JOURNAL OF VIROLOGY, Dec. 1980,p.847-859
0022-538X/80/12-0847/13$02.00/0 Vol.36,No.3
Defective
Interfering Influenza
Viruses and Host Cells:
Establishment
and
Maintenance
of Persistent Influenza Virus
Infection
in
MDBK and HeLa Cells
BARUN K. DE ANDDEBI P. NAYAK*
Department of Microbiology and Immunology, University of California-Los Angeles School ofMedicine, Los Angeles, California90024
WSN (HONI) influenza virus upon undiluted passages in different species of
cells, namely, bovine kidney (MDBK), chicken embryo (CEF), and HeLa
cells,
producedavarying amount ofdefective interfering (DI) viruswhich correlated
well with theability ofthe species of cell to produceinfectious virus. However,
thenatureof theinfluenzaDI viralRNA produced from a single clonal stock was
essentiallyidentical in allthreecelltypes, suggesting that these cells do not exert
agreatselectivepressurein the amplification of specific DI viral RNAs either at
early or late passages. DI viruses produced from one subtype (HON1) could
interfere with the replication of infectious viruses belonging to other subtypes
(HlNl, H3N2). DI viralRNAs could alsoreplicate with the helper function of
othersubtype viruses. The persistent infection of MDBK and HeLa cells could
be initiated by coinfecting cells with both temperature-sensitive mutants (ts-)
and DI influenza viruses. Persistently infected culturesat early passages (up to
passage 7) showed a cyclical pattern of cell lysis and virus production (crisis), whereas, at later passages (after passage 20), they produced little or no virus and
wereresistant toinfection byhomologous virus but not by heterologous virus.
Themajority of persistently infectedcells,however, contained the complete viral
genome since they expressed viral antigens and produced infectious centers.
Selectionof aslow-growing temperature-sensitive variant rather than the
pres-ence of DI virus or interferon appears tobe critical in maintaining persistent
influenzainfection in these cells.
Over the last fewyears we have studied the
properties ofvonMagnus influenza viruses (37)
produced in MDBK cells (25a). Like other
de-fective interfering (DI) viruses, these particles
appearnoninfectious, replicatein the presence
ofhelper infectious virus, and interferewiththe
replication ofhelpervirus. Furthermore, these
influenza DIparticlesalsopossess, inadditionto
viral RNA (vRNA)segments,smallerRNA
mol-ecules (DI RNA) of vRNA origin which are
absentininfectious viruspreparation (6, 17,25,
26).These DI RNA segmentsarise fromspecific
vRNAsegments(mostlyfrom Pgenes) by
inter-naldeletion(7, 8,25)andareresponsibleforthe
interferingproperty(18).
TheproductionofDIvirusesdependsonthe
natureof the host cell. With vesicular stomatitis virus (VSV), Holland et al. (14), and more
re-cently Kang et al. (19), have shown that the
sameclonal stockproduces different DI viruses
in different cell types, although the same DI
virus isproducedinagivencell type. Stark and
Kennedy (21, 36) also reported a strong host
influenceintheproductionofSemlik Forest DI
virus. The hostrange of influenza viruses varies
widely (2, 16, 32-34). WSN virus (HON1) has
been shown to possess adifferentialgrowth
po-tentialindifferenthostcells.Forexample, WSN
virus produceslargely infectious virus inMDBK
cells,predominantly incomplete virusinchicken
embryonic fibroblasts (CEF),andonlyabortive
infection with littleor novirus inHeLacells(5).
However, little information is available on the
role of different species of host cells on the
productionof influenza DI viruses.
Since DI viruses appearto reducethe
repli-cation ofinfectiousvirus andtherebyinhibit the
cellkilling effectofhomologouslytic virus, they
have been implicatedinestablishingand
main-taining persistentinfection both in vivo(15)and
in cultured cells for a number ofviruses, e.g.,
VSV(13), rabies virus(20),reovirus(1),measles virus(29),Japaneseencephalitisvirus(31), Sen-dai virus(30),lymphocytic choriomengitisvirus (28, 39), Sindbis virus (38), and Semliki Forest virus (23). In addition, temperature-sensitive
mutation andinterferon have been showntoaid
inestablishing thepersistently infected culture
in vitro (1, 11,41). Althoughthe mechanism of viral persistency has been studied extensively
847
on November 10, 2019 by guest
http://jvi.asm.org/
848 DE AND NAYAK
for a number of the viruses referenced above, verylittle information isavailable forinfluenza viruspersistent infections either innature orin cellculture.
Inthis paper we haveinvestigated the role of host cells, e.g.bovine (MDBK), chicken (CEF), and human (HeLa) cells, on both the amount and the nature of the influenza DI virus pro-duced in these cells. We have also analyzed the role of DI influenza virus in establishing and maintaining persistent infection of MDBK and HeLacells.
MATERLALS AND METHODS Cells and viruses. MDBK (bovine kidney), MDCK (caninekidney), and HeLa cellsweregrown
andmaintained inEagle minimal medium containing
10% heat-inactivated fetal calf serum (26). Chicken fibroblast(CEF) cultureswereprepared from 11-day-old embryonated eggs and grown in Eagle minimal mediumcontaining 10% heat-inactivated fetal calf se-rum and 10%tryptose phosphate broth. A/WSN/33 (HON1),A/USSR/90/77 (H1N1),A/PC/1/73 (H3N2), andA/Vic/3/75 (H3N2) influenza virus strains were
usedintheseexperiments. Cloneswereisolatedafter six successiveplaque purifications,in MDBKfor WSN
virusandinMDCKcells forotherviruses(26).Clonal stocksweregrownin MDBKcellsforWSN virus and
in MDCK cells for othervirusesasreported earlier
(17). Bothwild-type (ts+)and agroup II
temperature-sensitivemutant(ts52)ofWSN viruswereused(26). Production ofinfluenza DI virus in different cells.Samplesofclonal stocks of WSN virusprepared
in MDBK cells were passed serially, undiluted, in MDBK, HeLa,orCEFcellstoprepareDI virus (26). Clonal stocks ofts52initiallygrown in MDBKcells containednodetectableamountofDIvirus,as deter-mined by infectious center reduction assay and by RNAanalysisusing polyacrylamide gel electrophore-sis(17).
Long-term passage of DI virus. These
experi-ments were initiated with DI virus produced at the
undilutedpassage 2 (p-2).Subsequently,ateach pas-sage,cellswereinfectedwith DIvirusproducedatthe precedingpassageaswellasadditionalinfectious stock
virus. Briefly,MDBK, HeLa, and CEF cultures (150
cm2) wereinfected with 5 ml of DIvirusproducedat
theprecedingpassage, plus0.5mlofinfectious virus
(1PFU/cell).Viruswasharvestedat 14hpostinfection by subjectingthe cellsand culture supernatant to two
cycles of rapidfreezingandthawingandwasused as thesourceof DIvirus inoculum forthe next passage. Thisprocedureofinfectionwith DI andadded infec-tious viruses was continued forup to30 passages in
respectivecell lines. Themultiplicityof infection of DI virus in theselong-termpassages variedfrom2 to 20 defective interferingunits (DIU)per cell atdifferent passages.
