Copyright 0 1976 AmericanSocietyfor Microbiology Printed in U.SA.
Cell
Killing by Simian Virus
40:
Variation
in the Pattern
of
Lysosomal Enzyme Release, Cellular Enzyme
Release,
and
Cell
Death
During Productive
Infection of Normal
and
Simian Virus
40-Transformed
Simian Cell
Lines
LEONARD C. NORKIN* AND JANICE OUELLETTE
Department ofMicrobiology, UniversityofMassachusetts, Amherst,Massachusetts01002
Receivedforpublication22October1975
Simian virus40(SV40)growthonrhesuskidneycells andonthe T-22 line of
SV40-transformed green monkeykidney (GMK)cells islargelylimited bythe
lowplatingefficiency of SV40onthese cells. Inaddition,afraction of the rhesus
kidneyandT-22cellsareresistanttoinfection by SV40.Nevertheless,72-h viral
yields perinfected rhesus kidney and T-22 cell are nearly equivalent to that
obtained on normal GMK cells and are independent of the multiplicity of
infection. Despitethe production of high viralyields, infected rhesuskidney and
T-22cellsarekilled slowly by SV40. Monolayersof these cellsarealsorefractory
toplaqueformation by SV40. SV40 inducestherelease oflysosomal
N-acetyl-,8-glucosaminidase into the cytoplasmic fractionsof rhesus kidneyandT-22cellsto
an extentequaltothat observedduring infectionof rapidlykillednormal GMK
cells. Incontrast, damagetotheplasmamembrane, asindicatedbythe release
ofthe cellularenzymeslacticdehydrogenaseandglutamicoxaloacetic
transami-nase intothe overlay media, occurredtoamuchgreater extent in the normal
GMKcells than inthe rhesuskidneyorT-22cells. Neither alysosomal
hydro-lase mechanismnorviral releaseappear toberesponsible for this phenomenon.
Thedifferentratesandextentsofthe SV40cytocidalprocess onthesecells donot
resultfrom the differencesinthe viral platingefficiencyonthem.
The mechanisms by which cytocidal viruses
kill cells are largely unknown. This is so, in
part,becauseitisdifficulttodistinguish
cytoci-dal events from the many other phenomena thataccompanyproductive infection. The
stud-ies on cell killing by simian virus 40 (SV40)
reported herefollow from the observation that
host cell factorsdeterminetherateof cell
kill-ing during productive SV40 infection. Since
SV40 yields are nearly equivalent during the
rapidlycytocidal and slowly cytocidal infections
described below, phenomena associated with
thecytocidalprocess canbedistinguishedfrom
the epiphenomena that accompany productive
infection.
Intheoriginaldescriptions of therelationship
between SV40 and rhesus kidney cells in
cul-ture (9, 19), it was noted that growth of the virus and resultant cellular destruction took
muchlonger thanincultures of green monkey
kidney (GMK)cells. Results reported below
in-dicate that SV40growth on rhesus kidney cells
islargely limited by thelow plating efficiency
ofthis viruson thesecells. We find that SV40
yields on rhesus kidney cultures approach that
48
obtained onGMK cultures under conditions of
high virus-to-cell inputmultiplicity.
Neverthe-less, even under these conditions, cytopathic
effect is stillminimalonrhesuskidney cells. A
similar butnot sostrikingdissociation of viral
growth from cell deathwas alsoobservedwith
the T-22 line ofSV40-transformed GMK cells.
As describedbelow, we have taken advantage
of thepartial dissociation of SV40growth and
cell death on rhesus kidney and T-22 cells to
determine whether the virus-induced release of
lysosomal enzymes into the cytoplasm and/or
damage to the plasma membrane correlate
withcellkilling.
Lysosomal hydrolytic enzymes are released
intothe cytoplasm duringinfection by a
num-ber of cytocidal viruses including poliovirus
(13), mouse hepatitis virus (2), vaccinia virus
(2), Newcastle disease virus (15), herpes
sim-plex virus (1), and SV40 (1). It has been
sug-gested that this phenomenon, referred to as
"lysosomal activation," might lead to cellular
degenerationand death (2). We measured the
levelsoflysosomalenzyme release duringSV40
infection of normal GMK, rhesus kidney, and
on November 10, 2019 by guest
http://jvi.asm.org/
CELL KILLING BY SV40 49
T-22 cells. Our results indicate that lysosomal
activation follows the same time course and
occurs tothe same extent onall three celltypes.
Therefore, we suggest that lysosomal
activa-tion per seis notprimarily responsible forthe
death of SV40-infected cells.
Wealso examined the effect of SV40 onthe
integrityof theplasma membraneduring
infec-tionof normal GMK, rhesus kidney, and T-22
cells. Damageto theplasma membrane, as
in-dicated bythe release of cellularenzymes into
theoverlay media, occurred to amuch greater
extent on the normal GMK cells and was
corre-latedwithcell killing.
MATERIALS AND METHODS
Cell cultures. Normal GMK cell lines CV-1 and BSC-1 were obtained from the American Type Cul-ture Collection and T. L. Benjamin, respectively. The SV40-transformed GMK cell lines T-22 and
GMK/PARA-7-1 (particleaiding replication of ade-novirus) were kindly supplied by Janet S. Butel. The T-22 cell line was transformedby Shiroki and
Shimojo(17)usingaheavilyUV-irradiateddefective virionfraction of SV40(T-fraction). GMK/PARA-7-1 wastransformed by Butel using PARA-adenovirus 7. The LLC-MK2 line of rhesus kidney cells was obtained from the American Type Culture Collec-tion.A-31 BALB 3T3cells were obtained from H. L. Ozer. All cells were cultivated in Dulbecco modified Eaglemedium (DME, GIBCO), containing 10% fetal calfserum(FlowLaboratories), inahumidified5% CO2 atmosphere. Cultures did not contain myco-plasma asindicated by autoradiography (18).
