JOURNALOFVIROLOGY, Jan. 1983,p.332-342 0022-538X/83/010332-11$02.00/0
Copyright©1983,AmericanSocietyforMicrobiology
Vol.45,No.1
Characterization
of Herpes
Simplex
Virus
2
Temperature-Sensitive
Mutants
Whose
Lesions
Map
in
or
Near
the
Coding
Sequences for
the
Major
DNA-Binding Protein
ANNEE. SPANG, PAUL J.GODOWSKI, ANDDAVIDM. KNIPE*
Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Massachusetts 02115
Received 26 July1982/Accepted 4 October 1982
Bymarkerrescuewith clonedherpes
simplex
virus2DNAfragments,
wehave mapped thetemperature-sensitive mutations ofaseries ofherpes simplex virus 2 mutants to aregion
of the herpes simplex virus2 genomethatlies withinor near thecodingsequencesfor the majorDNA-binding
protein, ICP8.Incellsinfected withcertainofthese mutantsatthe nonpermissivetemperature,theassociationof themajorDNA-binding protein with the cell nucleuswasdefective.Inthese cells, theDNA-binding protein accumulated in the cytoplasmic and the crude nuclear detergent washfractions. At the permissive temperature, the maturation of the mutantICP8wassimilartothatof thewild-type viral protein. With the remainder of the mutants, the nuclear maturation of ICP8wassimilartothatencodedby the wild-type virusatthe nonpermissiveandpermissive temperaturesasassayed by cellfractionation.Herpes
simplex
virus(HSV)
encodesamajorDNA-binding protein
asabetaordelayed early
viral gene product (1, 12, 13, 23, 24, 29). This protein has been named ICP8 (29), VP130 (1), and ICSP 11/12
(24).
The geneencoding
this protein has been mapped at positions 0.38 to 0.41of the viralgenomeby
analysis
ofintertypic
recombinants (4, 18, 20), hybrid selection and in vitro translation ofmRNA (4), and analysis of defective viruses (10). The
major
DNA-binding
proteins encoded by both HSVtype 1 andHSV type 2
(HSV-1,
HSV-2)
bind moretightly
to denatured DNA than to native DNA(15,
23). Also, the protein encodedby
HSV-1 or HSV-2 can denaturepolydeoxyadenylic
acid-polyde-oxythymidylic acid, thereby
showing
DNA-melting activity
(22).Theprotein is transported
into
the cellnucleus (9, 21)througha seriesofstages andeventually binds to viral DNA in the infected cell (17).Initially
after synthesis the protein is found inthecytoplasm, but itquicklyassociateswiththe crude nuclear fraction. Detergent treatment of the crude nuclear fraction will
initially
release theDNA-binding protein, butlaterthe associa-tion becomesdetergent resistant. Atlatertimes, theproteincanbereleased fromthenuclei with DNaseI.TheconversiontotheDNase-sensitive form doesnotoccurwhenviral DNAreplication is blocked. Thus, the final stage of maturation appears to involvebinding
toreplicating
viral DNA.Wemappedatemperature-sensitive (ts)
muta-tion of
HSV-1,
tsHAl, withinor near the gene encoding ICP8 (4). Powell et al. (22)reported
that themajor DNA-binding proteinisdefective for theassociatedDNA-melting activity whenit is purified from cells infected with the HSV-2 mutant 186tsH9. Also, the cellular distribution of the DNA-binding protein detected by immu-nofluorescence maybe altered in cells infected with this mutant(22). Thus, these mutants
repre-sentthe bestcandidatesfor mutantsdefectivein the major DNA-bindingprotein.
In this
report,
we describe experiments in which we have physically mapped the ts muta-tions of a series of HSV-2 ts mutants to the regionofthegenomeencoding theDNA-binding protein. We also show that the association of ICP8 with the nucleus isdefective for certainof these mutants.MATERIALS ANDMETHODS
Cells and viruses. Vero cellmonolayer cultureswere used for the preparation and titration of virus stocks and for labeling infected cell proteins as described previously(17). Rabbit skin cells (16) were used for transfections. The origins of the HSV-2 virus strains were: 186 (25); 186tsB5 and 186tsAl (8); 186ts201 (D. Purifoy[UniversityofLeeds, Leeds, U.K.], per-sonalcommunication); IPB2tsl (31); andUW268ts19 (28). Seed stocks of these viruses were providedby Priscilla Schaffer, Harvard Medical School. Before use, the186ts+ strain of HSV-2 was plaque purifiedto give a uniform nonsyncytial population. The new clonal derivative wasdesignated HSV-2186syn+-1.
332
on November 10, 2019 by guest
http://jvi.asm.org/
HSV DNA-BINDING PROTEIN MATURATION 333
Molecular cloning of viral DNA. HSV-2 strain 186syn+-1 DNA was purified from infected cells by Nal equilibrium density gradientcentrifugation (16). HSV DNA and plasmid pBR322 DNAweredigested
withEcoRIandHindIlI (NewEnglandBiolabs, Bev-erly, Mass.). The endonucleases were inactivated at 70°C,and theDNAswereligated inareaction
involv-ing T4 DNA ligase(Bethesda Research Laboratories, Inc., Gaithersburg, Md.). The DNAswereintroduced by transformation (3) into the Escherichia coli K-12 strain MS372 obtained from M. Syvanen (Harvard Medical School).Ampicillin-resistant (Ampr), tetracy-cline-sensitive(Tets)transformantswereselected,and viralDNAinserts were screened with plasmidDNA preparations from small cultures(14).
Subclones of plasmidpEH60wereisolated by
cleav-ingpEH60 DNA withSall and ligatingtoSall-cleaved plasmid pBR325 DNA. This DNA was transformed into E. coli K-12 strain BC32 EndolP Thi- HsdR-HsdM+l(srl-recA)306,andchloramphenicol-resistant (Cmpr)Tets transformants wereselected.
Plasmid DNAfor markerrescue experiments was
obtainedbyCsClequilibrium density gradient centrif-ugation oflysates from plasmid-containing cells (6). Beforeextraction, plasmidDNAwasamplifiedin the cells by incubation in chloramphenicol (plasmid pBR322 vectors)orspectinomycin (pBR325 vectors).