Isolation and analysis of 32P-labeled viral RNA.MDBK, MDCK, CEF,andHeLacells(150-cm2 flasks)wereinfected with a mixture ofinfectiousvirus (1 PFU/cell) and DI virus (2 DIU/cell). The labeled viral RNA was isolated (27) and analyzed by slab gel
electrophoresis asdescribed before (17).
Electropho-resis wascarriedouteitherat4°Cor at room temper-aturefor21husing2.2%polyacrylamide-0.8%agarose
gelscontaining6Murea,0.036 MTris-hydrochloride (pH 8.1), 0.03 M NaH2PO4, and0.001 M EDTA as
described byFloydetal.(10).
Oligonucleotide mapping.32P-labeled infectious
orDI viral RNA (approximately 5 x 106to20x 106
cpm) waselectrophoresed inasingle lane ofaslabgel
asdescribed earlier(8). The positions of viral and DI
RNA segments werelocated fromanautoradiograph obtained after30min to 1hofexposure of Du Pont Cronexfilm. RNAwaseluted from thecorresponding gelsegmentsby theprocedure of Maxam and Gilbert
(22). Labeled RNA segments were digested with RNaseT,andanalyzedby two-dimensionalgel elec-trophoresisusingamodifiedprocedure of DeWachter andFiers(9). Thespecificconditions of electrophore-sis used foroligonucleotide mapping have been de-scribedpreviously (8).
Establishment of persistent infections in MDBK and HeLa cells. MDBK and HeLa cellswere
infected with either the infectious virus (1PFU/cell) aloneor amixtureof infectious (1 PFU/cell) andDI virus (p-2for MDBK and p-2forHeLa,2DIU/cell). At 14hpostinfection, infected cultures werewashed
thoroughly withphosphate-buffered saline (PBS) free
of Ca2" and Mg2" (PBS-) and overlaid with fresh
mediumwhich wasreplacedeveryday. Surviving cells eventually formed confluent monolayers of persist-ently infected (Pi) cells. These cellsweresubcultured twice a week at a ratio of 1:4. These Pi cells were
termedMP1,MP2, and MP3orHP1and HP2 when theywere ofMDBK orofHeLa cell origin,
respec-tively. Occasionally these Pi cells underwent crisis with extensivecytopathic effect andvirusproduction, butthey recovered after frequent changes of growth medium.
Challenge with infectious homologous and heterologous viruses. Picell cultures (MP1, MP2, MP3, HP1, and HP2) wereinfected with infectious virus(2to 10PFU/cell)atdifferentpassages. At14 h
postinfection the flaskswerechecked for cytopathic effect, and the supernatantswereanalyzed for PFU, DIU, andhemagglutinationunits(HAU)permilliliter
asdescribed earlier (17). For heterologous virus chal-lenge, Pi cellswere infected withNewcastle disease
virus (2to 10PFU/cell) andanalyzedforvirus pro-duction.
Infectiouscenterassay.Picellsweretrypsinized
and added at different concentrations to fresh MDBK
monolayers.After1 h at37°C, the agaroverlay me-dium was added andkepteither at34°Cor at39°C;
after4to5days, plaqueswerecounted (17).DIUper milliliterweredeterminedbyusingtheinfectious cen-terreductionassay as described previously (17).
Indirect fluorescent-antibody staining.
Unin-fected cells (MDBK and HeLa), acutely infected
MDBK and HeLacells, and a number of Pi cellsof different passages were grown onmicroscopic slides for 24 h and fixed with acetone in cold (-20°C) as
described by Roux and Holland (30). Subsequently theywerecovered with asolutionofanti-WSNrabbit serum(1:20dilutioninPBScontaining 500 hemagglu-tination inhibition units per ml) for 30min at room J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
DEFECTIVE INTERFERING INFLUENZA VIRUS 849
temperature, washed thoroughly, andfinally stained with fluorescein-labeled goat anti-rabbit 7S globulin (1:25in PBS-; GIBCO) for 30 min at room tempera-ture. Theselabeled cells were further washed twice with PBS- and examined under a microscope equipped withaUVlight source.
To detect the presence of viral antigen on the cell surface, live cells were used.Cells were trypsinized and incubated for 2 to 3 h at 37°C with the original conditioned growth medium. These cells were then washed with PBS- and incubated with anti-WSN an-tibody (1:20inPBS)at0°C for 30min.Subsequently they were washed in PBS- and stained with fluores-cein-conjugated goat anti-rabbit 7S globulin for 30min
at0°C. The suspended cells were washed twicefurther
withPBS- and examined for immunofluorescence.
RESULTS
Production of DI viruses in MDBK, CEF,
and HeLa cells. To determine the effect of
differentspecies of host cellsontheproduction
ofDIvirus,infectiousts52 WSN clones,sixtimes
plaque purified, were used to prepare clonal
stocks. Samples of clonal stock ofts52 (clone4;
2.8 x 107PFU/ml, 2,048HAU/ml) were
inocu-lated into MDBKandCEF (0.01PFU/cell)and
HeLa cells (1 PFU/cell). At low multiplicity
(0.01 PFU/cell), Helacellsproducedverylittle
virus. Totalvirus washarvested from these
in-fected cultures at 14 h postinfection and was
calledp-0. p-1viruswasproducedinrespective
cells by usingp-0 virus at 20 PFU/cell.
Subse-quently,5ml of undiluted viruspreparationwas
passedserially for the production ofDIvirus in
respective cells (26). Infectious virus (PFU per
milliliter), DI virus (DIU per milliliter), and
total virusproduction (HAUpermilliliter)were
assayedatdifferent passages by theprocedure
describedpreviously(17, 26).
DI virus was notdetectedat p-0 inMDBK,
CEF, or HeLa cells. At p-1, MDBK cells
pro-duced4 x 105DIU/ml,whereasboth CEFand
HeLa cells produced 1 x 105 DIU/ml.
Subse-quently,inseriallyundiluted passages(p-3to p-5), more DI virus wasproducedinall threetypes
of cells. However, both CEF and HeLa cells
produced only a moderate amount of DI virus
(CEF, 2 x 105 DIU/ml atp-4; HeLa, 2.5 x 105
DIU/mlatp-3) whencomparedtothe maximum
productionin MDBK cells(2.4x 106 DIU/mlat
p-5). Thetotalvirusparticleyield,asdetermined
by HAU,wasreduced inallcellsupon undiluted
passages. As the production of DI virus
in-creased,theoverallyield of infectious virus
de-creased in all threespeciesofcells. Atp-4,HeLa cells contained a detectable hemagglutination
titer(16HAU/ml),althoughnoDIorinfectious viruscould be detected. Theproductionof
hem-agglutinating antigen at p-4 may represent an
abortivereplicationofWSNvirus in HeLacells
(e.g.,intracytoplasmic inclusions[4]).