Virus. Small plaque SV40 (strain 777) was ob-tained from T. L.Benjamin.This virus was usedin all experiments unless otherwise indicated. Virus preparations were grownonCV-1cells fromplaque
isolates. SV40 was precipitatedfrom crudelysates
with methanol, extracted with
trichlorotrifluoro-ethane(AlliedChemical), pelletedat109,000xgfor 1 h,andresuspended in Hanks balanced salt solu-tion(HBSS) containing2%fetal calfserum.
Large plaque SV40 (Strain SV-L) was obtained from H. L. Ozer. Simian adenovirus 7 (SA-7) was obtained from T. L.Benjamin. Humanadenovirus5 (Ad 5) was obtained from the AmericanType Cul-tureCollection.
Plaque assays. Subconfluent CV-1 monolayers wereinfected with 0.05 ml of appropriate viral dilu-tions inHBSS containing 2%fetal calfserum. Ad-sorptionwasfor2hat37Cinahumidified5%CO2
atmosphere. The overlay contained DME, 0.09% agar (Difco), 10% fetal calf serum, and 0.01 M HEPES[(N-2-hydroxyethyl)piperazine] (Sigma). As-sayswereincubatedat 37Cinahumidified5%CO2
atmosphere and, unless otherwiseindicated,stained 8 to 10 days postinfection with the above overlay
(minus theserum)containing 0.009% neutral red. Antisera and immunofluorescence techniques.
Anti-SV40 horseserumwith ahomologoustiter of 1:640 versus 500 mean tissue culture dose ofSV40
strain 911 was obtained from Flow Laboratories. Incubation of 107 PFU/ml of SV40 strain 777 with an equal volume of a 50-fold dilution of this antisera for 1h at 37 C results in a 104-fold decrease in titer.
To detect production of the SV40 V or Tantigen by indirectimmunofluorescence cells were grown on 18-mm-square glass cover slips. Cover-slipcultures werewashed with HBSS, air dried, and fixed in cold acetonefor 10 min. Thehorse anti-SV40 sera diluted 1:5 in phosphate-buffered saline (PBS) followed by fluorescein-conjugated rabbit anti-horse globulins (Miles Research Laboratories) were used to detect V antigen. Hamster antiserum to SV40 T antigen (FlowLaboratories) diluted 1:5 in PBS followed by
fluorescein-conjugated rabbit anti-hamster globu-lins (Miles ResearchLaboratories) were used to de-tectthe SV40 T antigen. Incubation with all anti-bodies was for 1 h at 37 C. Cover slips were washed several times with PBS after exposure to each anti-body and then wet mounted with buffered glycerol andexamined under darkfield UV illumination.
Viable cell count. Attached cells were resus-pended with0.025%trypsinand addedtotheoriginal
growth media containing detached cells. Trypan blue (GIBCO) dissolved in HBSS was added to a final concentration of 0.08%. After 5 min a total cell count and a stained cell count were made using a hemocytometer.
Cellcloning. Cell suspensionswereexamined
mi-croscopicallytoconfirm theabsence of cell clumps. CellswerethenplatedinFalcon microtestplatesat 0.1cells per well. Any wells containing more than onecell,asrevealedby microscopeexamination24h later, were eliminated from further consideration.
Assays for infectious centers. Monolayers con-taining approximately 2 x 105 cells were infected with 0.05 mlof SV40 at the indicated MOIs. After
adsorption for 2.0 h at 37C the monolayers were washed five times withHBSS and remaining unad-sorbed virus was neutralized for 1 h at 37 C with anti-SV40horseserumdiluted1:50 inHBSS.
Mono-layerswerethen washedfivemoretimeswith HBSS.
Theinfectedmonolayerswerethenresuspendedwith 0.025%trypsin, diluted in DME containing10%fetal calf serum, andplatedasinfectious centers onto CV-1 monolayers. Results obtained with the A-31 cells indicatethat essentially all unadsorbed virus is re-movedbythese procedures (Table 1). At 22 h
postin-fectionthe liquid growth medium was removed and
replacedwith an agaroverlay as for a plaque assay.
One-step growth experiments, using these
proce-dures,indicate that insignificant amounts of extra-cellular virus are released by the time the agar
overlayisadded (Fig. 1).Assayswere stained with neutralredonday7asfor aplaqueassay.
Preparation ofhomogenates. Microscope exami-nation andmeasurementof latentenzymeactivity wereusedto assessthequality of homogenates
pre-pared in different ways. The procedure that was foundtogivemostrapidcellbreakagewithmaximal retentionoflatencywasthefollowing. Monolayers
were washed with HBSS and resuspended into a solutioncontaining40mMKCland10mMEDTAat pH 7. After 4 min on ice greater than 90% cell
18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 1. Platingefficiency and viralyieldsofSV40onLLC-MK2, T-22, CV-1,andGMKIPARA-7-1 cells Infectious centers Viralyield (PFU/infectious center")
MOI MK GMK/
LLC-MK2 T-22 CV-1 PARA-7-1 A-31 LLC-MK2 T-22 CV-1
PARA-7-0.01 40 103 0.6 x 103 6 x 103
0.1 4 x 102 2 x 102 6 x 103 0.8x 103 2 x 103 3 x 103 1.0 2 x 103 4 x 103 6 x 104 105 <40 0.8x 103 103 3 x 103 103
10.0 2 x 104 3 x 104 7 x 104 105 2 x 103 103 3 x 103 103
100.0 105 105 60 103 2 x 103 4 x 103
a Infectious centers weretiteredasdescribedinMaterials and Methods. Parallel infected cultureswere
harvested at 72 h, frozen andthawed, and furtherdisruptedby sonication. Viralyields weretiteredby plaqueassay on CV-1 monolayers.
-J -J w 0 w
;I-0
cr
0D
a-N.