Spectinomycin was provided by The Upjohn Co.,
Kalamazoo, Mich.
Marker rescue. Cotransfection of infected-cell ts mutantviralDNAs and uncleavedplasmidDNAs into rabbitskin cellswasperformedasreported previously (16).For theseexperiments, 0.5 ,ugeach ofmutantand plasmid DNAswereused.Thetransfection was
incu-bated at 37°C for 4to6days, and theprogeny virus wastitrated at33andat38or39°C.
Fractionation ofinfected cels. Infected cells were
labeledandfractionatedasdescribed previously(17).
Briefly, infected cellswerebroken byDounce
homog-enization. The crude nuclear pelletwascollected by
centrifugation; the supernatantrepresented the cyto-plasmic fraction. The crude nuclear pelletwastreated with a solution containing 1% Triton X-100-0.5% deoxycholate. After further centrifugation, the pellet represented the nuclear fraction, andthesupernatant represented the crude nuclear detergentwashfraction. Proteins were recovered from the supernatants by acetoneprecipitation. Proteins fromallfractionswere
analyzed on9.25% polyacrylamide gels asdescribed
(17).
RESULTS
Molecular cloning of HSV-2 DNA fragments. Toobtainaseries ofplasmids containing HSV-2
DNA inserts,wecleaved plasmid pBR322 DNA
with both EcoRI and HindIlI and religated the plasmid DNA withalargemolarexcessof
HSV-2 strain186 DNApreviously cleaved with EcoRI andHindIII (Fig. 1). The DNA molecules in the ligation mixture were introduced into E. coli by
transformation, and Ampr transformants were selected. Approximately 90% of the Ampr
trans-formants wereTets, indicatinga DNA insertor
alteration in the tetracycline resistance gene. Plasmid DNAwasprepared from the Ampr Tets
cellsandwascleaved with EcoRI andHindlllto
I B N H E A OJ M L K
J O G LTS H V F E I R K M OPUN
III I 11 ,,, ,
F J I G A P L H K NOM
0 0.2 OA 0.6 oA.
A
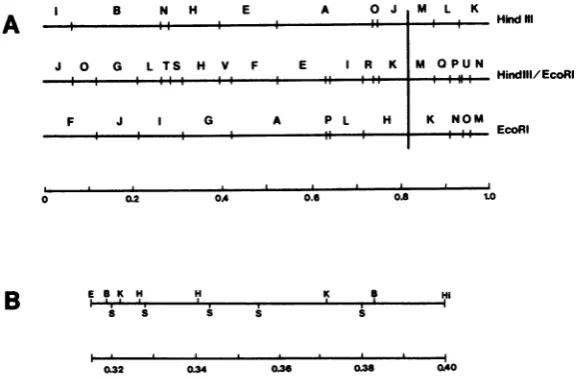
[image:2.489.101.390.397.586.2]B
FIG. 1. Restriction endonuclease cleavage maps of DNA of HSV-2 strain 186. (A) EcoRI and HindIll cleavagemaps,includingfragments generated by cleavagewithbothenzymes.Themapsarederivedfromthose of H.G. Hayward,T.G. Buchman,and B. RoizmanascitedbyMorseetal.(19)forHSV-2(G)DNA and from those ofCortini and Wilkie(5)forHSV-2(HG52)DNA. The HindIII-EcoRImappresentedwasshowntobesimilarto
thatpublished previously by Wilkieetal. (30). Mappositionsof 0to0.83representtheLcomponent;0.83to1.0 representtheScomponent.(B) CleavagemapoftheHSV-2 DNAfragmentinsert inplasmidpEH60 (0.31to0.40
mapunits).Abbreviations:S, SalI; B, BgllI; Hi, HindlIl; E, EcoRI; H, HpaI; K, Kpnl.TheSallcleavagesites definetheboundariesof theSallsubclonesdescribed in Table 1. The HSV DNAfragmentinplasmid pSGhas notbeenpositionedonthismap.
HindNI
lindlil/EcoRI
EcoRI
E B K H H K B HI
**, .I . IV
.
0.32 034 0.36 0.38 0.40
VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
334 SPANG, GODOWSKI, AND KNIPE
B
B 2:E ..A
B
\.t.
.
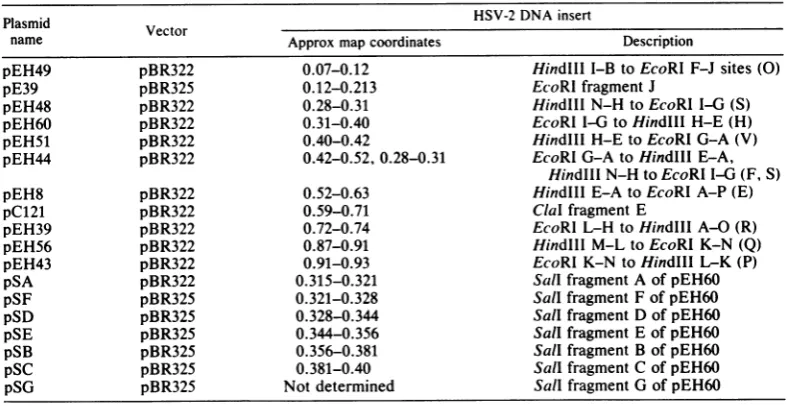
FIG. 2. Characterization of viral DNA fragments inserted in plasmids usedinthis study. (A)HindlIl-EcoRl fragmentinserts. DNAsfromHSV-2 strain186and fromplasmidsweredigested withHindlllandEcoRIand
weresubjectedtoelectrophoresisinan0.6%agarosegel. Lanes 1 and 10,strain 186 DNA; lane 2, pEH44; lane3, pEH60;lane 4,pEH49;lane5,pEH43;lane6,pEH56;lane7,pEH39;lane8,pEH48;lane9,pEH51.Note that plasmidpEH51 containsinsertsoftwosizes,aminoronecomigratingwith fragment Vandamajoroneslightly
smaller,probablyadeletedformof V (see text).(B)Sallsubclones ofplasmid pEH60. pEH60DNAand DNA
fromeach ofthesubclones containing Sallfragments ofpEH60weredigestedwithSall andweresubjectedto electrophoresisina1.2%agarosegel.Lanes 1 and 9,pEH60;lane2, pSA;lane 3,pSB;lane 4,pSC;lane 5,pSD;
lane6, pSE;lane7, pSF; lane8,pSG.
identify the plasmids with DNA inserts. Nearly all showed DNA inserts that comigrated with authentic HindIII-EcoRI DNA fragments of HSV-2 DNA (Fig. 2A). The identities of the
DNA inserts were verified by cleavage with other restriction endonucleases (data not
shown). Table 1 and Fig. 2 describe the series of plasmids used in this study.