Inexperiments involving long-tern passage of
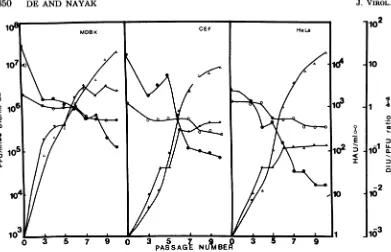
DI virus,cellswerecontinuously coinfected with DI virus andadditional infectious virus at each passage. Theresults (Fig. 1)show that the
over-all patterns of the production ofDI virus and
infectious virus, as well as the hemagglutinin
titer, were essentially similar inall three types
ofcells. Therewas agradual decrease of
infec-tiousvirusyield, accompanied by an increase in
DIvirus production, irrespective of the nature
ofthe cell. Again, the greatest amount of both
infectious and DI virus was produced in MDBK
cellsascomparedtothatproducedinCEFand
HeLacells.Finally, the DIU/PFU ratio showed
asteadyincrease in allcells(from0.125inp-3 to
19inp-10 for MDBK; from 0.005 in p-3 to 5.5 in
p-10for CEF; from0.007 in p-3 to 11 in p-10 for
HeLa cells). After passage 10, the same DIU/
PFUratiowasmaintainedup top-30 by
contin-ued coinfection with DI and infectious viruses
(data not shown). Thus, it appears that after
continued coinfection with infectious and DI
viruses over many passages, an equilibrium in
the production of infectious and DI viruses is
established.
Nature ofDI virus produced in MDBK,
HeLa, and CEF cells. The natureofRNA of
DIvirusreleased bythesecellsatdifferent
pas-sages was analyzed bypolyacrylamide gel
elec-trophoresisandoligonucleotidemapping. Figure
2showsthat the [32P]RNA ofpurified virus at
p-O contained only eightvRNA segments
with-out any detectable amount of DI virus in all
three cell species. However, the virus at the
undilutedp-3,atcoinfected p-9, andatp-30 from
all cell types contained a number of DI RNA segments in addition to the eight standard
vRNA's,althoughpresent invarying ratios.
Fur-thermore, we have rather consistently found
here(Fig.2and4), andpreviously (7,8, 18,26),
that the molar ratio ofM generelativetoother
viralgenesincludingthe NS gene isincreasedin DI particles. This would suggest either that someof the DIparticlescontainonlythe M gene or thatDIparticles arepolyploid in respect to the M gene. Anotheralternative, although
un-likely,isthepresenceofaDIRNAcomigrating
with the M gene. At presentwe have no idea which, if any, of thesepossibilitiesistrue.
The DI RNAsegmentsproducedatp-3, p-9,
and p-30 in all cell types were essentially the same in nature. At least six distinct DI RNA segments were found. At p-3, DIa migrating
faster than the NS genewas mostconspicuous
inall celltypes(Fig. 2A).Atp-30,DIadecreased
and a smaller DI RNA (DIb) became more
VOL. 36,1980
on November 10, 2019 by guest
http://jvi.asm.org/
850 DE AND NAYAK
I
E
LL U.
8
E
I-102
10
II
-1
al
10 X
-2 10
FIG. 1. Analysis oftotal infectious virus (PFUper milliliter, 0), defective interfering virus (DIUper milliliter,A)andhemagglutinin (HA Uper milliliter, 0) produced bythesameclonal isolate(no. 4)inMDBK, CEF, and HeLa cells during continued coinfection using infectious virus and DI virus frompreceding passagesasdescribed in thetext.ThecorrespondingDIU/PFU(A)ratioateach passage isalsogivenunder
thesameconditionsforthe threecell types.
prominent in all three types ofcells (Fig. 2B).
At p-9, anintermediate resultwasobtained(data
not shown). Thus it appears that after many
passages theearlyDI virus isgradually replaced byaDI viruswith asmallerDI RNA segment.
Further sequence relationship among the DI
RNAs wasdeterminedbyoligonucleotide
map-ping (8). DIa isolated from MDBK cells
con-tained spotscharacteristic ofthe P3 (polymer-ase) gene andtherefore originates from the P3
gene(Fig.3AandB).Similarly,DIafrom HeLa
cells and CEF cells containedspotsidentical to
those present in DIa of MDBK cells and is,
therefore, also of P3 origin (data not shown).
Both DIb RNAs isolated from p-30 ofMDBK
and HeLa cells produced essentially identical
oligonucleotide maps and are subsets of DIa
(Fig. 3C). These results suggest that both DIa
and DIb areinterrelatedand originate from the same P3 gene. These results also confirm our previous observation that the majority of influ-enzaDIRNAsoriginate from one of the polym-erasegenes (8).
DI virus-mediated interference against
different subtypes of influenza A viruses.
Interference mediatedbyDIviruses appears to
behighly specificandeffectiveagainst
homolo-gous or closely related viruses but not against
heterologous viruses. With VSV,heat-resistant
(HR)DIparticlesof the Indianaserotype
inter-fere poorly withNewJerseyMserotype.
How-ever, DI NewJerseyMvirusinterferesequally
with M and Ogden virus (24). Since a large
number ofserotypesof influenza virusare
avail-able, we tested the interfering ability ofWSN
DI (HON1) against USSR (HlNl), Port
Chal-mers(H3N2), and Victoria (H3N2) viruses. We
determined theinterfering titer of WSNDIvirus
against these virusesby usingadirect infectious
center reduction assay as described previously
(17). Theresults (Table 1) show thatessentially
thesametiter ofinterfering activityofWSNDI
virus (DIUper milliliter) wasobtained against
different subtype A viruses. Additionally, in a
separateexperimentwefound that the yield of
different subtypeviruses was reduced by
coin-fecting with WSN DI virus (data notshown).
Theseresults show thatWSNDI virusisequally
effective againstall influenzaAviruses.
Prelim-inarydatasuggest thatWSNDIis noteffective
againstinfluenza B viruses.
Tofurther determine whether WSN DI can
replicate by using thehelperfunctionof other influenza Aviruses,MDCKcellswerecoinfected with both DIandinfectiousviruses and labeled
with
32pO4
(8).Released virus was isolated andanalyzedinpolyacrylamide gels. Figure4shows
that inspiteofrepeated cloning allthree viruses containedvisibleDI RNAsegments. These re-sults
confirn
ourearlier findingthatmost,ifnot J. VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.496.64.455.57.309.2]DEFECTIVE INTERFERING INFLUENZA VIRUS
A OCO 0CO OC°
CL l a. a- a-Q
NS 2S
DHaA up
B 0
I--.
.ow.
_o~--f #
Nw
--
Dib-FIG. 2. Analysis of[3'P]RNAofstandard virusatp-Oand DI virusesproduced atp-3(A) andp-30(B) duringcontinuedcoinfectioninMDBK, CEF,and HeLa cellsusingthesameclonal stockvirus. Cellswere
infected with a mixtureofstandard virus(1 PFU/cell) and DI virus (2 DIUlcell). 32P-labeled RNA was
isolatedfrom purifiedvirus andanalyzed by polyacrylamide gel electrophoresis for21 hat4°C using180 V
(17).Arrows indicate thepositions ofsomeofthe DI RNA segments. DIa and DIbappear to be themost prominent DI RNAs in allthree typesofcellsatp-3and p-30, respectively. M(PO), M(P3), M(P30), C(PO), C(P3), C(P30), H(PO), H(P3), H(P30)representMDBK, CEF,and HeLa cells atp-0,p-3,andp-30, respectively.
all, wild-type viruses contain DI viruses.