0 0 -J -I
-2
24 48 72
TIME AFTER INFECTION (Hours)
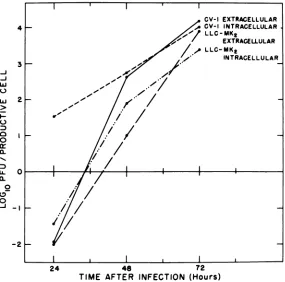
FIG. 1. One-step growth ofSV40onCV-1 andLLC-MK2cells.Cellswereinfectedat anMOIof100PFUI cell. Unadsorbed virus was removed and infectious centers were titered as described in Materials and Methods. Extracellularfluidandwashedmonolayers from parallel infected cultures, treatedinthesame way, wereharvestedseparatelyattheindicated times. Extracellularfluidwasclarifiedof any celldebrisby low-speedcentrifugation. Monolayers were frozen and thawedthree times and further disrupted by sonication. Virus yields weredetermined by plaque assay on CV-1 monolayers.
breakage was then achieved with 30 strokes of the Douncehomogenizer. An equal volume of a solution containing 0.24 M KCl and 10 mM EDTA atpH7 wasthenadded. Thehomogenate was sedimented at 20,000 xgfor 20 min at4C. Thesupernatant con-taining released lysosomal enzymes was removed with aPasteur pipette and the pelletcontaining in-tactlysosomes wasresuspended into an equal
vol-umeofasolution containing0.14MKCland10mM EDTA atpH 7. All sampleswere then frozen and thawedthreetimes.
Enzyme assays. Lysosomal
N-acetyl-P-glucosa-minidase(EC3.2.1.30)activitiesinsupernatantand pellet fractions were assayed by the procedure of Findlayet al. (10). Activityin eachfraction is as-sayed by measuring the release of p-nitrophenol
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.509.57.450.78.178.2] [image:3.509.115.400.222.504.2]CELL KILLING BY SV40 51 from the substrate
p-nitrophenyl-N-acetyl-,3-n-glu-cosamide(Sigma)asindicated bytheabsorbanceat 400 nm. Triton X-100 in the incubation mixture assuresthe activation of all particle-boundenzyme.
Proteinwasmeasured bythe method of Lowryetal. (16).
Lactic dehydrogenase (LDH) (1.1.1.27) was
as-sayed by monitoring therate atwhichpyruvate is reduced to lactate. This reduction iscoupled with the oxidation of NADH2 which isfollowed
spectro-photometrically in termsof reduced absorbance at
340nm. For thispurposeSigma kit number340-LD
wasused.
Glutamic oxaloacetic transaminase (GOT) (2.6.1.1) wasassayed using Worthington
Biochemi-cal Corp. determatube SGO.
RESULTS
SV40 plating efficiency and yields on T-22
andLLC-MK2cells.ResultsinTable1indicate
that the plating efficiency ofSV40 is
approxi-mately25-foldloweronboth LLC-MK2 andT-22
cells thanonCV-1 cells.This isindicated by the
relative numbers ofinfectiouscentersproduced
onthese cell linesatlow inputmultiplicities.If
the MOI is increasedto 10PFU/cell then
com-parable numbers ofinfectious centers are
pro-ducedoneach celltypewithinanorder of
mag-nitude. It is important to note that the viral
yieldsperinfected cellarenearly equivalenton
LLC-MK2, T-22, and CV-1 cells, andare
inde-pendent of the MOI (Table 1). AtanMOI of 10
PFU/cell SV40 growth on LLC-MK2 and T-22
cultures iscomparableto that obtainedonCV-1
cultures within an order ofmagnitude. These
resultsaretakentomeanthat viralproduction
onLLC-MK2and T-22 cells islargelylimitedby
theplating efficiency ofSV40onthese cells.
Results inFig. 1indicatethatrelease of SV40
from infected LLC-MK2 cells is comparable to
that on CV-1 cells. Similar results were
ob-tained with theT-22cell line.
A noninfectable cell fraction of T-22 and LLC-MK2 cultures. Assays for infectious
cen-tersindicatethat the number of infected
LLC-MK2 and T-22 cells approaches that of CV-1 cells at an MOI of 10PFU/cell. Nevertheless,
notall of the LLC-MK2and T-22 cellsare
sus-ceptibletoinfection. AsseeninTable2,assays
infectivity by the indirect immunofluorescent
antibody technique for the production of the
SV40Tand Vantigens indicate thatnotmore
than about50%of the LLC-MK2 and T-22 cells become infected atan input multiplicity of 50
PFU/cell. In contrast, 100o of the CV-1 cells
are infectedat this MOI.
It is notyet clearwhyso largeafraction of
the LLC-MK2 and T-22 cells are refractory to
SV40. TheplatingefficiencyofSV40is not
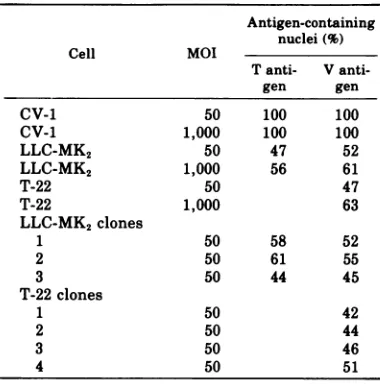
lim-TABLE 2. Production of SV40 T and V antigens on CV-1, LLC-MK2, and T-22 ceilsa
Antigen-containing nuclei(%)
Cell MOI
Tanti- V
anti-gen gen
CV-1 50 100 100
CV-1 1,000 100 100
LLC-MK2 50 47 52
LLC-MK2 1,000 56 61
T-22 50 47
T-22 1,000 63
LLC-MK2 clones
1 50 58 52
2 50 61 55
3 50 44 45
T-22clones
1 50 42
2 50 44
3 50 46
4 50 51
a Cover-slip cultures were fixed for
immunofluo-rescence at 48 hpostinfection. Clonal isolates were obtained as described in Materials and Methods. The rangeof errors in the above values due to sam-pling is + 5%to + 8%.
iting since notmore thanabout 60%of theT-22
and LLC-MK2 cells become infected at input
multiplicities of 1,000PFU/cell (Table 2).
Het-erogenicity of the LLC-MK2 andT-22 cultures
is notafactor because similar levels of
infectiv-ityareobtainedwithclonalisolates of these cell
lines(Table 2). The fraction of LLC-MK2 cells
producing T antigen correlates with the
frac-tionofproductively infected cells,asshown by
theproductionof viral antigen.Thus,the block
toinfection isearly, before theproductionofT
antigen.
