JOURNALOFVIROLOGY, Jan. 1978,p.104-114
Copyright © 1978 American Society for Microbiology Printed in U.S.A.Vol. 25, No. 1
Virus-Specific
DNA in
the Cytoplasm of
Avian
Sarcoma
Virus-Infected Cells Is a Precursor to
Covalently Closed
Circular Viral DNA in the Nucleus
PETER R. SHANK* AND HAROLD E. VARMUS
DepartmentofMicrobiology, University of California,San Francisco, California 94143
Received forpublication 9 August 1977
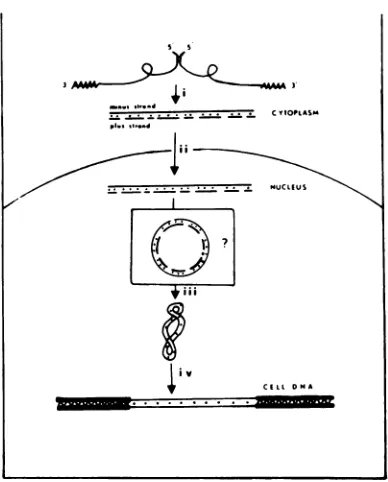
Threeprincipal forms of viral DNA have been identified in cells infected with aviansarcomavirus: (i) alinear duplexmolecule synthesizedin the cytoplasm, (ii) acovalently closed circularmolecule found in the nucleus,and (iii) proviral DNAcovalentlylinkedtohigh-molecular-weightcell DNA. To define
precursor-product relationships among these forms of viral DNA, we performed pulse-chase experiments using 5-bromodeoxyuridine to label by density the linear speciesof viral DNA in thecytoplasm duringthefirst4hafter infection. Aftera 4-to8-h chase withthymidine,aportion of thedensity-labeledviral DNA was
transported to the nucleus and converted to a covalently closed circular form. Weconclude that linear viralDNA, synthesizedinthecytoplasm,is theprecursor toclosed circular DNA observed in the nucleus.
Theability of RNAtumorvirusesto convert
theirsingle-strandedRNAgenomesinto double-strandedDNA isuniversallyrecognized (30, 37), butthemechanism bywhich this unusual form ofsynthesisoccurshasbeen
only partially
elu-cidated. Three major forms ofvirus-specific
DNA have beenidentifiedinculturedcellsafter productiveinfectionbyRNAtumorviruses: (i) linear duplex DNA comprised of a
genome-lengthminus strand(8.5to10kilobases,
comple-mentary to viral
RNA)
andsegmented
plus
strands (0.3 to 1.5
kilobases)
(10, 23,33); (ii)
covalentlyclosed circular
duplex
DNA(form
I) of subunitlength (9, 12, 23);and(iii)
viral DNA covalently integratedinto host cell DNA (pro-virus) (35). After infection by avian sarcomavirus(ASV),the linear
duplex
DNA is observed inthecytoplasm
within4 h(35);
form I DNA isfoundexclusively
inthe nucleusasearly
as5 h after infection (12); and integration ofviral DNAcan occur within9hafter infection (35). Basedupon thesefindings, inhibitor studies (11), andanalogieswithother viruses (1, 2), a hypo-thetical scheme for thesynthesisandintegration ofviral DNA canbe constructed (Fig. 1). The linear form of viralDNA issynthesized in the cytoplasm by virus-associated, RNA-directedDNA polymerase, transported to the nucleus, and then circularized and integrated into host
cell DNA, presumably with the assistance of
host enzymes.
Tovalidatethishypothesis,it is necessaryto
demonstrate that thecytoplasmic linear DNA
is a precursortonuclear form IDNA and that
form I DNA isaprecursor tointegratedprovirus.
In this report we present evidence favoring a
precursor-product relationship between
cyto-plasmic linear DNA and nuclear form I DNA.
Viral DNA constitutes too small a fraction of
totalcellular DNAtopermitlabelingwith
radio-activedeoxyribonucleotides, as in atraditional pulse-chase experiment. We have previously
shown, however,that viral DNAsynthesized in
the presence of 5-bromodeoxyuridine (BUdR)
hasahigh density attributabletothe
substitu-tion of BUdR forthymidineinbothstrands (32,
34).Weused BUdRtodensity label the precur-sorform in the cytoplasm during the first4 h
of infection by ASV; after blocking further
in-corporationof BUdR into viralDNA with
thy-midine, we followed the conversion of
BUdR-substituted DNA into form I molecules in the
nucleus.