TABLE 1. PlasmidscontainingHSV-2DNAinserts
Plasmid HSV-2 DNA insert
Vector
name Approx map coordinates Description
pEH49 pBR322 0.07-0.12 Hindlll I-BtoEcoRI F-J sites(0)
pE39 pBR325 0.12-0.213 EcoRlfragmentJ
pEH48 pBR322 0.28-0.31 HindIllN-HtoEcoRI 1-6 (S)
pEH60 pBR322 0.31-0.40 EcoRl1-6 toHindlIl H-E(H)
pEH51 pBR322 0.40-0.42 HindlllH-EtoEcoRI G-A(V)
pEH44 pBR322 0.42-0.52,0.28-0.31 EcoRIG-AtoHindlllE-A,
HindlllN-H toEcoRII-6(F,S)
pEH8 pBR322 0.52-0.63 HindIllE-A toEcoRI A-P(E)
pC121 pBR322 0.59-0.71 CialfragmentE
pEH39 pBR322 0.72-0.74 EcoRI L-H toHindlIlA-O(R)
pEH56 pBR322 0.87-0.91 Hindlll M-LtoEcoRl K-N(Q)
pEH43 pBR322 0.91-0.93 EcoRI K-N toHindIII L-K(P)
pSA pBR322 0.315-0.321 Sall fragmentAofpEH60
pSF pBR325 0.321-0.328 SallfragmentFofpEH60
pSD pBR325 0.328-0.344 SallfragmentDofpEH60
pSE pBR325 0.344-0.356 SallfragmentEofpEH60
pSB pBR325 0.356-0.381 SallfragmentBofpEH60
pSC pBR325 0.381-0.40 SallfragmentC ofpEH60
pSG pBR325 Notdetermined SallfragmentGofpEH60
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.489.107.394.70.308.2] [image:3.489.54.451.467.670.2]HSV DNA-BINDING PROTEIN MATURATION 335
Onesetofplasmidswarrantsfurthercomment
in that they all contained similar mixtures of inserted fragments. The largest insert fragment comigrated with the HindIII-EcoRI-V
frag-mentoriginating from 0.40 to0.42 map units in the L component. Theotherfragments were of various sizesslightly smaller than this fragment (seeplasmid pEH51 in Fig. 2A). Furtherpassage
of the cells caused a decrease in the amountof the insert comigrating with the authentic HSV fragment (datanotshown), and thus, the smaller fragments appearedtobe from plasmidsdeleted inthe HSV DNA insert. Othershave also identi-fieddeletions in cloned HSV-2 DNA fragments originatingnearmapposition 0.40 (G.Hayward,
personal communication).
We also cloned portions of the HSV-2genome
byusing other cleavage sites inaplasmidvector.
For the work reported here we have included
EcoRI fragment J and ClaI fragment E. Wehave
notdefined the entiremapforcleavage of HSV-2 DNA with ClaI. However, cleavage of the pC121 plasmid (ClaI fragment E) with BamHI yielded BamHI fragment A of HSV-2 DNA. This result, along with othercleavage data, has defined the approximate map positions of ClaI
fragment Eas0.59to0.71 mapunits.Thus, this
setof cloned DNA fragments (Table 1) contains nearly all of the uniquesequences of the HSV-2
genome.
Markerrescueof HSV-2tsmutantsin comple-mentationgroups2-2and 2-7.For markerrescue
studies, we selected a series of HSV-2 ts
mu-tants that were good candidates for mutants
having lesions in the ICP8 gene. As stated
above, others have reported that the mutant
tsH9 encodes a defective ICP8 molecule (22).
Thismutantfails tocomplement mutants inthe 2-2and 2-7complementationgroups(7, 26). We
therefore attempted to map the lesions in the
mutants of thesegroups. We used the plasmid
DNAs containing HSV-2 ts+ DNA inserts for marker rescue by the cotransfection protocol
(16).
We found that transfection of HSV-2 mutant
DNAs at thepermissive temperature, 34°C, led
toreiatively low titers of virus and ts+
recombi-nants. To determine the most efficient condi-tions forrescue of HSV-2 ts mutants, we incu-bated a set of transfected cultures at different
temperatures(Table 2). Markerrescuewas more efficient and resulted in higher titers of ts+ recombinants when the transfected cultures
were incubated at highertemperatures such as 37or38'C. For thisreason, mostofour transfec-tionswereperformed at37'C. Thistemperature
is permissive for some ts mutants and
nonper-missive for others.
We have not successfully mapped the tsH9 mutation because viral DNApurified from cells
infected with the mutant tsH9 had little or no infectivity by transfection. Infact, the addition of tsH9DNA to atransfection mixture contain-ing infectious HSV-2 ts+ DNA caused a de-creaseintheinfectivity ofthe ts+ DNA(datanot shown). Viral DNA from cells infected with strain UW268ts19 showed infectivity but could not be rescued by the addition of ts+ DNA fragments.
Wemapped the tsmutation in strain 186tsAl by markerrescue.The mutant wasconvertedto
Ts'
by cotransfection of mutant DNA with plasmid pEH60 DNA containing the HSV-2 DNA sequences from 0.31 to 0.40 map units (Table 3). Themutation wasfurther mapped by the use of the Sall subclones of the pEH60 plasmid. Only the pSB plasmid (0.356to 0.381 map units) rescued the tsA1 mutant (Table 3). Similarly, the pEH60 plasmid rescued the mu-tant strains 186ts178 and 186ts39(Table 4). The pSB plasmid rescued both mutants ts178 and ts39(Table 4). Themutations have been placed intheorderts39-ts178-tsAlby two-factor genet-ic crosses (7). These mutations lie within map coordinates 0.356to0.381 (Table 1).The HSV-2 IPB2tsl mutant was rescued
by
thepEH60plasmid (Table 3). However, we did observe inoneexperimentamuch less efficient rescue by the pEH51 plasmid, which yielded very small plaques at the nonpermissive tem-perature (datanot shown). This appearedto be duetoselection ofapartialrevertant. Thus, the lesion in tsl maps between 0.31 and 0.40 map units. Thetsl mutant was alsorescued by plas-mid pSC (0.386 to 0.40 map units), so this mutation maps to the right of the others de-scribed above.