Dissociation of viralgrowth and cell death
onT-22 and LLC-MK2cells. When high input
multiplicity infectionsare studied(MOI = 100
PFU/cell), the viral yieldson CV-1, T-22, and
LLC-MK2cultures arenearly equivalent.
Nev-ertheless, the cytocidal process occurs much
more slowly on LLC-MK2 and T-22 cultures
than on CV-1 cultures under theseconditions
(Table 3).
Itisimportant to notethe total cell number
perculture every time the nonviablecell
frac-tion is measured since those cells that were
refractory at the time ofinfection may be
in-creasing in number during the experiment.
This would result in an underestimate ofthe
rate atwhich infectedcellsarekilled.
In the experimentpresented in Table 3 the total cellnumber perculture was determined
VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.509.265.455.88.281.2]52 NORKIN AND OUELLETTE
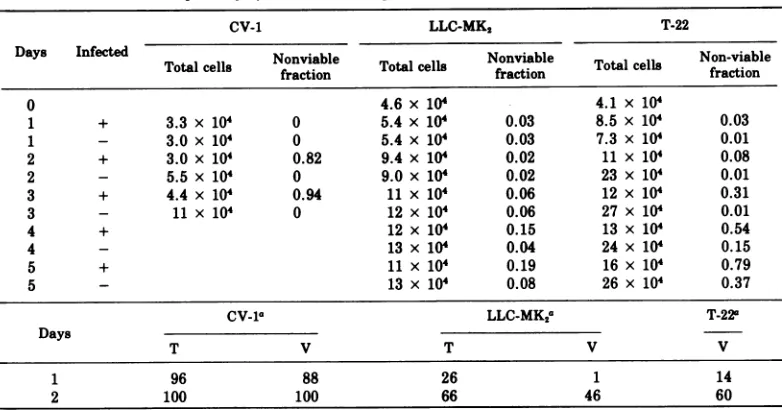
TABLE 3. Susceptibility of CV-1,LLC-MK2, and T-22cells to the SV40 cytocidal effect
CV-1 LLC-MK2 T-22
Days Infected Nonviable l Nonviable Total cell Non-viable
Totalcells fraction Totalce fraction fraction
0 4.6 x 104 4.1 x 104
1 + 3.3 x 104 0 5.4 x 104 0.03 8.5 x 104 0.03
1 - 3.0 x 10 0 5.4 x 10' 0.03 7.3 x 10' 0.01
2 + 3.0 x 104 0.82 9.4 x 104 0.02 11 x 104 0.08
2 - 5.5 x 104 0 9.0 x 104 0.02 23 x 104 0.01
3 + 4.4 x 104 0.94 11 x 104 0.06 12 x 104 0.31
3 - 11x 104 0 12 x 104 0.06 27 x 10' 0.01
4 + 12 x 104 0.15 13 x 10' 0.54
4 - 13 x 104 0.04 24 x 104 0.15
5 + 11x 104 0.19 16 x 104 0.79
5 - 13 x 104 0.08 26 x 104 0.37
CV-la LLC-MK2a T-22a
Days
T V T V V
1 96 88 26 1 14
2 100 100 66 46 60
aPercentageof antigen-containing nucleic. Cellswereinfectedat anMOI of 200 PFU/cell.
every time the nonviable cell fraction was
measured. In this experiment anattemptwas
also madeto limitcell growth by usingmedia
partially depletedofserumgrowthfactors.The
fractionof infected cellswasdeterminedby
im-munofluorescent staining. Sixty-sixpercentof
thecells of the infected LLC-MK2 cultures
ex-pressed the SV40 T antigen at 2 days
post-infection. It is difficulttodetermine the fraction ofinfected LLC-MK2 cells before 48 hby immu-nofluorescence because the latent period on
these cells is somewhat longer than on CV-1
cells(Fig. 1, Table3).Atanyrate, littleifany
further cell growth occurred in the infected LLC-MK2 cultures between day2andday5.By
5days onlyabout 11% of the cells of the infected
LLC-MK2culture were killed bySV40.
There-fore, onlyabout 17% of the infected LLC-MK2
cells(0.11/0.66)arekilledbySV40in5days.
Therate ofSV40-induced killingof the T-22
cells is intermediate between that of the CV-1 andLLC-MK2 cells (Table3). Sixty percent of thecellsof the infected T-22 culture expressed
the SV40 V-antigen at 2 days postinfection. Therewaslittle, ifany, further cellgrowth in
this cultureduringthenext2days. By4 days only39%of thecellsof theinfected T-22 culture
were killed by SV40. Therefore, about 65% of
the infected T-22cells (0.39/0.60) arekilled by
SV40in 4days. Incontrast, 82%of theinfected
CV-1cellsare killedby2days.
AttemptstouseT-22 andLLC-MK2 cells in
SV40 plaque assays. Numerous unsuccessful
attempts weremade to produce SV40 plaques
on T-22 and LLC-MK2 monolayers using both
smallplaque and largeplaquestrainsofSV40.
Inthese experimentsmonolayerswereinfected
at either 50 or 90% confluency. Various
over-layswere used containing either DME, MEM,
or F-12 media supplemented with either 5 or
10%calforfetalcalfserum.Infected
monolay-ers were kept alive for as long as 35 days.
Nevertheless, plaques were neverobserved on
these cell lines, even on plates receiving 104
PFUs. In contrast, plaques were readily
ob-served on CV-1 and BSC-1 monolayers by 8
days postinfection under these conditions. It
maybe noted that pretreatment of theT-22and
LLC-MK2 monolayers with DEAE-dextran (25
,ug/ml
for10min)and/or the presence ofDEAE-dextranin the overlay (10 ,ug/ml) did not
en-hance plaque formation in monolayers kept
alivefor 35 days. This treatment is known to
enhanceuptakeofSV40 (5).
Passage of both small and large plaque
strains ofSV40onT-22andLLC-MK2 cells did
not resultinviruscapable of producing plaques
onthese cells.