MATERIALS AND METHODS
Celsandvirus. Wehaveused the QT-6 ceH line
(derived fromamethylcholanthrene-induced fibrosar-coma ofJapanese quail [21]) to study synthesis of ASV DNA (12).Cellswere propagated as monolayers at41°Cinmedium199supplemented with10% tryp-tosephosphate broth,0.1%(wt/vol) sodium bicarbon-ate, 5% fetal calf serum, 1% heat-inactivated chick serum,and 1%dimethyl sulfoxide (growth medium). Monolayers of QT-6 cellswereinfected with the B77 strain ofASV(B77-ASV)at amultiplicity of infection
(MOI) of1focus-forming unit/cell in the presence of
4yg ofpolybreneperml. B77-ASVwasgrown in and titeredon chicken embryo fibroblasts as previously
reported (19). QT-6 cellular DNA was radioactively
104
on November 10, 2019 by guest
http://jvi.asm.org/
CYTOPLASMIC ASV-SPECIFIC DNA 105
, s
3
i3'
1']_ii_ 1
-^^ ."..*
- **LI. . "I -,- CVIOPLASIA
,."...,.^d ~ ~ ~
~~~~~~~~~~~~~~~~~~~
iiv
_~ ~ ~ ~~~~~~~~~~ILLt D "
FIG. 1. Modelforthe synthesis ofASVDNA. (i) The70SviralRNA molecule isadimercomposedof two35Ssubunitsjoinedattheir 5'ternini(18). Syn-thesisoftheminus strandofASVDNA bythe virion-associatedRNA-dependentDNApolymerase is ini-tiated near the 5' end of the viralgenome on a
tRNA"Pmolecule (5, 29). Using a terminal
redun-dancy (3, 14,24) in the viralRNA, thepolymerase "leaps"tothe 3' endofthesameor adifferentRNA subunit and completes the synthesis ofa
genome-lengthminus strand.Before completion ofthe minus
strand,plus-strand DNAsynthesis commences
(un-publisheddataoftheauthors)intheopposite
direc-tion, possibly using fragmented genome RNA as
primer. (ii)The linearviralDNAmolecule, synthe-sized in the cytoplasm and comprised of
genome-length minus strands andsegmented plus strands, istransported into the nucleus. (iii) Within the
nu-cleus, theplus-strand fragmentsarecompleted and
joined(possibly bycellularenzymes)immediately
be-forethemoleculeiscovalentlyclosed.Thisconversion
may occur via an asyet unobservedopen circular
intermediate. (iv) Covalently closed circular DNA
presumably integrates covalently into the cellular
genomeat unknown sitesbyarecombinational event
(1).Toallowsynthesis ofgenomicviral RNAdirectly fromatemplateofproviralDNA, atleastonecopy must beorientedsothatit is colinearwith the viral RNA.
labeledbythe inclusionof0.1 ytCi of[tH]- or
["C]-thymidinepermlinnormalgrowthmedium. To label
QT-6 cellular DNAradioactively inthe presence of BUdR, [3H]deoxyguanosine (0.1 to 0.4tiCi/ml) was
includedin the normalgrowthmedium.
BUdRlabeling and washprocedure.To label DNA withBUdR, QT-6 cellsweregrownin normal
growth medium supplemented with 10,g of BUdR
(Calbiochem) per ml. To stop BUdR incorporation,
the cells were washed twice with Tris-glucose (0.14 M NaCl, 5 mM KCI, 5.5 mM glucose, and 25 mM
Tris-hydrochloride, pH 7.4) containing 50 ,ug of
thy-midine perml and then incubated in normal growth medium containing 50,Ug ofthymidine per ml.
Cellfractionation and DNAextractions. Cells
were fractionated into nucleus and cytoplasm after disruption in 1% Nonidet P-40(ShellOil) aspreviously described (12). The nuclei were thendissolved at 1 x 107 per ml in 20 mM Tris (pH 7.2)-10 mM EDTA (TE buffer) andlysedby theaddition of sodium
do-decyl sulfate (SDS) to 1%, and chromosomal DNA
wasprecipitated by the addition of 5 M NaCl to 1 M according to the method ofHirt (15). Afterovernight
incubation at 4°C, the chromosomal DNA was pel-letedbycentrifugation of 13,000 x g for 30 min. DNA
waspreparedfromthecytoplasmandtheSDS-NaCl
supernatantbyconventional methods (32, 35). In brief,
thesampleswereincubated withself-digestedPronase
(500
ig/ml)
at37°Cfor 1 hfollowed byphenol extrac-tionand ethanol precipitation. The DNA wasresus-pended inTE.buffer and digestedfor 1 h with
pan-creatic RNase (100,ug/ml) at 37°C followedby Pro-nasedigestion (250
Ig/ml)
at 37°C for 30min, phenolextraction,andethanolprecipitation.
Gradient sedimentation. Fractionation of DNA
according to the extent of substitutionof BUdR for
thytnidinewasaccomplished byequilibriumbanding
indensitygradientsof CsCl.Samplesof DNA in TE
buffer were sheared by three passages through a 19-guage needle and mixed with solid CsCl (Harshaw radiotracergrade)togiveafinaldensityof 1.75g/cm3. Centrifugationwasfor 60 hat200Cin either the type 40rotor(at33,000rpm) orthe type 42.1 rotor (Beck-man) (at31,000 rpm), dependingon the number of
cellsextracted. Densitiesweredeterminedboth from measurementsof refractive index (with a Bausch and Lomb refractometer) and from the bandingposition
ofradiolabeledcellularDNA.