Whether these visible DI RNA segments are
newly formed during plaque isolation and
pro-duction ofclonal stockor arepre-existing
con-taminants is being currently investigated. Our
gel analyses showed that WSN DI RNA can
replicateefficientlyin the presence ofHlNland
H3N2helperviruses andcaninterfere withthe
replicationof vRNA segments.AnalysesofRNA
by polyacrylamide gel electrophoresis at two
temperatures (room temperature and 4°C)
clearlyshowed that themajority ofvRNA
seg-ments,asexpected,arepredominantlyofhelper
virus origin (Fig. 4). A reduction of specific
vRNA segments ofchallenge virus as reported
previously (5, 17, 25) was also observed (e.g.,
band2from top ismissinginUSSR xDI,lane
6,Fig. 4).When thehelpervirus also contained
DI RNA segments, both WSN DI RNA and
helper virus DI RNA could replicate in
coin-fected cells. Similarly,DIvirusmade from Port
Chalmers(H3N2)virus couldalso interfere with
WSN, USSR,and Victoria subtypes, and Port
Chalmers DI RNA could replicate with the
helper function of these viruses (data not
shown). These results suggest that WSN DI
virus can interfere with as wellas replicate in
the presence of other subtypes ofinfluenza A viruses.
Establishment ofPi cultures. Since
per-sistentinfectionof cultured cellswith manylytic
viruses has been established, itwas of interest to determine whether influenza virus can also
establishpersistent infectionandtoanalyzethe
conditions thatfavortheestablishment of
per-sistent infection.
No Pi cultures of MDBK (Pi MDBK) and
HeLacells(Pi HeLa)could be established when
cells were infected with either wild-type (ts+)
virus (0.1 to 1 PFU/cell) or temperature-sensi-tivemutantalone(0.1to1PFU ofts52percell).
Picultures couldnot beestablishedeven when cellswerecoinfected with ts+ and DI virus. On
the other hand, Pi MDBK (MP1, MP2, and
MP3)and Pi HeLa cell(HP1andHP2) cultures could be routinely obtained by infecting cells
VOL. 36,19860 851
0 0.0
CO CY) CY)
13- a_
a_-m
C)
2
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.496.96.383.68.343.2]852 DE AND NAYAK
with both an infectious temperature-sensitive
(ts52) virus and DI virus simultaneously. The source of DIvirus, whether obtained from ts+ or ts-mutants, did not affect the establishment of Pi cultures.
At earlypassages (<p-7),cells showed a
typi-calcyclicalpattern of cytopathiceffect and virus
production (crisis) usually immediately after
subculture. Pi cultures subsequentlyrecovered
upondaily washing with fresh medium and
be-*
tel.
*w
-~~
_0 _17
. .
A
%.*4-H11
!-
7
Iee%ft\1 0-
Ell
came confluent again. These Pi cultures
ap-peared more rounded and refractile than the
uninfected cells and wereeasily dispersed with
trypsin.
Presenceand expression of viralgenome
in Pi cultures. Pi cultures, once established,
can grow and multiply as well as produce a
relativelysmallamount of virus. There are two
likely explanations fortheseresults: (i) asmall
percentage ofthecells in cultures are infected
*;s
J8--J
4-
t-f
1
1
E
7-a
1.1x
P3
X
DDI
a
(MDBK
)
a
x
*-H11
*
Fi
Ix
Dlb(MDBK)
FIG. 3. Oligonucleotidemaps of P3, DIa,and DIbRNAs. Individual[32P]RNA segments were isolated,
digestedwithT,ribonuclease,andanalyzed by two-dimensional gel electrophoresis (8). Detailed oligonucleo-tideanalyses ofindividual genes have been reportedpreviously (17, 25a). Only the spots common among P3,
DIa,and DIb areidentified. x's (bottomandtop)represent xylene cyanol and bromophenol blue,respectively.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.496.80.457.186.611.2]DEFECTIVE INTERFERING INFLUENZA VIRUS
andare producing virus at a given time or (ii)
the majority, if not all, of the cells are infected
andcontain viralgenome, but either some orall
of them areproducingsmall amounts of virus. A number ofexperiments describedbelowsupport
the latter explanation. First, the Pi cultures
(MP1, MP2, MP3, and HP1, HP2) were resistant
tosuperinfectionbyhomologousbut not
heter-ologous viruses. Both early- (<p-16) and
late-passage (>p-30) Pi MDBK and Pi HeLa cells
showed little cytopathic effect (<15%) after
su-perinfection with WSN virus. Control MDBK
celLs, wheninfected with either ts52 or ts+ WSN
virus, produced 2,048 HAU/ml and 300 x 106
PFU/ml, whereas Pi MDBK cells (MP2 and
MP3)produced only32HAU/ml and3 x 103to
5 x 103PFU/ml uponsuperinfection (Table 2).
Similarresults were obtained with HP1 and HP2
cells as compared tonormal HeLa cells. How-ever, whencells were challengedwith a
heter-ologous virus (Newcastle disease virus), both
normal cells and Pi cellswere equally sensitive.
Over 80% of cells showed cytopathic effect
within24hafterinfection, and high HAU(4,096
to8,194HAU/mlinMDBK;512to1,024HAU/
mlin HeLacells) and infectiousvirus titers (50
x 106to 70x 106PFU/ml for MDBK and 3 x
105 to 5 x 105 PFU/ml for HeLa cells) were
obtainedinbothnormal andPicultures.
Second, the expressionof viralantigensin Pi
cellswasexaminedbyimmunofluorescence and
electron microscopy. Accordingly, MP2, MP3,
HP1, and HP2 cultureswere examined forthe
TABLE 1. Titerof WSN DI virus measured against challengeinfectiousvirusesof different Asubtypesa
Challengevirus DIU/ml
WSN(HONi) ... ... 16.5x106
PortChalmers(H3N2) .... 7.5 x106
USSR(HlNl) ... .... 10.5x106
Victoria (H3N2) ... 7.8x 106
'Titerof WSN DI viruswasdeterminedby infec-tiouscenterreduction assay against different A sub-type viruses. WSN DI viruswas prepared by high-multiplicity passages as discussed previously (26). MDCK cellswereinfected with different dilutions of WSN DI virus andsuperinfected with4PFU/cellof challenge viruses of different subtypes. Cells were trypsinized and platedonmonolayers of MDCK cells for infectious center assay. Reduction of infectious
centersof eachchallengevirus atdifferentdilutionsof
WSN DI virus was determined and the number of DIUofWSNDI viruspermilliliterwascalculatedin
comparison with each challenge virus as described previously (17). Clonal stocks of infectious WSN
(HON1),PortChalmers(H3N2),Victoria(H3N2),and
USSR(HlNl) viruspreparationscontained30x 106 PFU/ml and 1,024 HAU/ml, 15 x 106PFU/ml and
512 HAU/ml, 17x 106PFU/mland1,024 HAU/ml, and10x106PFU/mland512HAU/ml,respectively.
Z xCCzz
co) z x cn) co D
cp Q , CL)D
D)
-5- n L
FIG. 4. Replication of WSN DI RNA with the helperfunctionof other influenzaAsubtypes: USSR (HINI),Port Chalmers(H3N2), and Victoria (H3N2). WSN DIwaspreparedfromasix-times-cloned virus by three consecutive passages. Subsequently, cells
wereeitherinfected with standard virusorcoinfected with both standard virus and WSN (p-3) DI virus. 32P-labeledviral RNAwasisolatedandanalyzed by polyacrylamide gel electrophoresis as described in Fig.2.Arrowsshowanumberofprominent DI RNA segments.In spite of repeated cloning, standard Port Chalmers and Victoria viruses also contain DI RNA segments.