There is no inherent inability of T-22 and
LLC-MK2 cells to produce virus plaques since
these cells gave rise to SA-7 plaques as
effi-cientlyasBSC-1andCV-1 cells. Inaddition,
T-22 cells gave rise to human adenovirus 5
plaquesasefficientlyasthehuman cell line,
L-132. These results with the T-22 cell line are in
agreement withearlier observations that T-22
cells express the human adenovirus helper
function as shown by their ability to support
on November 10, 2019 by guest
http://jvi.asm.org/
CELL KILLING BY SV40 53
growth of human adenovirus7(8).
Lysosomal enzyme redistribution. In the
following experiment, wetakeadvantage of the
partial dissociation of viral growth from cell deathonLLC-MK2 and T-22cellstodetermine
whether lysosomal activation is correlated with the SV40-induced cytocidalprocess. The
distri-bution of lysosomal N-acetyl-f3-glucosamini-dasewasfollowed since this hydrolase displays a greater degree of latency than either acid phosphataseor f8-glucuronidase (7).
Lysosomal enzyme redistribution resulting
from infection is expressed as the difference
between infected and control cultures with
re-spect tothepercentagesof cellular
N-acetyl-,f-glucosaminidase in cytoplasmic fractions and
as the specific activity of the enzyme that is
achievedinthecytoplasmic fractions(Table 4). Viewed in eitherway the results indicate that
the release of lysosomal enzymeinto the
cyto-plasmoccurstothesameextentonCV-1,
LLC-MK2, and T-22 cells. The levels of activity in thecytoplasmic fractions of the control cellsare
notunusual (7) and probably reflect disruption ofsomelysosomes during homogenization.
Thefractions of nonviable cellsat48and 72 h inparallel infected cultures arealsopresented
inTable4.The fractions of nonviable cellsat48
hdonotcorrelate with the levels of
N-acetyl-,8-glucosaminidase in the cytoplasmic fractionsat
this time. Furthermore, the accumulation of the lysosomal hydrolase in the cytoplasmic fractions of the infected LLC-MK2 and T-22
cul-tures at48hdoesnotcorrelate with cell death
overthe next24 h. These results indicate that
the SV40-induced activation of lysosomal
hy-drolasesper seisnotresponsible for cell death.
Wealso studied the timecourseoflysosomal enzyme releaseduring6V4U infectionotCV-1
experiments
lysosomal
activationwasobserved-nai intecte hutrsv inone
exgeri-mentlysosomalactivationdidnotoccuronanX
ce However, in experimeints
similar levels oflysosomal activation were ob-servedoneach of the infected culturesby48h. Insomeexperimentsasomewhathighershift of enzyme activity to the cytoplasm resulted
from infection atanMOIof 100 PFU/cell than from infection at 1,000 PFU/cell. Thiswas ap-parently not due to interference at the higher
MOI since equivalent viral yields were
ob-tainedatthetwoinputmultiplicities (datanot
shown).
Release ofN-acetyl-,3-glucosaminidase into the overlay media has not been considered in
theexperimentsreportedthus far. As described
below in a differentcontext, infected and
con-trol cells release notable amounts of activity
into the overlay fluid by 48 h (Table 5). The resultsreportedinTable 5 wereobtained with cells incubated under serum-free conditions
after infection. Serum-free conditionswere
nec-essaryintheseexperimentsbecause the levels
ofthese enzymesin serum arehigh enoughto obscure virus-inducedchanges. Wetriedmany
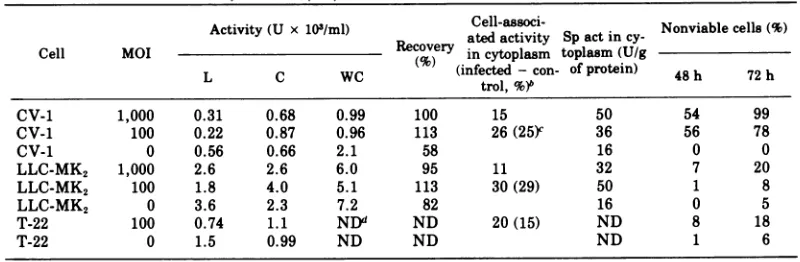
TABLE 4.
N-acetyl-,B3glucosaminidase
activityinlysosomal (L), cytoplasmic(C),and whole cell(WC)fractions of infected and control cellsat48ha
Activity (U x 103/ml) atedCell-associ-activity Spact Nonviablecells (%) in
cy-Cell MOIRecovery in cytoplasm toplasm (U/g
L C WC (infected - con- ofprotein) 48 h 72 h
L C WC
~~~~~~~trol,%48)b2
CV-1 1,000 0.31 0.68 0.99 100 15 50 54 99
CV-1 100 0.22 0.87 0.96 113 26(25Y 36 56 78
CV-1 0 0.56 0.66 2.1 58 16 0 0
LLC-MK2 1,000 2.6 2.6 6.0 95 11 32 7 20
LLC-MK2 100 1.8 4.0 5.1 113 30(29) 50 1 8
LLC-MK2 0 3.6 2.3 7.2 82 16 0 5
T-22 100 0.74 1.1 NDd ND 20(15) ND 8 18
T-22 0 1.5 0.99 ND ND ND 1 6
a Resultsarethe average ofduplicatesamples. One unitofactivityisdefinedastheamountof enzyme
necessarytorelease 1 gmolofp-nitrophenol permin.
bDifference between infected and control cultures with respect to the percentages of cell-associated activityinthecytoplasmic fraction.
cThe dataintheparentheses represent the differences between the infected andcorrespondingcontrol cultures withrespecttothepercentagesof total activated enzyme (cytoplasmicactivityplusextracellular activity). Percentages of total activated enzyme were obtained by addingthe fractions of extracellular activities (Table 5, 48 h) to the corresponding products ofthe fractions of cytoplasmic cell-associated activities(above)by the fractions of cell-associated activities(Table5,48h).