Covalentlyclqsedcircular DNA(formI)was
sepa-rated from relaxed forms (linear or nicked circular forms IIIandII)by bandinginCsCl density gradients
containing intercalatingdyes. Each gradient contained
300ugof ethidium bromide (EB) (22) orpropidium diiodide (PI2) (16) per ml, and the sample was dis-solved in TE buffer to give final densities of 1.62
g/cm3. The gradients were centrifuged for 60 h at 33,000 rpm in a type 40 rotor at
200C.
Covalentlyclosed circular DNA was also separated
fromopen circular or linear forms bysedimentation in alkaline sucrosegradients (35). Gradients of 5 to 20% sucrosecontaining0.3NNaOH,0.7NNaCl,and 0.001 M EDTAwere prepared inpolyallomer tubes andcentrifugedat 17,000 rpm for 16 h at
200C
in an SW27.1 rotor (Beckman). 3H-labeled plasmid DNA, pML21 (formsIandII;molecularweight, 6.7 x 106) servedasanextemal sedimentation marker.Hybridizationreagents. 32P-labeled
complemen-tary DNA(cDNA) waspreparedinvitro from
deter-gent-activatedB77-ASV in thepresence of
actinomy-cin D asdescribed (8). Theproduct synthesized was fractionated intosingle-anddouble-stranded fractions
on hydroxyapatite, and the single-stranded fraction
wasused forannealing.This32P-labeled cDNA, which
-moor,.--.-- . . . . .t
VOL. 25, 1978
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.505.52.246.62.303.2]SHANK VARMUS
containedsequencesprimarily complementarytothe 5' end of the viral genome (6, 13), had a specific
activity of1x 108 cpm/ug.
"2514abeledASV RNAwaspreparedaspreviously
describedby Commerford (4)and hadaspecific
activ-ity of1x108 cpm/uAg.
Hybridization assay for ASV specific DNA. Hybridization of labeled ASV cDNAtounlabeled viral DNA was performed as previously described (12).
After the addition of100,ug ofcalfthymus DNAas
carrier, sampleswerediluted to1.0ml in0.3 NNaOH,
heatedto80°C for2h(to fragment and denature the DNA), neutralized, ethanol precipitated, and
resus-pended in 20ptlof TE buffer.After addition of1,000 to 1,500 cpm of[nP]cDNA and adjustment of the
NaCl concentrationto0.6M, the sampleswere
over-layered with mineral oil and incubatedat680C for60 h.Annealing of the cDNAwasassayed by resistance
to the single-strand-specific nuclease Si (27). The amountof unlabeled viral DNA ineachsamplewas
then calculated fromacalibration curveconstructed
inparallel with each hybridization experiment by
an-nealing 32P-labeled cDNAto increasing amounts of DNA from XC cells (rat cells transformed by the
Prague C strain of ASV) (28). Since the standard used (DNA from XCcells) is double stranded, the calibra-tion isaccurate only whenbothstrands of theviral DNAarepresent.Whenonlyonestrand of viral DNA
ispresent,i.e.,atthetopofanalkalinesucrosegradient where thelong minus strandsareseparated from the
shortplus strands, this calibrationcannotbe used.
["25I]RNA wasusedtodetect the minus strand of viral DNA. Annealings with [luI]RNA were
per-formedexactlyasdescribed abovefor[nP]cDNA
ex-cept that 100,ig ofyeast RNA carrier per ml was
addedtoeach reaction. Theannealingof[125I]RNA
wasassayed by resistance to digestion byamixture
ofpancreatic and Ti RNase (50,tg/ml,5 U/ml) in 2
xSSC (SSC=0.15M NaCl+0.015Msodium citrate)
at370C for 1 h. The nuclease resistance (ca. 5%) of the cDNA and RNAafterincubation with calfthymus DNAwasconsidered background for the assay and
wassubtracted from eachanalysis.
RESULTS
BUdRlabeling ofviralDNA. We have pre-viously shown (32, 34) thatviral DNA
synthe-sized in duckembryo fibroblasts infected with
ASVcanbedensitylabeled with BUdR. When
cells have been labeled foranappropriatetime,
viral DNAthat isfullysubstituted with BUdR
(HH DNA)canbeseparatedfrompartially
sub-stituted (HL) and unsubstituted (LL) cellular
DNAby banding in CsCl density gradients.