expression of viral antigens by indirect
immu-nofluorescence using antisera against WSN
vi-rus.Approximately70 to80% of thecells showed
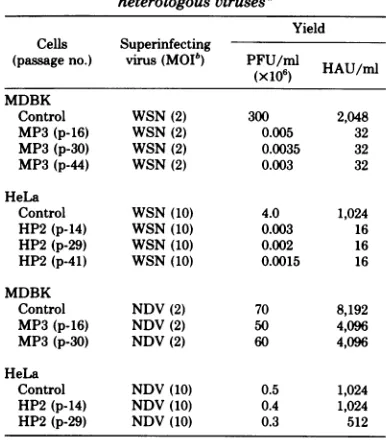
apositive reaction. Both acetone-fixed (Fig. 5)
andlive cells(Fig. 6) demonstrated thepresence
of viral antigen in the cytoplasm and cellular
membrane ofinfectedcells.Tofurtherconfirm
the expression of viral genes, thin sections of
bothacutelyinfected and Picellswere studied
by transmission electron microscopy. Results
confirmed that althoughno viruswas released
in the supernatant,buddingvirusparticleswere
present on the membrane of Pi cells (data not shown). Finally,infectious centerassay further
demonstratedthepresence of the viral genome
in Picells. PiMDBK and Pi HeLacells,when
platedonmonolayersofuninfectedMDBKcells,
showed a 70 to 80% plating efficiency as
com-paredtothe 100% plating efficiencyofacutely
infectedMDBK and HeLacells (Table3).This
slightlylowerplatingefficiencyof Pi cellscanbe due toeither DI virus present insome Pi cells
853
VOL. 36,1980
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.496.268.417.72.302.2] [image:7.496.41.232.435.488.2]854 DE AND NAYAK
TABLE 2. Virusproductionafter superinfection of persistentlyinfected cellsbyhomologousand
heterologous virusesa
Yield Cells Superinfecting
(passageno.) virus(MOjb) PFU/ml HAU (X106)
HUml'
MDBK
Control WSN(2) 300 2,048
MP3(p-16) WSN(2) 0.005 32
MP3 (p-30) WSN(2) 0.0035 32
MP3 (p-44) WSN (2) 0.003 32
HeLa
Control WSN (10) 4.0 1,024
HP2(p-14) WSN(10) 0.003 16 HP2 (p-29) WSN(10) 0.002 16 HP2 (p-41) WSN (10) 0.0015 16 MDBK
Control NDV (2) 70 8,192
MP3 (p-16) NDV (2) 50 4,096
MP3 (p-30) NDV(2) 60 4,096
HeLa
Control NDV(10) 0.5 1,024
HP2(p-14) NDV(10) 0.4 1,024
HP2(p-29) NDV (10) 0.3 512
aNormalMDBKandHeLa cellsand anumber of persist-ently infected cells at different passages were superinfected withWSN virus or Newcastle disease virus (NDV) for 14 h as described in thetext.Thesupernatantswerethenharvested andanalyzed for HAU as well as PFU (for WSN virus we used MDBKmonolayersat34°C,and for Newcastledisease virus weused CEFat37°C).
bMOI,Multiplicity of infection.
preventing plaque formation (17) or somecells
notproducinganyinfectious virusatall. These
above experiments suggest that the virus
ge-nomeispresent andexpressed (at leastpartly)
in the majority of Pi cells, although complete
infectiousparticlesarenotproduced.
Nature ofvirus expressed by the Pi
cul-tures.Asdiscussedabove,littleornodetectable
infectious viruswasreleasedby the Picultures
(Table 4),althoughPicellsproduced infectious
centers onmonolayersofMDBKcells(Table 3).
Todetermine thenatureof the virusreleasedby
the establishedPicells,anumber ofexperiments
were done. In one, persistently infected cells
were trypsinized and plated on monolayers of
MDBKcells for infectious centerassay at
340C
(permissive) and
390C
(nonpermissivetempera-ture). Results show that little or no wild-type
(ts+)viruswasreleased from Picultures (Table
3). This was not surprising since Pi cultures could beestablishedonlywith ts-mutants, and wedid notdetectanywild-typerevertants in Pi
cultures. In thesecond type ofexperiment, the
virus released byPi cellswas amplifiedby
co-cultivating with monolayers of MDBK cells.
Again,virus was releasedonly at34°C andnot
at
390C.
Additionally, the viruses released at340C
were also ts- mutants and did notform
plaques at 390C. Viruses isolated from Pi
cul-turesformed smallerplaqueseven at340C than
did the original clone4 ofts52virus.
Addition-ally, the clones isolated from Pi cultures
pro-ducedaloweryieldat340C.
Role ofts- mutants in establishing and maintaining persistent cultures. Our data
suggestthat both DI virus andts- mutants are
needed inestablishing Pi cultures with influenza
virus.Two typesof experimentswereperformed
tofurther evaluate the role ofts mutantsinboth
establishingandmaintainingthepersistent
cul-tures. First, MDBK and HeLa cells were
in-fected with thets- mutantalone and incubated
eitheratpermissive (340C) andsemipermissive
(3700)or atrestrictive(390C)temperatures. We
found thatover99% of thecellsweredestroyed
in 3 dayseither at 34 or37°C,whereasonly40
to 50% of cells were killed at the restrictive
(390C) temperature, using the ts- virus alone.
However, eventually all cells underwent lysis,
andno Picultureswereobtainedateither
per-missive orrestrictive temperatures. Inthe
sec-ondtype ofexperiment, Pi cultures (MP2, MP3,
HP2,HP3)that wereestablishedby coinfection withts- mutantand DIvirus and maintainedat
370C were shifted to either 34 or 390C. After
shiftdown, thesePiculturesdemonstrated
nei-ther crisisnor anyincreasedamounts of
infec-tiousvirus, as comparedtoPicultures keptat
370C.Norwasthereanyobservedchange when
thePicultureswereincubatedat390C.
Absence of detectable interferon in Pi
cultures. Culturesupernatants fromPiMDBK
and PiHeLa cells were used for preparing
ex-ogenous interferon andwereassayed for
inter-feronbyplaquereductionassay inhomologous
cells with VSV (3). There was no detectable
amount of interferon ineitherPiMDBKorPi
HeLacells. Additionally, both of these Pi
cul-tures, although resistant to homologous
infec-tion, were sensitive to heterologous infection,
namely Newcastle disease virus (Table 2) and
VSV (datanotshown). Thesetwoexperiments
suggest that neither exogenous nor endogenous interferon was amajorfactor in themaintenance
ofpersistent influenza virus infection ofHeLa
andMDBK cells.