dND,Notdone. VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.509.56.456.433.564.2]NORKIN
times to measure the activities of
N-acetyl-f3-glucosaminidase in lysosomal, cytoplasmic,
and extracellular fractions under serum-free
conditions to determine the total fraction of
"activated" enzyme. However, wehavenotyet
been able to obtain an acceptable level of
la-tencyand/or recovery whenfractionating cells
that have been incubated under serum-free
TABLE 5. Percentage of total cellular enzyme activitiesreleased into the overlay mediaof infected
and control cultures
Activity in overlaymedia (%)
Cell MOI Hours
N-acetyl-LDH GOT
0-glucos-amini-dase
CV-1 100 24 0 0 15(0P)
0 24 0 0 15
100 48 82(60) 54(50) 49 (25)
0 48 22 4 24
LLC-MK2 100 24 0 0 7(1)
0 24 0 0 6
100 48 6(6) 1(1) 13(5)
0 48 0 0 8
100 72 32(32) 19(13) 22(9)
0 72 0 6 13
T-22 100 24 0(-5) 3(-3) 16(-3)
0 24 5 6 19
100 48 39(10) 21(4) 33 (3)
0 48 29 17 30
100 72 66(20) 29(7) 45 (11)
0 72 46 22 34
aData in parentheses are the differences between
in-fectedand control cultures. All resultsaretheaverageof duplicate samples.
vl
z
0
--J
w
cr
0
0
-J
-I
conditions. To "correct" for the SV40-induced
leakageofN-acetyl-p8-glucosaminidase we
com-bined data obtained in different experiments
carriedout in the presence and absence of
se-rum. This "corrected" data appears in the
pa-renthesis of Table 4. It was obtained as
de-scribed in the legend to that table. We donot
know whether leakage into the overlay fluid
occurs to a similar extent in the presence of
serum.At any rate, whenviewing the corrected data the overall impression is that the levels of
virusinducedactivation oflysosomal
N-acetyl-,B-glucosaminidase on CV-1, T-22, and
LLC-MK2 cultures are similar and not correlated
withthe rates or extent of cellkilling.
Leakage of cellularproteins. The release of
cellular proteins from virus-infected cells has
been noted previously (6, 11, 15). The damageto
the plasma membrane that this phenomenon
reflects is followed below by monitoring the
release of the cytopi. .L ic enzymes, LDH and
GOT, as well aslysosomal
N-acetyl-/3-glucosa-minidase into the overlay media.
Since serum contains high levels of these
enzymes, cultures were washed and overlayed with serum-free media at 4 h postinfection.
As seen in Table 5, infected CV-1
cells,
incomparison to control cells, release notable
amounts ofthecytoplasmic enzymes, LDH and
GOT,
as well aslysosomalN-acetyl-f3-glucosa-minidase into the overlay media by 48 h. Much
lower levels of enzyme are released into the
overlay fluid by the infected LLC-MK2 and T-22
cultures relativetothe controlcultures, atthis
-O ViableCellFraction
o---a T Antigen Negative Cell Fraction
_
---- VAntigen NegativeCell Fraction.~~~~~
I°1
I I I
0 2 3 4 5 6 7
INPUT MULTIPLICITY (PFU/CeIl)
I I I II
8 9 10
FIG. 2. Relationship betweencytopathogenicityandantigen-forming ability of SV40onCV-1cultures as a function of theMOI. Antigen-producing cell fractions were determined at48h and viablecellfractionswere determinedat72 h.
n r ...
.V.
-0.21
-0.41
-0.61
-
0.81
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.509.64.457.364.613.2]CELL KILLING BY SV40 55
time. Even at 72 h the virus-induced enzyme
leakagefrom LLC-MK2 and T-22 cells is much
less than that found with CV-1 cells at 48 h.
This is most clear in the case of GOT. These
results aretakento mean that damage to the
plasma membrane, as measured by enzyme
leakage from the cell, is correlated with the
rate ofcell killing.
MOI andcytopathic effect.Since the plating
efficiency of SV40 is more than 20-fold higher
on CV-1 cells thanoneither LLC-MK2 or T-22
cells (Table 1), the effective MOI withagiven
viral inoculum may be considerably higheron
the CV-1 cells than on theother two cell lines.
For this reason it is necessary to determineif
therateof cell killingand/or plasma membrane
damagearemultiplicity dependent.
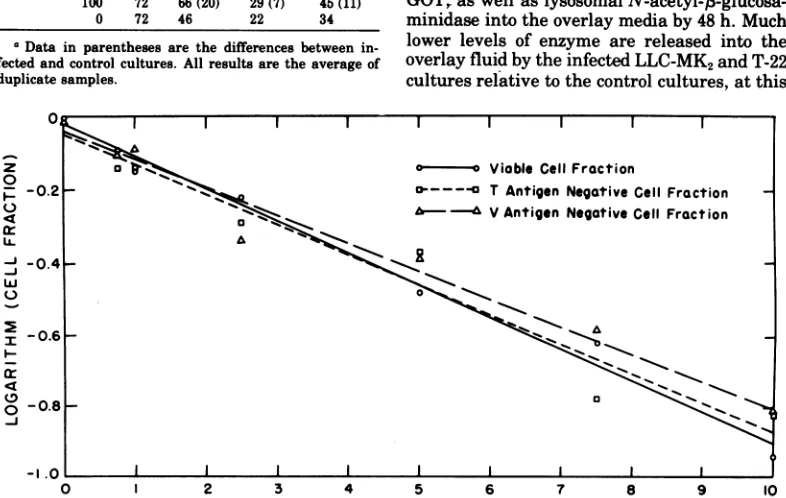
The experiment illustrated in Fig. 2 shows
the relationship between the fraction of
in-fected CV-1cells and the fraction of cells
subse-quently killed at input multiplicities ranging
from0.75 to 10PFU/cell. The fractions of CV-1
cells producing T and V antigen were
deter-minedat 48h. The nonviablecell fractionswere
determinedat 72 h. The resultswereanalyzed
asfollows.Accordingtothe Poisson distribution
the relationship between the surviving cell
fraction,
f/,
and thevirusdosage, x,isgivenbyf,
=e-r,ortheequivalentlnf,
= -x, assumingone-particle-to-kill kinetics. The graph of the
values of
1nf,
versusthe virusdosageshould bea straight line. A similar relationship should
existbetweenthe logarithmof the nonantigen
producing cell fraction and the virus dosage.