Marker rescue of strain 186tsB5. The mutant 186tsB5 is in complementation group 2-3 (26), the same group as strain HG52ts6, which con-tainsamutationdefinedasencodinga thermola-bileDNA polymerase (11) and mappingat posi-tions0.4 to0.41mapunits (2).Torelate themap
positions
of thepolymerase
gene in HSV-2 toTABLE 2. Effect of temperature of incubationon
efficencyofrescue
Temp of Titer
DNA incubation
(OC) 33°C 380C
tsB5 33 3.7 x 104 <101
37 2.5 x 103 <101
38 <lo <10'
tsB5 +HsuI 33 2.1 X 103 3.6 x 102
digestofts+ 37 3.6 x 105 1.6 x 105
DNA 38 2.8 x 105 1.4 x 105
HsuI digest 37 <10l <101
of ts+ DNA VOL.45, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.489.252.447.545.674.2]336 SPANG, GODOWSKI, AND KNIPE
TABLE 3. Marker rescue of strains 186tsAl and IPB2 tsl Titers Clonedfragment added
tsA1 tsl
Name Approx map 39°C 330C 39°C 330C
coordinates
None <lo 6.2 x 105 <102 107
pEH49 0.07-0.12 <lo 7.4 x 105 <102 1.6 x107
pE39 0.12-0.21 <103 108 <102 1.3 x 107
pEH48 0.28-0.31 <10' 7.2 x 10' <102 7 x 106
pEH60 0.31-0.40 5.4 x 106 6x 107 1.9 x 105 5.7 x 106
pEH51 0.40-0.42 <lo 5.6 x 104 <102 5.6 x 106
pEH44 0.42-0.52 <lo 1.8 x tO' <102 7 x 106
pEH8 0.52-0.63 <lo 3 x 104 <102 1.1 X 107
pC121 0.59-0.71 <lo 2.4 xtO' <102 1.5 x
107
pEH39 0.72-0.74 <lo 2.6 x104 <102 6 x106
pEH56 0.87-0.91 <lo 1.6x 10' <l12 1.3 X 107
pEH43 0.914.93 <lo' 3 x 10' <102 8 x106
pSA 0.315-0.321 <lo 4.2 x104 <102 107
pSF 0.321-0.328 <lo 9x 102 <102 4 x 106
pSD 0.328-0.344 <lo 1.5 x 104 <103 1.5 x 107
pSE 0.344-0.356 <lo 2.2 x 103 <102 107
pSB 0.356-0.381 3.4 x 104 1.6 x 106 <lo2 1.2 x 107
pSC 0.381-0.40 <lo 4.8 x 104 2.1 x 104 8 x 106
pSG Notdetermined <lo 1.1 x 10' <102 9x 106
those of the mutations described above, we HG52ts6(2)showsanoverlapbetweenthemap mapped tsB5 by marker rescue. The tsB5 mu- positions of these two mutations. Thus,themap tantwasrescued onlybytransfection withplas- positions of these putative polymerase muta-midpEH51 DNA (0.40 to 0.42 map units; Table tions agree, and the physical mapping of the 5). Comparison ofthe restriction endonuclease entire groupof mutations approximates the ge-cleavage sites bounding the pEH51 insert with netic mapping of the mutations in having the the map position of the sequences rescuing order(ts39, ts178,
tsAl)-tsl-tsB5.
TABLE 4. Markerrescueof strains 186ts39 and186tsl78 Titer Clonedfragmentadded
ts39 ts178
Name Approx mapcoordinates 380C 330C 380C 330C
None <lo <lo <101 <10'
pEH49 0.07-0.12 <lo
<lo,
<10' 10'pE39 0.12-0.21 <10' <10' <10' 10'
pEH48 0.28-0.31 <lo <lo <lo' lo
pEH60 0.31-0.40 5.1 x 106 1.0 x 107 7 x10' 6 x 106
pEH51 0.40-0.42 <lo <lo <lo <10'
pEH44 0.42-0.52 <10' <lo' <lo' <10'
pEH8 0.52-0.63 <10' <lo' <lo' <10'
pC121 0.59-0.71 <10' <lo' <10' <10'
pEH39 0.724.74 <10' <lo <lo <10'
pEH56 0.87-0.91 <10' <lo <lo <10'
pEH43 0.91-0.93 <10' <o1' <10' <10'
pSA 0.315-0.321
<lo'
<lo'
<lo' <10'pSF 0.321-0.328 <lo <10' <lo' <10'
pSD 0.328-0.344 <10' <10' <10' <10'
pSE 0.344-0.356 <10' <10' <10' <10'
pSB 0.3560.381 1.4 x 107 2.3 x 107 3.8x 106 1.4 x 107
pSC 0.3814.40 <10' <10' <10' <10'
pSG Notdetermined <10' <10' <10' <10'
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.489.54.446.442.674.2]HSV DNA-BINDING PROTEIN MATURATION 337
TABLE 5. Markerrescueof strain 186tsB5
Cloned fragmentadded Titer
Approx
Name map 39°C 330C
coordinates
None <10' 1.3 x 105
pEH49 0.07-0.12 <lo 4.6 x 104
pE39 0.12-0.21 <lo 9.5 x 103
pEH48 0.28-0.31 <lo 1.2 x 105
pEH60 0.31-0.40 <lo 3.4 x 103
pEH51 0.40-0.42 1.2 x 106 3.6 x 106 pEH44 0.42-0.52 <lo' 4.8 x104
pEH8 0.52-0.63 <lo' 4.2 x 104
pC121 0.59-0.71 <lo 4.8 x 104
pEH39 0.72-0.74 <lo 8.6 x 104
pEH56 0.87-0.91 <lo 3 x 104
pEH43 0.91-0.93 <lo' 6 x 104
Nuclear association of ICP8 in cells infected with mutant viruses. As the first step in the phenotypic characterization of these mutants, weexamined the nuclear accumulation ofICP8 in cells infected with the mutant viruses. We usedacell fractionation protocol described pre-viously (17) and above. We examined nuclear
ts+ tsAl ts178 ts39 A
localizationin the presence ofphosphonoacetate because (i) some of the mutant viruses are defective for DNA replicationatthe nonpermis-sivetemperature (26) and(ii) inhibitionof DNA replication increasesthe rateofnuclear localiza-tion of ICP8 (17; A. Spang and D. Knipe, unpublished results). Thus, inhibition of viral DNA replication by phosphonoacetic acid in cells infected with wild-type and with mutant viruses allows a valid comparison of the two typesofinfected cells.