Role ofDIvirus in themaintenance ofPi
cultures. Since DI particles have been
impli-cated inpersistentinfection,anumber of
exper-imentsweredone todetermine whether DI
vi-ruses werepresent inpersistently infected cul-tures. Accordingly, the releasedvirus was
am-plified and labeled by plating Pi cells on the
monolayerof MDBKcellsandincubating in the
presence of32PO4 medium.
[32P]vRNA
wasan-alyzedinpolyacrylamide agarose gels (26). The
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.496.64.257.95.315.2]FIG. 5. Presence of viral antigen inpersistently infectedMDBK (MP3) andHeLa (HP2) afteracetone fixation. Cultures (24 h old) grown on coverslipswerefixed in acetone and examined for viral antigens by indirectimmunofluorescenceasdescribed in the text. (A)UninfectedMDBK; (B) acutelyinfectedMDBK; (C)
persistentlyinfected MDBK, MP3; (D) uninfectedHeLa; (E) acutelyinfectedHeLa; (F)persistentlyinfected
HeLa, HP2.
855
on November 10, 2019 by guest
http://jvi.asm.org/
I
[image:10.496.72.458.51.623.2]I
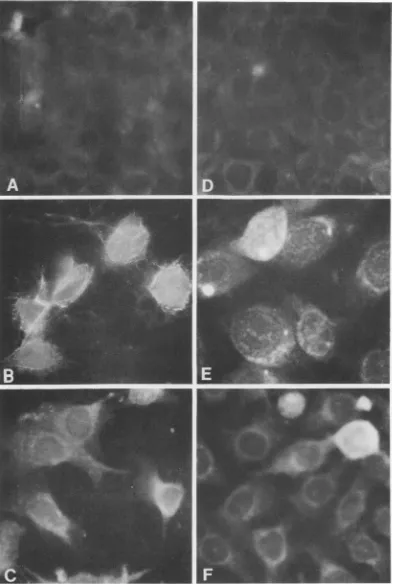
FIG. 6. Thepresence of viral antigen on the membranes of persistently infected cells. Live cells were examined for the presence ofviral antigen by indirect immunofluorescence as described in the text. (A) UninfectedMDBK; (B) acutelyinfected MDBK; (C) persistently infected MDBK, MP3; (D) uninfected HeLa;
(E)acutelyinfectedHeLa; (F) persistently infected HeLa,HP2. 856
on November 10, 2019 by guest
http://jvi.asm.org/
DEFECTIVE INTERFERING INFLUENZA VIRUS 857
TABLE 3. Infectious center assay of Picellsa
No. of infectivecenters per Cells(passage no.) 100cells
34°C 390C
MDBKxts52b 100 1
MP1 (p-16) 70 0
MP2 (p-17) 75 0
MP3 (p-17) 71 0
MP1(p-30) 55 0
MP2(p-31) 50 0
MP3 (p-35) 60 0
HeLaxts52b 70 0
HP1 (p-14) 90 0
HP2 (p-16) 80 0
HP1 (p-41) 78 0
HP2 (p-43) 70 0
aResults were calculated from duplicate assays us-ing 400, 200, and 100 trypsinized cells. Pi cells of different passages were trypsinized, plated on fresh MDBKmonolayers(5-cmplates)indifferent numbers (400, 200,and100cellsperplate), and incubated for1
hat37°C after the addition of 5 ml of agar overlay. Theseplates were then incubated at34and390C,and plaques were counted on days 3and4.
bControl MDBK and HeLacellswereinfected with infectious ts52 virus (4 PFU/cell) for 1 h at 370C, trypsinized, and assayed by thesameprocedure.
results showthatonlystandard vRNA segments
couldbedetected,withno appreciable amount
of specific DI RNA segment in the released
virus. Thiswassurprising becauseDIvirus was
usedinestablishing the persistent infection. The
lackof DI RNAcannotbe duetotheinabilityof
DI virustoreplicateineither of thesecells, since
DI viruses can be produced and amplified in
both of these cells.
Tofurtheranalyze thepresenceofDIvirusin
Picultures, culture supernatant obtained after
amplification was analyzed for interference by
deterniining the reduction of virusyieldand the
reduction of infectious center after coinfection
with infectious virus (17). Although the virus
released from Pi cultures had a lower PFU/
HAU ratio, it did not possess any interfering
activity againsthomologous virus eitherin the
yield reduction (Table 4) or in the infectious
center reduction assay (data notshown). This
would suggest that the virus released by Pi
cultures was defective but noninterfering. We arein theprocess of further characterization of
thisvirus.
DISCUSSION
Effects of host on DI virus production.
The production and amplification of DI virus
depend partly onthe parental infectious virus
andpartlyonthe hostcells(14, 19).WSN virus
replicates most efficiently in MDBK cells and
least efficiently in HeLa cells (5). Experiments
reported here show thatboth infectious and DI
viruses are produced most in MDBK cellsand least in HeLa cells. CEFcellsproduce an inter-mediate amount of both infectious and DI vi-ruses.Such a result would suggest that the steps required in the replication for influenza DI virus
areessentiallythe same as those needed for the
replication of infectiousvirus.
The nature of DI RNA(s) generated in all
three species of cells fromasingle clonal stock
appearsidentical bypolyacrylamide gel
electro-phoresis and oligonucleotidespotanalysis. The
results, although apparently different, are not
-totally inconsistent with those observed for VSV
(14, 19). The host factor(s) influencing the
na-ture of VSV DI virus does not appear to be
absolutely selective because (i) someofthe DI
viruses produced in different species of cells
appear identical and (ii) any predominant DI virus, irrespective of its source of origin, can
replicatein agiven cell. Our results suggest that
the nature of influenza DI virus is primarily
determined by those thatarealreadypresentin
the clonal stock and that MDBK, CEF, and
HeLacells donot exert agreatselectivepressure
intheamplificationofinfluenzaDIvirus.
DI virus-mediated interference among
different subtypes of influenza viruses.DI
virus prepared from WSN virus (HON1) can
effectively interferewith thereplicationofother
influenza A viruses, namely HiN1 and H3N2
viruses. Equally, WSN DI RNAs can replicate
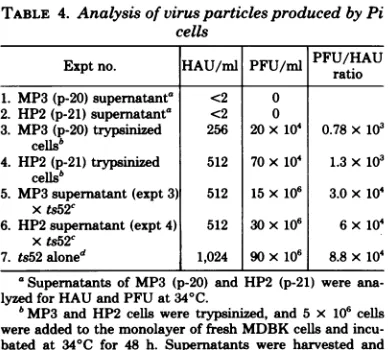
TABLE 4. Analysis of virus particles produced by Pi cells
Exptno. HAU/ml PFU/ml PFU/HAU ratio 1. MP3(p-20) supernatanta <2 0
2. HP2(p-21)supernatanta <2 0
3. MP3(p-20)trypsinized 256 20x 10' 0.78x 103 cellsb
4. HP2(p-21)trypsinized 512 70x104 1.3x 103
cells'
5. MP3supernatant(expt 3) 512 15x106 3.0x 104
xts52c
6. HP2supernatant (expt 4) 512 30x106 6x104 xts52c
7. ts52aloned 1,024 90 x106 8.8x 104
aSupernatantsof MP3 (p-20) andHP2(p-21) were
ana-lyzedfor HAUandPFU at340C.
bMP3and HP2 cellsweretrypsinized, and5x 106cells
wereaddedtothemonolayerof fresh MDBK cellsand incu-batedat 34°C for 48 h. Supernatants wereharvested and analyzed for HAU and PFUat34°C.
cMonolayersof MDBK cells (150cm2) were coinfected with 5 ml of the supernatant (experiments 3 and 4) and infectious ts52virus(1 PFU/cell). The virusyieldwas har-vestedat 48hand34°CandassayedforHAU and PFU.
dMDBK cellswereinfectedwith ts52 virusalone(1PFU/ cell)for 48 h at34°C.