We usedQT-6cells in thisstudybecausethey
supportthree- tofivefoldmoreviral DNA
syn-thesis than duckembryofibroblasts after
infec-tion with B77-ASV (12). In addition, one to
three copiesof form I viralDNA are regularly
synthesizedpercellafterinfection ofQT-6cells
with B77-ASVathigh MOI (12). To document
our abffity to density label viral DNA in the
QT-6 line, cells were infected with B77-ASV
(MOI = 1) in the presence of 10 ,Ig of BUdR and0.1,uCiof
[3H]dG
per ml. After4h thecells were fractionated by the SDS-NaCl precipita-tion of Hirt (15), and DNAwasextracted from thesupernatantfraction and bandedto equilib-rium in aCsCl gradient. Thepositionof LL and HLcellularDNAin thegradientwasdetermined from the 3H radioactivity within a portion of each fraction, and viral DNAwas detected by annealing aportion of each fraction of the gra-dient with virus-specific [32P]cDNA, as de-scribed in Materials and Methods (Fig. 2). All the viral DNA banded at a density of ca. 1.78 g/cm3,consistent with the replacement of 80 to 100% ofthymidine residues by BUdR in both strandsduringinfection ofQT-6cells, as in duck embryo fibroblasts. Cellular DNA banded at densitiesof1.70and1.74g/cm3, appropriatefor LL and HL DNAs.Efficacy of chasing BUdR from QT-6 cells. To use BUdR labeling in a pulse-chase experiment,weneededtodocument our ability toblockincorporation of BUdR into DNA dur-ing the chaseportion ofthe experiment. Wein-traub (38) has shown that incorporation of BUdR into chicken embryo fibroblast DNA is decreasedby90% within1to 2minby washing the cells in medium containing thymidine. We therefore designed a chase procedure in which QT-6 cells were washed twice in Tris-glucose buffercontaining50,ugofthymidineper ml and thenincubated in growth medium containing 50
,ug
ofthymidine per ml.Wefirst assessed the effect of this chase pro-cedure on the labeling of cellular DNA under
ourexperimentalconditions.Cellswereinfected with B77-ASV in the presence of10,ugof BUdR perml. Onesetofcellswaslabeled from2to 4
hafter infection with[3H]dGand harvested4h postinfection; this procedure mightbeexpected
togenerate3H-labeledHL cellularDNA. Four hoursafterinfection,twoothersetsofcellswere washed and incubated with mediumcontaining 50 ug ofthymidine per ml as described above. One ofthese sets was labeled with
[3H]dG
for15 min immediately after the wash, and the
other set waslabeled from 15 to 30 min after
the wash. After extraction and shearing, the DNA from all threesets wasbandedto equilib-rium in CsCl gradients
containing
an intemal "4C-labeled marker of LL QT-6 DNA. As ex-pected, the[3H]DNA
radiolabeled in thepres-ence of BUdR banded at the position of HL DNA (1.74
g/cm3)
(Fig. 3A), whereas the aver-age density of DNA synthesized 0 to 15 min after the chase was onlyslightly
greater than that ofLL DNA(Fig. 3B);allthe DNA synthe-sized 15 to 30 min after the chase co-bandedwiththe14C-labeledLL DNA(Fig. 3C),
on November 10, 2019 by guest
http://jvi.asm.org/
CYTOPLASMIC ASV-SPECIFIC DNA 107
.20
C
z
J
>l
.15 h .10 .05
Density-I
* (9/crn3) 1.80
_ ~ (g/cm') 11.75
1.70 elS CELL HLCELLLL
- ,0 \
I\
- I %O.&.-4.-4.O
5 10 15 20
FRACTION
FIG. 2. BUdR density labeling of viral DNA in QT-6 cells. 2 x10C QT-6 ceUs were infected with B77-ASV(MOI=1)in thepresenceof10pg ofBUdR, 4 pgofpolybrene,and0.1utCi of [3H]dGperml.Four hours after infection the cells were trypsinized,
washedtwice inThis-glucose, andlysed bythe
SDS-NaCiprocedure described by Hirt (15). DNA
ex-tractedfromtheSDS-NaCIsupernatant was
centri-fugedto equilibriumina CsCIdensity gradient at
33,000 rpm in atype40 rotorfor60h at200C. After
collection from below, 10% of each fraction was
counted directly todetermine the banding position
of 3H-labeled LL and HL cellular DNA. Densities
weredeterminedfromtherefractiveindex. After de-naturation andfragmentationby incubation at80fC
in 0.3 NNaOH, the sample waspreciitated with
ethanol, and viral DNA was detected by annealing
32P-labeled ASV-specific cDNA (1,000 cpm) to the
remainderofeachfractionas described in Materials and Methods.Annealingwasassessedby resistance
ofthecDNAtoSinucleasedigestion,andthe amount
ofviral DNA was determined by comparison to a calibration curvegenerated by annealing the/p]
cDNAtoincreasingamountsofXC DNA as described
inMaterials and Methods.
ing that the chase was completely effective within15min.