Thus, best fit straight lines were drawn
throughthe datainFig. 2using the method of
least squares. The results are consistent with
one-hit kinetics. The coincidence of the lines
indicatesthefollowing. Thefraction of T
anti-gen-producingcellscorrelates with the fraction ofV antigen-producing cells. This shows that
once infection is initiated, asindicatedby the
expression of the viral T-antigen, productive
infection ensues. The correspondence between theantigen-producingcellfractionsat48hand
the nonviable cell fractions at 72 h indicates
statisticallythat infected cellsarekilledby72h
regardless of theMOT.Inthiscontext itmay be
noted thatviralyieldsperinfected cellare
inde-pendentof the MOI (Table 1).
We also measured the extent to which
en-zyme leakage isdependent on theMOT.
Leak-ageofLDHand GOT from CV-1cellswas
some-what multiplicitydependent over arangeof1
to 100PFU/cell. Nevertheless, moreenzyme is
released fromCV-1 cellsinfectedatanMOI of1
PFU/cell than fromLLC-MK2 orT-22 cells
in-fected at an MOI of 100 PFU/cell (data not
shown).
DISCUSSION
The results reported above indicate that
SV40 growth on both rhesus kidney and T-22
cells is largely limited by the relatively low
plating efficiency of SV40 on these cells. In
addition, afraction of the rhesus kidney and
T-22 cells are refractory to infection by SV40.
Despite thepresence of resistant cellfractions,
we were able to show that infected rhesus
kid-ney and T-22 cells are killed more slowly than
infected CV-1cells, even though the viral yields
perinfected cell are nearly equivalent on each
cell type. In the original descriptions of the
interactionofSV40with rhesuskidneycells in
culture(9, 19),these infections were
character-ized by slow viral growth as well as byminimal
cell killing. It appears that low input
multiplic-ityinfections were being followed in these
stud-ies.
The dissociation of viral growth from cell
death thatwehave observedduringSV40
infec-tionof rhesus kidney cell culturesisquite
strik-ing. This is surprising in view of the fact that
DNA virus production is generally correlated
withcell killing. Although it is known that host
cell factorscaninfluence the killing of cellsby
RNAviruses, ourresultsindicate that host cell
factors can also determine the time or rate at
which the cytocidal process is set in motion
during infection by DNA viruses as well.
Todate, nosinglehypothesiscan explain the
mechanism by which viruses kill cells.
Gins-berg(12)suggested that virus-inducedcell
dam-agemight result fromapassive role of the virus
suchasdepletion of cellular components
essen-tial for lifeormechanicalharm due to excessive
production of virus and its components. Our
studies suggestthat thisis notthe basis for cell
killing by SV40 since infection of rhesus kidney
and T-22 cells results in normal virus yields
accompanied by slow cell death.
Very littleisknown aboutthe virus-induced
biochemical derangements that might lead to
cell death. Incaseswhere virus-induced
meta-bolic depressions are recognized, such as the
inhibition of host RNA and protein synthesis
whichaccompanies infection by many viruses
including vaccinia virus, mengovirus, and
poliovirus, there is little evidence to connect
these events to the virus-induced cell death
(i.e., 3, 4, 14). In the case ofSV40-infected
sim-iancells there is no known inhibitionof cellular
metabolic functions.
One promisingsuggestion thathas received
much attention is that virus-induced redistri-bution oflysosomal hydrolases might lead to
cellular degeneration and death (2). As noted
above, "activation" of
lysosomal
hydrolases
VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
56 NORKIN AND OUELLETTE
occurs during infection by a number of
cyto-pathic viruses (i.e., 1, 2, 13, 15). In
particu-lar, Allison andBlack(1)havenotedlysosomal
activation by histochemical methods in
SV40-infected GMK cells. Nevertheless, our studies
comparing lysosomal activation during SV40
infection of rhesus kidney, T-22, and normal
GMK cells indicate thatlysosomalactivationis notcorrelated with cellkilling.
It may be noted that Allison and Black (1)
alsoobservedalessextensive, transient
lysoso-mal activation during SV40 infection of 3T3
mouse cells. These infections are neither
pro-ductivenorcytocidal. Wewereunabletodetect
lysosomal activationonSV40-infected 3T3 cells
using the procedures described above (datanot
shown). Therefore, we suggestthat lysosomes
participate inSV40growthorrelease, insome
as yet unspecified manner, without directly
causing cell death.
Damage to the plasma membrane, as indi-catedby the release of cellularenzymesinto the overlay medium, was found to be correlated
with the cytocidal process. It occurred to a
muchgreater extentonthe CV-1 cellsthanon
either the rhesuskidneyorT-22 cells.
Although we do not yet know the
mecha-nism by which SV40 induces the observed
permeability changes, a lysosomal hydrolase
mechanism is not consistent with ourresults.
Also, enzymeleakageisnot simply associated
with virus release (see Table 5 and Fig. 1).
Muchmoreenzymeisreleased into theoverlay
media by CV-1 cells at 48 h than by rhesus
kidney cellsat72 h. Nevertheless, much more
virusisreleased byrhesus kidneycellsat72h
than by CV-1 cellsat48h. From thiswe
sug-gest that theleakage ofenzymesandvirus from cells reflect different types of injury. As
sug-gested byGilbert(11),cellulardamagemaybe
reversible atthe stageof virusrelease, butnot
afterthe releaseofenzymes.
Therelatively slow SV40 cytocidalprocess on
LLC-MK2 and T-22 cells is not sufficient to account for the inability of SV40 to produce
plaqueson monolayers of these cells. No SV40
plaques are produced on these cell lines inas
manyas35days.Our results inno waysuggest
that virus-producing cells remain viable for
this lengthoftime. Therelatively lowplating
efficiency of SV40 on these cells is also not a
sufficient explanation since no SV40 plaques
are produced on monolayers infected with as
many as 104 PFUs. The inability of SV40 to
produce plaques on these cells might involve
therefractory cell fractionaswellas some
com-bination of theabove factors.