The ICP8 encoded by mutants tsAl, ts178, andts39 accumulated in the nuclearfraction at 33or39°Catapproximately thesamelevelsasin cells infected with ts+ virus (Fig. 3). Thus, the maturation appeared to benormalfor the ICP8 protein encodedby these mutants.
Incontrast, in cells infected with strain tsH9 at 39°C, very low levels of nuclear associated ICP8 were observed (Fig. 4). ICP8 was largely observed in the cytoplasmic and nuclear deter-gentwashfractions (Fig.4). By microdensitom-etryandplanimetry,we determinedthe amount of ICP8 in each subcellular fraction. The per-centages of the total ICP8 at 39°C were as follows. ts+: Nuclear,
55%;
cytoplasmic, 19%;ts tsAl tsl78 ts39 B
-5 -8 -5
T*..'.
_1_ -._ -20
-20
-26
*-36
-36 .i.S-...-...
iN vv
*rf
'$t
..<wiz.nX...rt
K\|2 :.<,;>4.e>**'',,,*w#0,..;i...
_w
N C D N C D N C D N (: CX N C
D N C D N C D N C D
FIG. 3. Nuclear localization ofICP8 encodedbystrainstsAl, ts178,andts39.Cells infectedat39°C (A)or
33°C(B) with strain186syn+-1orthemutantviruses inthe presenceof300
p.g
of sodiumphosphonoacetateper ml were labeled with[35S]methionine for 15 min at 4hpostinfection. The cultures were subjected to chase conditions for1 handthenwerefractionated. Abbreviations: N, nuclearfraction;C,cytoplasmic fraction; D,crudenucleardetergent wash fraction. Shownaretheautoradiogramsofthegelsinwhich thevariousfractions weresubjectedtoelectrophoresis.
VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
338 SPANG, GODOWSKI, AND KNIPE
390
30
0 here. In cells infected with strain tsH9, the ICP8
---tsH9protein
did chaseslowly
into the crude nuclear_.ts
ts_
tsH9 ts detergent wash fraction butonly
chased to alimited degree into the nuclear fraction. These IcP results wereconsistent with thewild-type
HSV-2 ICP8 following the pathway: cytoplasm crude nuclear
detergent
wash fraction -* nucle-us, with the mutant protein being slow in the first transitionand blocked in the second. These results are consistent with those of Powell et al. (22), who reported that in cells infected with* b b - 5 strain
tsH9,
ICP8 showsan altered distribution.. -8 by immunofluorescence.
A verydifferent situationwasobserved inthe analysis ofthe maturation of ICP8 encoded by - 20 strains tsl andtsl9. We observed that fraction-ationof cells infected at39°C with these mutants ledto analtereddistribution ofanumberof viral proteins in the subcellular fractions at 5 to 6 h
..
-6
postinfection (Fig. 6A). Several virus-specificproteins normally in the cytoplasmic fraction, e.g., ICP26 and 36, were isolated with the
nucle-36 arfraction from the mutant-infected cells
(Fig.
-_<x_36 6A). We alsoobserved that fractionation of cells
at 5 to 6 h
postinfection (39°C)
with theseN C D N C D N C NN C D
FIG. 4. Nuclear localization of ICP8 encoded by strain tsH9. Cells infected with strain 186syn+-1 or tsH9 at 33 or390C were labeled and fractionated as described in the legend to Fig. 3. Shown is the autoradiogramof the gel in which the various fractions weresubjected toelectrophoresis. Abbreviations are explained in thelegendtoFig. 3.
detergent wash, 23%. tsH9:Nuclear, 20%; cyto-plasmic, 50%; detergent wash,20%. At330C the maturation of the tsH9 ICP8 to the nuclear fraction was slightly less than thatofthe wild-type protein but appeared to be nearly normal. Thedistributionof ICP8at33°Cwas asfollows. ts+: Nuclear,61%; cytoplasmic, 25%; detergent wash, 15%. tsH9: Nuclear, 44%; cytoplasmic, 28%; detergentwash,28%.
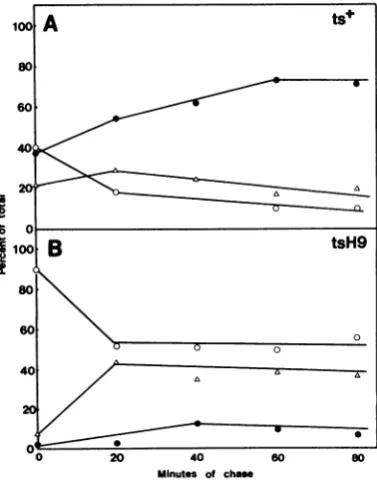
Toexamine the kinetics ofthe maturation of the tsH9 ICP8protein at390C, we fractionated cellsatvarioustimesduringachaseperiod (Fig. 5). After a 15-min chase period, we observed someofthewild-typeviralICP8 already associ-ated with the nuclearfraction. During the chase period, the amount in the cytoplasmic fraction declined, the amount in the detergent wash fraction increased transiently, and ICP8 then accumulatedinthe nuclearfraction. Thispattern is very similar to thatpreviously described for HSV-1 ICP8(17), the differencespossiblybeing attributable to the longer labeling period used
"S 0
Z
o-ioo
B
tsH9
80
60
40 f A
20 /
40
0 20 40 60 80
Minutes of chase
FIG. 5. Kinetics of ts+ andtsH9 ICP8 maturation at390C.Cultures of cellsinfectedat39°Cwere labeled for 15min, chasedforthe times indicated, and frac-tionated. Theproteins in each fraction wereanalyzed by electrophoresis. Theresulting autoradiogramwas scanned with a Joyce-Loebl microdensitometer, and peakareasweredeterminedby planimetry. Shownare thepercentages of totalICP8in thefractions fromthe nucleus (0), cytoplasm (0), and nuclear detergent wash(A).