VOL. 36,1980
on November 10, 2019 by guest
http://jvi.asm.org/
[image:11.496.244.438.427.602.2]858 DE AND NAYAK
in the presenceof infectious virus of other
sub-types.These resultssuggestthat thegene
prod-uctsrequired for thereplication of DIRNAs,as
wellasthose for the interference ofvRNA's,can
functionequallywell amongthe viralsubtypes.
These results are not surprising since genetic
reassortmentexperiments amongdifferent
sub-types suggest that the genes among different
influenza A subtypes can be exchanged freely
and thatgeneproducts ofonesubtypeare
func-tional with othersubtypes. Our results extend
the idea offunctional genetic exchange among
the viralsubtypestotheDIvirus-mediated
in-terference.
Establishment and maintenance of
per-sistent influenza infection. The results
re-ported here show that influenza viruses under
certain conditionscanestablishpersistent
infec-tion ofMDBK and HeLacellsin culture. The
presence of both DI virus andts- mutants ap-pears to be critical in establishing the initial
condition favorable for persistent infection. As
yet we have been unable toobtain Picultures
byusing eitherts+ virusalone, ts- virusalone,
or ts+ virus with DIvirus. A similar initial
re-quirement of bothDIvirus andts- mutanthas
been reportedfor VSVand BHK-21 cells (13),
whereas DI virus in thepresenceofts+virus has beenshown toestablishpersistentinfection ofa
numberof other cell-virussystems (23, 30,38).
Whether different ratios of DIvirustots+virus
orother conditions suchasinterferontreatment
could help in establishing persistent influenza
infection incellculturesisbeingcurrently
inves-tigated.
Persistent influenza infection in MDBK and
HeLacellsappears not tobemaintainedat the
population level (i.e., a small fraction of cells
being infectedat agiventime)since themajority
ofcellscontainandexpressviralantigen(s).Nor
doesinterferonappear toplayasignificant role
in the maintenance of persistent infection of
influenzavirus in thesecells. Our resultssuggest
thatthe selection ofvirus variantswithgreatly
reduced growthcapacityat
370C
withoutkillinghost cells is probably required for maintaining
persistent influenza infection. Repeated crises
observed in the early passages would assist in
the selection of a viruspopulationwith agrowth behaviorcompatibletocellulargrowth. The vi-ruspopulation obtained from stable Pi cultures
producedsmallplaquesand areducedyield at
340C
compared tothe original clone 4 ofts52.Whether these virus clones isolated from Pi cultureswouldbelesscytolyticand establish Pi
infectionmoreeasily, andprobably without the
helpof DIviruses,isbeingstudiedcurrently.
AlthoughDI virusappears to be critical in the
initialphaseofestablishingpersistent infection,
we wereunabletodetect either the DI virusor the DIRNAinreleased virus from Pi cultures.
During the initial phase ofinfection, DI virus is
needed to suppress the cytopathic effect of
in-fectious virus and to help the survival of host
cells during crisis. In addition, DI virusesmay
accelerate the selection ofanappropriate variant
of infectious virus (12, 35). However, once the
variant isselected andastable cell-virus
associ-ation isestablished, DI virusmay notbe needed
and thus may be eliminated from Pi cultures.
The presence of viral antigens and defective
budding particles on the cellmembrane would
block thereceptors forsuperinfecting
homolo-gous infectious virus but not for heterologous
virus.
In nature aswell as in cell culture there ap-pears tobe acontinuous evolution of influenza
viruses (40).DIviruses, which appear to occur
commonly in influenza virus replication (17),
mayfurther aid inthe selection ofvariants and
thushelpin theevolution of the virus and the
creation ofdiversityamong theviruspopulation.
ACKNOWLEDGMENTS
A/USSR/90/77, A/PC/1/73, A/Vic/3/75 wereobtained from Alan Kendall, Center for Disease Control, Atlanta, Ga. We thankCharles Samuel, University ofCalifornia, Santa Barbara forperforming the interferon assays of supernatants ofpersistentlyinfected HeLa cells. Wealso thank Zane Price forelectronmicroscopyand forphotographs.
These studiesweresupportedin partbyaPublicHealth Service grant from the National Institute of Allergy and Infectious Diseases (AI-12749) andby the National Science Foundation(PCM 7823220).
LITERATURE CITED
1. Ahmed,R., and A. F.Graham.1977.Persistent infec-tions in Lcells with temperature-sensitive mutants of reovirus. J.Virol. 23:250-262.
2.Almond, J. W. 1977.Asingle gene determines the host range ofinfluenza virus. Nature(London) 270:617-618. 3. Baron, S.1969.Interferonproductionassay and charac-terization, p.399-410.In K. Habeland N. P. Salzman (ed.),Fundamental techniques invirology. Academic Press,Inc., New York.
4. Caliguiri,L.A.,and K. V. Holmes.1979. Host-depend-entrestriction of influenza virus maturation. Virology 92:15-30.
5.Choppin,P.W., andM.W. Pons.1970.The RNAs of infective and incomplete influenza virions grown in MDBKand HeLa cells.Virology 42:603-610. 6. Crumpton,W. M., N. J.Dimmock,P. D.Minor, and
R.J.Avery. 1978.TheRNAsofdefectiveinterfering influenza virus.Virology90:370-373.
7.Davis,A.R.,A.L.Hiti,and D. P.Nayak.1980. Influ-enzadefective interfering viral RNA is formed by inter-nal deletion of genomic RNA. Proc. Natl. Acad. Sci. U.S.A.77:215-219.
8. Davis,A.R., and D. P. Nayak. 1979. Sequence relation-ships among defectiveinterferinginfluenzaviralRNAs. Proc. Natl. Acad. Sci. U.S.A.76:3092-3096.
9. DeWachter, R., and W. Fiers. 1972. Preparative two dimensionalpolyacrylamidegelelectrophoresis of 'P-labeledRNA.Anal. Biochem.49:184-197.
10. Floyd,R.W.,M. P. Stone, and W. K. Joklik. 1974. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
DEFECTIVE INTERFERING INFLUENZA VIRUS 859
Separation of single stranded ribonucleic acids by acryl-amide-agarose urea gelelectrophoresis. Anal. Biochem. 59:599-609.