Although the chase procedure appeared highly effective with respect to cellular DNA, viral DNA, synthesized in the cytoplasm bya
viralpolymerase, mightpossiblyutilize different nucleotide pools. We therefore also tested our
abilitytopreventBUdRincorporationintoviral DNA.UninfectedQT-6cellswereincubated for
4 hwith10
jig
ofBUdRperml, washedtwice, and then infected with B77-ASV in thepresenceof50
Ag
ofthymidine perml. After 8 h, viral DNAextracted from theSDS-NaClsupernatantwasanalyzedinaCsClequilibriumdensity
gra-dient (Fig. 4). All viral DNA detected by
an-nealing with 32P-labeled cDNA banded in the
region of LL DNA, indicating that our wash procedure was effective in blocking BUdR
in-corporation into viral DNA. Since viral DNA
synthesisisdetectablewithin 1 h after infection
underthese conditions (unpublished data ofV.
Smith andauthors), the chase procedure rapidly
stops BUdRincorporationintoviralDNA.
How-ever, it was notpossibletoexamine thekinetics of the chase procedureforviralDNA as strin-gently asforcellularDNA.
Pulse-chaseexperiments with density-la-beled viral DNA. (i) Linear viral DNA is precursor to form I DNA. To test and illus-trate our experimental plan without the com-plexities introduced by cell fractionation, we
per-80
60
40
20
0
O 20
x
a-u 10
z a
I--30
20
10
18
b
x U
u
z a
u
[image:4.505.84.232.59.180.2]5 10 1 5 20 25
FIG. 3. Efficiency of chase with QT-6 ceUular DNA. Three sets of QT-6 cells were infected with
B77-ASV(MOI=1)in thepresenceof10gofBUdR
and4pgofpolybreneperml. Twohoursafter
infec-tion,onesetofcells(A)waslabeled with 0.2,iCiof
pH]dGper ml in the presenceofBUdR. Four hours
afterinfection,setAwasharvested and total cellular
DNA was extracted At thesametime,theremaining
two sets ofcells (B and C) were washed twice in
Tris-glucose containing50pg ofthymidineand
in-cubated ingrowthmedium containing50pg of thy-midine perml.The cells insetBwerelabeledfrom 0to 15minafterthe wash with 0.4uCi of[3H]dG
per ml, and the other ceUs (C) were labeled with
[3H]dG from 15 to 30min afterthe wash. Celular
DNA extractedfromeach setofcells was sheared and bandedtoequilibriuminCsCIdensity gradients,
asdescribedin thelegend ofFig.2,aftertheaddition
of14C-labeledunsubstitutedQT-6ceU DNA. The
gra-dientfractionswerecounteddirectlyin anaqueous
scintiUationfluortodetermine thebanding position of3H(0)-and14C()_-labeledcelularDNA. VOL. 25, 1978
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.505.266.458.167.454.2]
~0.8-
<0.6-z
a
_J
0.4-
>0.2-5 10 15 20
FRACTION
FIG. 4. Efficiencyof chase with viral DNA. QT-6
cellswereincubatedfor4hwith10pgof BUdR and 0.1,uCiofPH]dGper ml. The cellswerethenwashed
twice withTris-glucose containing
50jg
of thymidinepermlandinfected with B77-ASV (MOI=1) in the
presenceof4pgofpolybrene and50pgof thymidine
perml. Eight hours after infection, the cells were
harvested, andthedensity of viral DNA in the
SDS-NaCisupernatantwasdetermined bybandingina
CsCldensity gradientasdescribed in thelegend of
Fig. 2.
formed the preliminary experiment illustrated inFig.5.Ourpreviousresults(12) demonstrated
that little or no form I viral DNA would be
present4hafterinfection. Ourexperimentwas
therefore designed to density label the linear form of viral DNA during the fit 4 h after
infection, wash out the BUdR, and determine
whether density-labeled DNA was converted
into form I molecules.
QT-6 cellswereinfected with B77-ASV in the presence of BUdR and 0.1 ,uCi of [3H]dG per
ml;after 4h,aportionofthe cells(pulse sample)
washarvested,andasecondportion (chase sam-ple) wassubjected tothethymidinechase
pro-cedure and allowedtoincubate foranadditional
4hbeforecollection. ViralDNA,extractedfrom
theSDS-NaCl supernatantofthepulsesample,
banded in the position expected for HH DNA
inaCsClequilibriumdensity gradient (Fig. 5A)
and in the position expected for linear DNA
when rebanded inaCsCldensity gradient
con-tainingpropidiumdiiodide (CsCl-P12) (Fig.50). (Binding ofP12, abuoyantintercalating dye, is
restricted in superhelical [form I] DNA
com-pared with linear ornicked circular molecules,
causingformI DNA to exhibitagreaterdensity
than do the other forms inCsCl-PI2gradients.)
In contrast, viral DNA extracted from the
SDS-NaCl supernatantofthe chasesample
ex-hibitedabroad spectrum of densities after
equi-librium centrifugation in CsCl gradients (Fig.