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.489.63.230.77.368.2] [image:7.489.257.446.340.583.2]HSV DNA-BINDING PROTEIN MATURATION
A
ts
tsl
B
ts+
C
ts
' .4_
"niw -5
= -8
-26
-36
r_-N C D N C D
.C C.. 0. _
N C D N C D N C D N C D
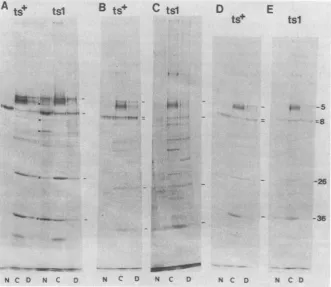
FIG. 6. Maturationof ICP8in cellsinfectedwithstraintsl. Cells infected withts+ortslvirusesatdifferent temperatureswerelabeled for15min, chased for60min,andfractionatedatdifferenttimesasfollows. (A)
ts+-and tsl-infected cells (39°C), fractionated at 5 h postinfection. Note the additional bands (U) and high backgroundin thetsl nuclearfraction. (B)ts+-infectedcells(39°C), 2.5 hpostinfection. (C)tsl-infected cells (39°C), 2.5 h postinfection. (D) ts+-infected cells (33C), 6h postinfection. (E)tsl-infected cells (33°C), 6h postinfection.
mutants yielded cell nuclei with irregular edges
as shown by phase microscopy (data not
shown). This suggested that these nuclei had
cytoplasmic fragments still attachedtothem. In contrast, the nuclei of cells infected with wild-type virus showeda smooth sphericalstructure
after detergent treatment. Therefore, the struc-ture or fractionation of these mutant-infected
cells appeared to be abnormal at 5 to 6 h
postinfection. However, the structure of the
nuclei from mutant-infected cells at 2 to 3 h
postinfection appeared similar to that of nuclei
from cells infectedwithwild-type virus (datanot
shown). We therefore labeledcellsinfectedwith
wild-type and tsl mutantvirusat1.5 h postinfec-tion and chased for1 h. Undertheseconditions, we observed comparable patterns of viral pro-tein bands in the subcellular fractions of cells infected with wild-type and with tsl viruses,
except that the tsl ICP8 did not accumulate in thenuclei(Fig. 6BandC). Instead, itwasfound
in thecytoplasmic and crude nuclear detergent washfractions. These resultssuggestthat thetsl
ICP8 protein accumulated in the nonnuclear
fractions and that this localizationorsomeother factor led to abnormal fractionation ofcells at
later times postinfection. Upon fractionationof
cells infected at33°C,we observed similar
pro-teinpatterns in cellfractions from cells infected with ts+ ortsl virus (Fig. 6D and E). Nuclear transport of tsl ICP8 may be slightly less than
that of ts+ virus but is nearly normal at 33°C. Similar results to those for strain tsl were
ob-served with cells infected with strain tsl9(data
notshown). However, nucleartransportof ICP8 comparable with that of the wild-type protein was observed in cells infected with strain tsAl and fractionated at 3 h postinfection (data not
shown).
We have observed that nuclear transport of
ICP8 is comparable with that of the wild-type proteinat39°Cin cells infectedwith straintsB5
or tsA8 (6 h postinfection; data not shown). Thus, the only mutants which are defective in
nuclearassociation of ICP8 as assayed by cell
fractionationare tsH9, tsl, andtst9.
D
E
tS+
tsl
_~~~~~~~~
."r- *ft"
_.-..W.
339 VOL.45, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.489.78.410.77.364.2]340 SPANG, GODOWSKI, AND KNIPE
DISCUSSION
Wehave mapped by markerrescuethelesions ofa numberof HSV-2tsmutantsin complemen-tation groups2-2 and 2-7.These mutations map between positions 0.356 to 0.40 of the viral
genome. These map positions overlap the map
positions previously reported for the coding
sequences of the major DNA-binding protein,
ICP8, and for the mutant tsHAl (4). We have alsoshownthat thenuclearlocalization of ICP8 is defective in cells infected with three of these
mutants atthenonpermissivetemperature. Pow-ell et al. (22) have shown that the cellular distribution of the DNA-binding protein is al-tered and that DNA-melting activity of the DNA-binding protein is defective with one of
these mutants, tsH9. Thus, the simplest inter-pretation is that some or all of these mutants
encodea defective DNA-binding protein.
Physical mapping of mutations. Dixonetal. (7) have recently shown that themutantsin comple-mentationgroups2-2 and2-7 failtocomplement and show a complex pattern of overlapping complementation withaseriesof othermutants.
Bytwo-factor geneticcrossesthey haveordered
themutations into a lineararray: tsB5-group 1
(tsH9, tsl, tsl9,ts42082)-group 2 (tsAl,
tsA8)-group3 (ts178, ts201)-group 4 (ts39). We have mapped mutations from groups 2 (tsA1), 3
(ts178), and 4 (ts39)tosequenceswithinacloned DNA fragment of molecular weight 2.6 x 106
(plasmid pSB; 0.356 to 0.381 map units). We
rescued the tslmutation(group 1) withacloned
fragment arising from 0.381 to 0.40 map units
and the tsB5 mutation with a cloned DNA fragmentfrom 0.40to0.42mapunits.Thus,our
physical mapping has yielded the order (ts39, ts178, tsAl)-tsl-tsB5 from left to right on the
prototype form of the genomic DNA (Fig. 1). This is consistent with the order of the genetic
mapfrom right toleft asdescribed above.
