 2'-Deoxy-2'-azidocyti-dine,anewinhibitor of DNA replication in
mamma-lian cells. Eur. J. Biochem. 72:371-378.
21. Tegtmeyer, P. 1972. Simian virus 40 deoxyribonucleic acidsynthesis: the viral replicon. J. Virol. 10:591-598.
22. Tegtmeyer,P. 1974.Alteredpatternsofprotein synthe-sis in infectionbySV40mutants.ColdSpring Harbor Symp. Quant. Biol. 39:9-16.
23. Tegtmeyer, P. 1975. Function of simian virus 40geneA
intransforming infection. J. Virol. 15:613-618. 24. Tegtmeyer, P., C. Dohan, and C. Reznikoff. 1970.
Inac-tivatingand mutagenic effects of nitrosoguanidineon
simian virus40.Proc.Natl. Acad. Sci. U.S.A. 66:745-752.
25. Tegtmeyer, P., M. Schwartz, J. K. Collins, and K. Rundell.1975.Regulation oftumorantigen synthesis by simian virus 40geneA. J. Virol. 16:168-178. 26. Tenen, D. G., P. Baygell, and D. Livingston. 1975.
Thermolabile T (tumor) antigen from cells trans-formedbyatemperaturesensitivemutantofsimian virus40. Proc.Natl. Acad. Sci. U.S.A. 72:4351-4355.
27
on November 10, 2019 by guest
http://jvi.asm.org/