5B).The DNArangedfrom HH molecules
pre-sumably completed duringthe 4-hlabelingwith
BUdRtoLLmolecules synthesized onlyduring the chase period; the species of intermediate densitywerepresumablymolecules of different "ages," dating from the time of the thymidine chase. Theheterogeneous densityof viral DNA observed after the thymidine chase contrasted sharplywith thehomogeneity of cellular DNA synthesized after a similar chase (Fig. 3B and C). Although it is possible that the chase pro-cedure is lessefficient with respect to viral DNA, the observed data are also consistent with the hypothesis that viral DNA is elongated at a relatively slow rate. This would mean that many incomplete molecules were present 4 h after infection and completed during the chase; this idea issupportedby kinetic analysis of the syn-thesis of viral DNA (seeDiscussion).
When the HH DNA recovered from thechase sample was centrifuged in CsCl-PI2 gradients (Fig. 5D), approximately 25% of the viral DNA banded in the position of form I DNA, demon-strating that linear DNAsynthesized during the first4h ofinfection could be found in a
cova-lently closed circular form4h later.
(ii) Linear viral DNA in the cytoplasm is precursor to form I DNA in the nucleus. To determine whether the linear precursor to form I DNA was present initially in the cyto-plasm of infected cells and then transported into the nucleus, a pulse-chaseexperiment including cell fractionation was performed. The DNAs from the nuclear and cytoplasmic fractions of the pulse and chase cells werebanded to equilib-rium in CsCldensitygradients,andthe position of cellular DNA and viral DNA was detected asdescribedpreviously (legend toFig. 2). All of the viral DNApresent in the 4-hpulsesamples banded at the position of HH DNA (Fig. 6A andC). A small amount of viral DNA(less than 2% of the total) was detected in the nuclear fraction 4 h after infection and probably
re-flectedcontamination of the nuclei with cyto-plasmic material, since the nuclei were not washed after cellfractionation.
Both the nucleus and cytoplasm from the chase cells contained viral DNA banding in a
broadspectrumof densities(Fig.6BandD),in
agreement with the previous experiment (Fig. 5B). Thedensity of each molecule presumably reflects thedegreeof itscompletionatthe time of the chase procedure; therefore thegradient
ofdensitiescorrespondstoagradient of agesof
viral molecules. Most if not all of the oldest (HH) viralDNAinthenucleus must have been fully synthesized in the cytoplasm during the
pulseofBUdR andtransportedto thenucleus duringthe chaseperiod,since therewas atleast
five times more HH DNA inthe nuclei at 8h
thanat 4h after infection.
Similarly,
themole-J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.505.72.223.47.210.2]CYTOPLASMIC ASV-SPECIFIC DNA
.15 .10 m).05
z a
< .15
.10 .05
Pulse Chose
0.8 0.6 0.4 0.2 .25 .20
.15 .10
.05
z a
5:i
5 10 15 20 5 10 15 20
FRACTION
FiG. 5.Analysisofpulse-labeled viral DNA in CsCl and CsCl-P1 gradients. Twosetsof QT-6cells (7x 101 each)wereinfectedwith B77-ASV(MOI=1)inthepresenceof10 pgof BUdR,4ugofpolybrene, and 0.1
itCiof[3H]dGperml.Fourhoursafter infection,thepulsecellsweretrypsinizedandsubjectedtoan SDS-NaCifractionation; thechase cells werewashed as described in the legend toFig. 3 and incubated in growth mediumcontaining50pgofthymidineperml. Eighthoursafterinfection, the chasesample was
harvestedasdescribedforthe 4-hsample. AfterDNAextraction,thepulseand chasesampleswerecentrifuged
toequilibrium in CsCldensity gradients asdescribedin the legend of Fig.2. Viral DNA wasdetectedby annealing20%ofeachgradientfractionwith3P-labeledASVcDNA. Theposition ofHLand LL cellular
DNA, indicatedbythearrows,wasdetermined bydirectly counting1%ofeachgradientfraction.Pools of HH viralDNA,indicatedbythebars,weremade,and the DNAwasrecoveredbyprecipitationwith ethanol. Thesecondarystructureofthe viral DNA wasthen assayed by bandingtheDNAfrom each poolin CsCl densitygradients containingtheintercalating buoyant dyePI2.One-thirdofthe HH DNA recoveredfrom thepulseand chasesampleswasbanded toequilibriuminaCsClgradient (startingdensityof1.62g/cm3) containing300(pgofPI2perml. Thedensityatwhichopencircular(formI)orlinear(form III)HH DNA would bandisindicatedbythearrow. TheCsCl-PI2gradientswerecentrifuged for60 h at33,000rpm ina
type40 rotor at20°C. Viral DNA wasdetected in the CsCl-P1 gradients by annealing with 3P-labeled cDNAafterdenaturation and ethanolprecipitationasdescribedin thelegendtoFig.2.
cules of intermediatedensitywereatleast
initi-ated in thecytoplasm during the pulse period.