11. Friedman,R.M.,and J. M. Ramseur. 1979. Mecha-nisms ofpersistent infections by cytopathic viruses in tissue culture. Arch. Virol.60:84-103.
12. Holland, J. J., E. A. Grabau, C. L. Jones, and B.L. Semler.1979.Evolution ofmultiple genome mutations during long-term persistent infection by vesicular sto-matitis virus.Cell 16:495-504.
13. Holland, J. J., and L. P. Villarreal. 1974. Persistent noncytocidal vesicular stomatitis virus infections me-diated by defective T particles that suppress virion transcriptase. Proc. Natl. Acad. Sci. U.S.A. 71:2956-2960.
14.Holland, J. J., L. P. Villarreal, and M. Breindl. 1976. Factorsinvolved in the generation and replication of rhabdovirus defective T particles. J. Virol. 17:805-815. 15. Huang,A.S., andD.Baltimore. 1970.Defective viral particles and viral disease processes. Nature (London) 226:325-327.
16. Israel, A., M. Semmel, and J.Huppert. 1975. Host-range mutants offowl plague virus (FPV): comparison of the genome and virus proteins. Virology 68:504-509. 17. Janda, J. M., A. R.Davis, D. P. Nayak, and B. K. De. 1979.Diversity and generation of defective interfering influenza virus particles. Virology95:48-58.
18.Janda, J. M., and D. P. Nayak.1979.Defective influenza virus ribonucleoproteins cause interference. J.Virol. 32: 697-702.
19. Kang,C. Y., T.Glimp,J. P. Clewley, and D. H. L. Bishop. 1978. Studiesonthegeneration of vesicular stomatitisvirus(Indiana serotype) defective interfering particles.Virology84:142-152.
20. Kawai,A.,S.Matsumoto, and K. Tanabe.1975. Char-acterization of rabies viruses recovered from persist-ently infected BHKcells. Virology67:520-533. 21. Kennedy,S.L.T.1976.Sequencerelationships between
the genomeand theintracellular RNA species of stand-ard and defectiveinterferingSemliki Forest virus.J. Mol. Biol.108:491-511.
22. Maxam, A.M., andW.Gilbert.1977.A newmethodfor sequencing DNA. Proc. Natl. Acad. Sci. U.S.A. 74:560-564.
23. Meinkoth, J., and S. L. T. Kennedy. 1980. Semliki Forestviruspersistencein mouseL929cells. Virology 100:141-155.
24. Metzel,P.S., W. M. Schnitzlein, and M. E. Reich-mann. 1978.Characterization of distinct vesicular sto-matitisvirus, NewJersey serotype, isolates with respect tonucleic acidhomologies, interferenceby DI particles andprotein structure, p.515-526.In B.W.J.Mahy and R. D. Barry (ed.), Negative strand viruses and host cells.AcademicPress, Inc., New York.
25. Nakajima,K.,M.Ueda,and A.Sugiura. 1979.Origin of small RNA in von Magnusparticles of influenza virus.J.Virol. 29:1142-1148.
25a.Nayak, D. P.1980.Defectiveinterferinginfluenza viruses.
Annu. Rev.Microbiol. 34:619-644.
26. Nayak, D. P., K. Tobita, J. M. Janda, A. R. Davis, and B. K. De. 1978. Homologous interference mediated bydefective interfering virus derived from a tempera-turesensitivemutant of influenza virus. J. Virol. 28: 375-386.
27. Palese, P., and J. L. Schulman. 1976. Differences in RNA patterns of influenza A viruses. J. Virol. 17:876-884.
28. Popescu, M., and F.Lehmann-Grube. 1977. Defective interfering particles in mice infected with lymphocytic choriomeningitis virus. Virology77:78-83.
29. Rima, B. K., B.W.Davidson,and S. J. Martin. 1977. The role of defective interferingparticles in persistent infection of Verocellsbymeaslesvirus.J.Gen. Virol. 35:89-97.
30. Roux, L., and J. J. Holland. 1979. Role of defective interferingparticles ofSendai virus in persistent infec-tions.Virology 93:91-103.
31. Schmaljohn, C., and C. D. Blair.1977.Persistent infec-tion of cultured mammalian cells by Japanese enceph-alitis virus. J.Virol.24:580-589.
32. Scholtissek, C.,L.Koennecke, and R. Rott. 1978. Host range recombinants of fowlplague (influenza A) virus. Virology 91:79-85.
33. Scholtissek,C.,and B. R.Murphy. 1978.Hostrange mutantsofaninfluenzaAvirus.Arch. Virol. 58:323-333.
34. Schulman, J.L.,andP.Palese.1977.Virulence factors of influenza A viruses: WSN virus neuraminidase re-quired for plaque productioninMDBKcells. J. Virol. 24:170-176.
35. Semler, B. L., and J. J. Holland.1979.Persistent vesic-ular stomatitis virus infection mediates base substitu-tionsinviral termini. J. Virol.32:420-428.
36. Stark,C.,and S.L.T.Kennedy.1978.Thegeneration and propagation of defective interfering particles of Semliki Forestvirus indifferentcelltypes.Virology89: 285-299.
37. von Magnus, P. 1954. Incomplete forms of influenza virus. Adv.VirusRes. 2:59-78.
38. Weiss, B., R. Rosenthal, and S. Schlesinger. 1980. Establishment andmaintenanceofpersistent infection by Sindbis virusinBHKcells.J.Virol. 33:463474. 39. Welsh, R. M.,P.A.Burner, J. J.Holland,M. B. A.
Oldstone, H. A. Tompson, and L. P. Villarreal. 1975. Acomparison of biochemical andbiological prop-ertiesof standard and defective lymphocytic chorio-meningitis virus. Bull. W.H.O.52:403-408.
40. Young, J.F., U.Desselberger,and P. Palese. 1979. Evolution of human influenzaA virusesinnature: se-quentialmutation inthe genomes ofnewHlNl isolates. Cell 18:73-83.
41.Youngner, J.S.,andD.0.Quagliana. 1975. Temper-ature sensitive mutantsisolatedfromhamster and ca-ninecelllinespersistently infectedwith Newcastle dis-easevirus. J.Virol.16:1332-1336.
VOL. 36,1980
on November 10, 2019 by guest
http://jvi.asm.org/
![FIG. 2.prominentduringinfectedisolated(17).C(P3), Analysis of [3'P]RNA of standard virus at p-O and DI viruses produced at p-3 (A) and p-30 (B) continued coinfection in MDBK, CEF, and HeLa cells using the same clonal stock virus](https://thumb-us.123doks.com/thumbv2/123dok_us/1487990.101464/5.496.96.383.68.343/prominentduringinfectedisolated-analysis-standard-viruses-produced-continued-coinfection-clonal.webp)
![FIG. 3.digestedDIa,tide Oligonucleotide maps of P3, DIa, and DIb RNAs. Individual [32P]RNA segments were isolated, with T, ribonuclease, and analyzed by two-dimensional gel electrophoresis (8)](https://thumb-us.123doks.com/thumbv2/123dok_us/1487990.101464/6.496.80.457.186.611/digesteddia-oligonucleotide-individual-isolated-ribonuclease-analyzed-dimensional-electrophoresis.webp)