The presence of SV40-resistant fractions
among clonal isolates of both LLC-MK2 and T-22 cells indicates that there can be
hetero-geneity of susceptibility to infection among
presumably homogeneous cells. Our results
suggestthat resistant cellsoccuratrandom in the population and that resistance is
tran-siently expressed.
A comparisonofthe interaction of SV40 with
thetwoSV40-transformedpermissivecelllines,
GMK/PARA-7-1 and T-22, indicates the
follow-ingdissimilarities. There isnoevidence foran
SV40-resistant fraction of GMK/PARA-7-1
cells. Also, SV40 produces plaques on GMK/
PARA-7-1 monolayers with anefficiencyequal
tothatonnormalGMK cells(datanotshown).
InsofarasGMK/PARA-7-1 cellscanbe
consid-eredtobetransformedby SV40,the comparison
with T-22 cells is of interest withrespect tothe
wide variation that has been observedinother
aspectsof the interaction ofSV40 with various
SV40-transformed GMK cells (i.e, 8). In this
context, we did not detect a significant
differ-ence between T-22 and GMK-PARA-7-1 cells
with respectto therateof cell killing by SV40
(data notshown).
It maybe noted thatrhesus kidney cultures
infected at anMOI of100PFU/cell, and
subse-quentlypassaged, remain viable. In thecourse
of3months these cultures progressto a stable
carrierstate.Concurrently, all the cells acquire
several properties of the transformed
pheno-type and express the SV40 T antigen
(manu-scriptinpreparation).
ACKNOWLEDGMENTS
Thisinvestigation wassupported by Public Health Ser-viceresearchgrant 5ROI CA12948 from the National
Can-cerInstitute,agrantfrom theAmerican CancerSociety, MassachusettsDivision, Inc.,andbyagrantfrom the Re-search Council ofthe University ofMassachusetts, Am-herst.
The excellent technicalassistanceof James Bruno and Cheryl Goguenisgratefully acknowledged.
Wethank Donald L. Schneider forhis advice on the preparationof cellhomogenatesand the assay for
N-acetyl-B-glucosaminidase.
WearegratefultoJanetS. Butel for kindly providing celllines T-22 andGMK/PARA-7-1.
LITERATURE CITED
1. Allison, A. C., and P. H. Black. 1967. Lysosomal changesinlytic and nonlytic infections with the sim-ianvacuolatingvirus(SV40). J. Natl. Cancer Inst. 39:775-787.
2. Allison, A. C., and K. Sandelin. 1963. Activation of lysosomalenzymes invirus-infected cells and its pos-sible relationshipto cytopathic effects. J. Exp. Med. 117:879-887.
3. Bablanian,R. 1970.Studieson the mechanism of vacci-nia virus cytopathic effects: effects of inhibitors of RNA and protein synthesis on early virus-induced celldamage.J. Gen. Virol. 6:221-230.
4. Bablanlan,R., H. J. Eggers, and I. Tamm. 1965. Stud-ies onthemechanismof poliovirus-induced cell
on November 10, 2019 by guest
http://jvi.asm.org/
CELL KILLING BY SV40 57 age.I.Therelation betweenpoliovirus-induced
meta-bolic andmorphologicalalterations in cultured cells. Virology26:100-113.
5. Barbanti-Brodano, G., P. Swetly, andH. Koprowski. 1970.Superinfection of simian virus 40-transformed permissive cells with simian virus 40. J.Virol. 6:644-651.
6. Blackman, K. E.,and H. C. Bubel. 1969. Poliovirus-induced cellularinjury.J. Virol.4:203-208. 7. Bowers, W. E., J. T. Finkenstaedt, andC. de Duve.
1967. Lysosomesin lymphoid tissue. I. The measure-ment ofhydrolytic activities in whole homogenates. J. Cell Biol. 32:325-337.
8. Butel, J. S., L. S.Richardson, and J. L.Melnick.1971. Variation inproperties of SV40-transformed simian cell linesdetectedby superinfection with SV40 and humanadenoviruses. Virology46:844-855.
9. Easton,J. M. 1964. Cytopathic effect ofsimian virus 40 onprimarycell cultures ofrhesus monkeykidney.J. Immunol. 93:716-724.
10. Findley,J.,G.A.Levvy, and C. A. Marsh. 1958. Inhibi-tionofglycosidases by aldonolactonesof correspond-ingconfiguration.II.Inhibitors of P-N-acetyl-glucos-aminidase. Biochem.J. 69:467-476.
11. Gilbert, V. E. 1963. Enzyme release from tissue cul-tures as anindicator of cellularinjurybyvirus. Virol-ogy 21:609-616.
12. Ginsberg,H.S. 1961.Biological and biochemical basis
for cell injury by animal viruses. Fed. Proc. Fed. Am. Soc. Exp. Biol. 20:656-660.
13. Guskey, L. E., P. E. Smith, and D. A. Wolff. 1970. Patterns of cytopathology and lysosomal enzyme release in poliovirus-infected HE4-2 cells treated with either 2-(a-hydroxybenzyl)-benzimidazole or guanidineHCl. J. Gen.Virol. 6:151-161.
14. Haase, A. T., S.Baron, H. Levy, andJ. A. Kasel. 1969. Mengovirus-induced cytopathic effect in L-cells; pro-tectiveeffectofinterferon. J. Virol. 4:490-495. 15. Katzman, J., and D. E. Wilson. 1975. Newcastle disease
virus-inducedplasmamembrane damage. J. Gen. Vi-rol.24:101-113.
16. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J., Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. 17. Shiroki, K., and H.Shimojo. 1971.Transformation of
greenmonkey kidney cellsby SV40genome:the es-tablishmentof transformedcell lines andthe replica-tion of humanadenoviruses and SV40 in transformed cells.Virology 45:163-171.
18. Studzinski, G. P., J. F. Gierthy, and J. J. Cholon. 1973. Anautoradiographic screeningtestformycoplasmal contamination ofmammalian cell cultures. In Vitro 8:466472.
19. Sweet, B. H., and M. R. Hilleman. 1960. The vacuolat-ing virus SV40.Proc. Soc.Exp. Biol. Med. 105:420-427.
VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/