However, the siteatwhich thesemoleculeswere
completed and the site of synthesis of the
young-est (LL) molecules found in the nucleus after
the chasecannotbedetermined fromthis
exper-iment. Assuming thatall viral DNA synthesis
is initiated in thecytoplasm, thepresenceof LL
viral DNA molecules in the nucleus at times
when HH and HL moleculesare presentinthe
cytoplasm (Fig. 6B and D) implies thattransport
of viral DNA into thenucleus isnotdependent
ontheageof themolecules.
Fractions from the indicated regions of the
CsClgradientsshown inFig.6AthroughDwere
pooled, and thestructure of DNAineach pool
wasanalyzed by centrifugation inCsCl-EB
gra-dients (Fig. 7). Inaccord with ourprevious
re--ilts (12),noform Iviral DNAwasdetectable in thecytoplasmateither4or8hafter infection
(Fig. 7A). In addition,noneof thesmallamount
ofviralDNAassociated with thenucleusat4h
postinfectionwasformI DNA(Fig. 7B);asnoted
earlier, the small amount of DNA present in
the nuclear fractionat4hprobablyrepresented
cytoplasmic contamination. By 8 h after
infec-tion, when therewasfivetimesmoreHHDNA
in the nucleus than at 4 h (Fig. 6C and D),
approximately 50% of the HH DNA in the
nu-cleus consisted of form I molecules (Fig. 70).
Similar analysis in CsCl-EB gradients of DNA
recovered from the HLregion of theCsCl
gra-dient shown in Fig. 6D indicated that 5to 10%
consisted of form I molecules (datanotshown).
Theseresults indicate that the majority of form
I viral DNA molecules present in the nucleus
musthavecomefromthepool of
BUdR-substi-tutedcytoplasmic viral DNA. Sincewe
encoun-teredsignificant variability in recovering
BUdR-substituted DNA fromthepreparativeCsCl
gra-dients (Fig. 6), ourconclusions arebased upon
theproportionsofforn I DNA intherecovered
DNA(Fig. 7) andnotupontheabsoluteamounts
ofform IDNA.
Since the density of DNA used in these
ex-perimentswasalteredbyBUdRsubstitutionas
wellasby the binding ofintercalating dyes,we
confirmedourobservationsby sedimentation in
I~~~~~ '
C
E L HL CEL LL
/CL'L(EL
D
's
1
CeCI-Pl, CiCI-Pp,
A
A I.1 \\
i
_11A4
109
VOL. 25,1978
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.505.122.360.69.251.2]110 SHANK
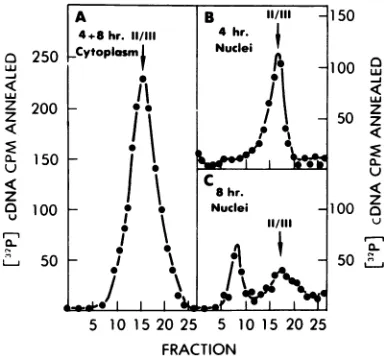
alkaline sucrose gradients, a method independ-ent of the density of DNA. In alkaline sucrose gradients, form I molecules partially denature and collapse into a fast-sedimenting form (36), permitting both the detection of form I mole-cules and an estimation of their size (26). Ge-nomelength strands derived from linear mole-cules should sediment as 18 to 20S in these gradients (26); since there is no evidence for multimeric linearmolecules,viralDNA migrat-ing faster than 20S was presumed to be conva-lently closed circles. In addition, these gradients permitmeasurementof the size of each strand present in the linear viral DNA molecules (see below).
HH viral DNA from the various pools shown
in Fig. 6 was sedimented in alkaline sucrose
gradients and assayed by annealing with
[32P]-cDNA (Fig. 8). These gradients confirmed the observationsmade by equilibrium density cen-trifugation inCsCl-EB gradients (Fig. 7). There was no evidence of rapidly sedimenting (form I) DNA in the cytoplasm at either 4 or 8 h.