Defects in nuclear localization ofICP8. Three
of themutantsin thegroup1clusterwereshown to have a defect in the maturation of ICP8 to
the cell nucleus at the nonpermissive tempera-ture. Because the tsl mutation maps to a
posi-tion close to or within the ICP8 coding
se-quences, it seems likely that this phenotype is
dueto adefect inthe ICP8molecule itself. We
have been unable to map the tsH9 or tsl9
mutations physically; however, two-factor
crosses have shown that these mutations map
close to the tsl mutation (7). Thus, these also appear to have defects in the ICP8 molecule. Thegroup2mutantsshownoapparentdefectin
ICP8 localization as assayed by cell
fraction-ation. These mutantsclearlyfailtocomplement thegroup1mutants;therefore,bothgroupsmay
have defects in the ICP8protein.Further studies
must be performed by immunofluorescence or otherfractionation techniquestobe certain that the ICP8 proteins encoded by the group 2 mu-tants are localized to the same site in the nucleus asthewild-type protein. In this regard, Powellet al. (22) have reported that the DNA-binding proteinislocatedin thenucleus of cells infected withgroup 2 mutants at39°C. Our fractionation studies are consistent with this.
This analysis suggests that there may be at least two clusters of mutations which define different mutant phenotypes associated with ICP8. Three of the group 1 mutants show a defect in ICP8 localization, whereas those in group 2 show no defect in ICP8 localizationas determined by cell fractionation. We have simi-larresults withHSV-1 ts mutantsof complemen-tation group 1-1 in that some show defects in transportbutothers donot.Some of themutants whichare not defective for nuclear association exhibitadefect in thefunction of ICP8 in the cell nucleus (C. Lee and D. Knipe,
manuscript
in preparation). Thus,mutantswhicharedefective inICP8functionmayshow different phenotypes ordefects in differentstagesofnuclear localiza-tion.We havepreviously reported the pathway for maturation of the HSV-1 ICP8moleculeas
cyto-plasm
-*crude nucleardetergent
wash -+nucle-us (17). A similar result was observed for the maturation of the HSV-2 ts+ ICP8 protein. However, this pathway has been altered in cells
infected
with strain tsH9. In thesemutant-infect-ed
cells,
thechaseof
ICP8 from the cytoplasmicfraction is slow and
incomplete,
and the transi-tion from the detergent wash fractransi-tion to the nuclear fraction is almost nonexistent. Thus, ICP8maybealmosttotally extranuclear in cells infected with strain tsH9 at39°C. These kinetic studies with tsH9-infected cells support thepathway previously
reported (17) withablock innuclear
localization.
These resultsareconsistent with those of Powell et al. (22), who observed cytoplasmicfluorescence
of ICP8 in thesemu-tant-infectedcells at39°C.
Abnormal fractionation of cells infected with mutant tsl. Subcellular fractions derived from cells infected at 39°C with strain tsl or tsl9 exhibited abnormal distributions of viral pro-teinsfrom cells fractionatedatlatertimes(5 to 6 h)postinfection. Many proteins notusually as-sociated with the nuclearfraction of cells infect-edwithwild-typevirus werefound inthe nucle-arfraction of the mutant-infectedcells.Thiswas observedonlyatthenonpermissivetemperature andthusappearedtobe relatedtothetslesions. Analysis ofts+ revertants is neededtoconfirm this correlation. However, at early times of
infection,
the mutant-infected cells fractionatedsimilarly
to cells infected withwild-type
virus,
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
HSV DNA-BINDING PROTEIN MATURATION 341
exceptthat themutantICP8 was notlocalizedto
the cell
nucleus. In the mutant-infected cells atthe nonpermissive temperature, nuclear local-ization of ICP8 appears to be defective, and thereis aprogressive change in the structure of themutant-infected cellswhich alters their frac-tionation. Further work is in progress to exam-ine the locationof ICP8 in these mutant-infected cells at 39°C. Other experiments have shown that the HSV-1 ICP8 isextracted with the deter-gent-insoluble cellular framework (M. Quinlan and D. Knipe, manuscript in preparation). Thus, itis possible that the tsl ortsl9 ICP8 molecule may accumulate on the cytoplasmic framework ina solubleor insoluble formand alterthe way infectedcellsfractionate intonucleus and cyto-plasm. Immunofluorescencestudies of the cellu-larlocation of the ICP8 coded by mutants tsH9, tsl, and tsl9 should allow us to define further thecytoplasmic maturation sites for ICP8 within theinfected cell.
ACKNOWLEDGMENTS
We thank Priscilla Schaffer for communication of unpub-lished results,Abbott Laboratories, North Chicago, Ill., for a gift ofphosphonoacetate, and Rosemary Bacco for prepara-tion of the manuscript.
These studies were supported by Public Health Service grantCA26345 from the National Cancer Institute. P.J.G. is a predoctoraltrainee supported by National Institutes of Health training grant5T32GM071%. D.M.K. is a Cancer Research Scholarof the Massachusetts Division of the American Can-cerSociety.
LITERATURE CITED
1. Bayliss, G. J., H. S. Marsden, and J. Hay. 1975. Herpes simplex virus proteins: DNA-binding proteins in infected cells and in the virus structure. Virology 68:124-134. 2. Chartrand, P., N. D. Stow, M. C.Timbury, and N. M.
WUlkie. 1979. Physical mapping of paar mutations of herpes simplex virus type 1 and type 2 by intertypic markerrescue. J. Virol. 31:265-276.
3. Cohen, S. N., A. C. Y. Chang, and L. Hsu. 1972. Nonchro-mosomal antibiotic resistance in bacteria: genetic trans-formation of E. coli by R-factor DNA. Proc. Natl. Acad. Sci. U.S.A.69:2110-2114.
4. Conley,A.J.,D. M.Knipe,P.C. Jones,and B.Rolzman. 1981. Molecular genetics of herpes simplex virus. VII. Characterization of atemperature-sensitive mutant pro-duced by in vitro mutagenesis and defective in DNA synthesisand accumulation of y polypeptides. J. Virol. 37:191-206.
5. Cortini, R., andN. M. WLUkie. 1978. Physical maps for HSV type 2 DNA with five restrictionendonucleases. J. Gen.Virol. 39:259-280.
6. Davis, R., J. Roth, and D. Botstein. 1981. Advanced bacterial genetics. Cold Spring Harbor Laboratory, Cold SpringHarbor, N.Y.
7. Dixon,R. A.F., D. J.Sabourin,and P. A.Schaffer.1983. Geneticanalysis oftemperature-sensitive mutants which define the genes for the major herpes simplex virus 2 DNA-bindingprotein and a new late function. J. Virol. 45:343-353.
8. Esparza, J., D. J. M. Purifoy, P. A. Schaffer, and M.
Benyesh-Melnick. 1974. Isolation, complementation and preliminary phenotypiccharacterization of temperature-sensitive mutants ofherpes simplex virus type 2. Virology 57:554-565.
9. Fenwick,M. L., M. J. Walker, and J. M. Petkevich. 1978.
On the association of virus proteins with the nuclei of cells infected with herpes simplex virus. J. Gen. Virol. 39:519-529.
10. Frenkel,N., H. Locker, and D. A.Vlazny.1980. Studies of defective herpes simplex viruses. Ann. N.Y. Acad. Sci. 254:347-370.
11. Hay, J., H. Moss, A. T. Jamieson, and M. C. Timbury. 1976. Herpesvirus proteins: DNA polymerase and pyrimi-dine deoxynucleoside kinase activities in mutants of her-pes simplex virus type 2. J. Gen. Virol. 31:65-73. 12. Honess, R. W., and B.Roizman. 1973. Proteinsspecified
by herpes simplex virus. XI. Identification and relative molar rates of synthesis of structural and nonstructural herpes virus polypeptides in the infected cell. J. Virol. 12:1347-1365.
13. Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus macromolecular synthesis.I.Cascade regula-tion of the synthesis of three groups of viral proteins. J. Virol. 14:8-19.
14. Isberg, R., and M. Syvanen. 1981. Replicon fusions pro-moted by the inverted repeats ofTn5: the right stem is an insertion sequence. J. Mol. Biol. 150:15-32.
15. Knipe, D. M., M. P. Quinlan, and A. E. Spang. 1982. Characterization of two conformational forms of the ma-jor DNA-binding protein encoded by herpessimplexvirus
1.J. Virol. 44:736-741.
16. Knipe, D. M., W. T. Ruyechan, and B. Roizman. 1979. Molecular genetics of herpes simplex virus. III. Fine mapping of a genetic locus determining resistance to phosphonoacetate by two methods of marker transfer. J. Virol.29:698-704.
17. Knipe, D. M., and A. E. Spang. 1982. Definition of a series of stages in the association of two herpesviral proteins with the cell nucleus. J. Virol. 43:314-324.
18. Marsden, H. S., N. D. Stow, V. G. Preston, M. C. Tim-bury, and N. M.Wilkie.1978. Physical mapping of herpes simplexvirus-induced polypeptides. J. Virol.28:624-642. 19. Morse, L. S., T. G. Buchman, B. Roizinan, and P. A. Schaffer. 1977. Anatomy of herpes simplex virus DNA. IX. Apparentexclusion of some parental DNA arrange-ments in the generation of intertypic (HSV-1 x HSV-2) recombinants. J. Virol. 24:231-248.
20. Morse, L. S., L. Pereira, B.Roizman, and P. A. Schaffer. 1978.Anatomy of herpes simplex virus (HSV) DNA. X. Mapping ofviral genes by analysis of polypeptides and functions specified byHSV-1 x HSV-2 recombinants. J. Virol.26:389-410.
21. Pereira, L., M. H. Wolff,M. Fenwick, and B. Roizman. 1977. Regulation of herpesvirus macromolecular synthe-sis. V. Properties of alpha polypeptides made in HSV-1 and HSV-2infected cells.Virology77:733-749. 22. Powell, K. L., E. Littler, and D. J. M. Purifoy. 1981.
Nonstructural proteins of herpes simplex virus.II. Major virus-specific DNA-binding protein. J. Virol. 39:894-902. 23. Powel, K.L., and D. J. M. Purifoy. 1976. DNA-binding proteins of cellsinfected by herpes simplex virus type 1 andtype 2. Intervirology 7:225-239.
24. Purifoy,D. J. M., and K. L. Powell. 1976. DNA-binding proteins induced by herpes simplex virus type 2 in HEp-2 cells.J. Virol. 19:717-731.
25. Rawls, W.E., D. Laurel, J. L.Melnick,J. M.Glicksman, and R. H. Kaufman.1968. A search for viruses in smeg-ma, premalignant and early malignant cervical tissues. The isolation of herpesviruses with distinct antigenic properties. Am. J. Epidemiol. 87:647-655.
26. Schaffer, P. A., V. C.Carter, and M. C. Timbury. 1978. Collaborativecomplementation study of temperature-sen-sitive mutants ofherpes simplex virus types 1 and 2. J. Virol.27:490-504.
27. Spear, P., and B.Roizman. 1980. Herpes simplex viruses, p. 615-745. In J. Tooze (ed.), DNA tumorviruses. Cold SpringHarborLaboratory, Cold Spring Harbor, N.Y. 28. Takahashi, M., and K.Yamanishi. 1974. Transformation
ofhamster embryo and human embryocells by ts mutants ofherpes simplex virustype 2. Virology 61:306-311. VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
342 SPANG, GODOWSKI, AND KNIPE 29. Wilcox, K. W., A. Kohn, E. Sklyanskaya, andB. Roizman.
1980. Herpes simplex virus phosphoproteins. I.
Phos-phate cyclesonandoffsomeviralpolypeptides andcan
alter their affinity for DNA. J. Virol. 33:167-182. 30. Wilkie, N. M., A. Davison, P. Chartrand, N. E. Stow,
V.G.Preston, and M.C. Timbury.1978.Recombination
in herpes simplex virus: mapping of mutations and analy-sis of intertypic recombinants.Cold Spring Harbor Symp. Quant. Biol. 43:827-840.
31. Zygraich,N., andC. Huygelen.1973. Invivobehavior of
atemperature-sensitive (ts)mutantofherpesvirus hominis
type2.Arch. GesamteVirusforsch. 43:103-111. J. VIROL.