(Insufficient viral DNA was recovered from the 4-hsample of nuclear DNA to allow analysis in an alkaline sucrose gradient). The HH sample from the nuclear fraction after 8 h of infection contained twopopulations of forn I viral DNA. The large population sedimented at 65S, con-sistentwith amolecularweight of about 6.5 x 106.The other componentsedimented at around 45S, consistentwithamolecular weight of about
2 x 106 to 3 x 106. This smaller population
presumably represents the highly defectiveform I viral DNA we have previously observed in QT-6 cells infected with B77-ASV (12). Al-thoughcDNAannealed much moreextensively tolinearDNAatthetop ofthese gradients than tothe closedcircularDNA, therelative propor-tionsofform Iandlinear DNAcannot be esti-mated from these analyses. The efficiency of annealing of cDNA to plus strands fromlinear DNA was exaggerated because the minus strands, which normally compete with the cDNA, wereseparatedfrom the plus strands on thebasis of size. This accounts for the apparent
Ts
.28,-4hr Cytoplasm
A CELL CELI
f\ HL LL
-
0 \0
I**I I
4hrNuclei C
.20k CELL CELL HL LL
.12k
.04
;-9
5 10 15
FRACTION
8hr Cytoplasm
B CELL CELL
4'
H.L
LL0 0
I I
4,0ot
8hrNuclei CELL CELL
D ILL
*urii@4' I I
5 10 15
[image:7.505.148.390.331.547.2]FRACTION
FIG. 6. CsCI gradient analysis ofpulse-labeled viral DNA in thenucleus andcytoplasm. QT-6cells(2x
1(P')wereprelabeled for24 h with0.I#CiofPHJthymidineperml. CeUswerethen infectedwithB77-ASV
(MOI= 1) in thepresenceof10 pgofBUdR per mlasdescribedinMaterials and Methods. Four hours
after infection, halfthecellswereharvested and theotherhalfweresubjectedto thechaseprocedureand incubatedforanadditional4 h ingrowthmedium containing50 pgofthymidineperml. Thecells were fractionatedintonucleiandcytoplasm;DNAwasprepared fromthecytoplasm,andanSDS-NaClsupernatant wasprepared from the nuclei as described in Materials and Methods. The DNA was then banded to equilibriuminCsCI density gradientsinatype42.1rotor,and viral DNAwasdetectedby annealing10%of eachfractionwith32P-labeledcDNAasdescribed in thelegendtoFig.2. Thepositions ofthecellularHL
andLL DNA, indicated by thearrow, were determined by directly counting 3Hin 1%ofeachgradient
fraction. Thepools ofHHDNAindicatedbythe barsweremade,and DNA wasrecoveredby precipitation withethanol andanalyzedinCsCI-EB gradients (Fig. 7)oralkalinesucrosegradients (Fig. 8).
1.6
0)
C
< 1.2 z
-J 0.8
> nA
z
a
5:
V.4
I
on November 10, 2019 by guest
http://jvi.asm.org/
CYTOPLASMIC ASV-SPECIFIC DNA
a
LU)
z z z
u
z
u
CL
.n
0 250
200
150 [
100 50 1
[image:8.505.54.246.61.239.2]5 10 15 20 25 5 10 15 2025 FRACTION
FIG. 7. Analysis of pulse-labeled viral DNA in CsCI-EBgradients. HH DNA from the pools indi-cated in Fig. 6 was concentrated byprecipitation with ethanol and banded in CsCl-EB density gra-dients(startingdensity=1.62g/cm). The 4- and 8-h cytoplasmicsampleswerepooled to reduce the
num-ber of analyses, sinceprevious data (12) indicated
the absenceof formIDNAfromthe cytoplasm. The arrowindicates the bandingposition oflinear (III) oropen circular(I) DNA. Fivepercentof the cyto-plasmicpoolwas analyzed, whereas allof the 4-h nuclear and one-thirdofthe 8-h nuclearpoolswere
analyzed.Viral DNA wasdetected byannealingeach
fractionofthegradients with
132P]cDNA
asdescribedin Materials and Methods.
a
-I
z z
z
a-u
zK
0a-1200
1000
800
600
400
200
discrepancy betweenFig. 7C and 8B.
Size ofthe strands ofviralDNApresent in linear forms. We have reported that the linear viral DNA present in the cytoplasm of ASV-infected duckcells contains a long minus strand(complementarytothe viralgenome) and ashortplus strand (same polarity as the viral genome) (31). The same form of viral DNA is present inASV-infectedQT-6cells (H. E. Var-mus, S. Heasley, H. J. Kung, H. 0. Oppermann, V. C. Smith, J. M. Bishop, and P. R. Shank, submittedforpublication),andsimilar forms of viral DNA have been found inmurineleukemia virus-infectedcells(10)and inmousemammary tumorvirus-infectedcells(23).This unusual lin-earduplex DNA appears to be thecharacteristic form of DNA transcribed in vivo from the ge-nomeof RNAtumorviruses.
We therefore designed a pulse-chase experi-mentwith the BUdR density-labelingprotocol to determine whether elongation of the plus-strand fragments was detectable during the chase. Viral DNA recovered from the HH region of CsCl density gradients from the cytoplasm aftera4-hpulsewithBUdR and from both the nucleus and cytoplasm after an additional 8-h chase withthymidine (cf. Fig. 6) wassedimented in alkaline sucrose gradients (Fig. 9). Minus-strand viral DNA extracted from both the 4-h and 12-hcytoplasmand the 12-hnuclei, detected by annealingwith
"MI-labeled
viral RNA, sedi-mented at 10 to 20S (Fig. 9). Theplus strand,1200 a 1000 < z
z
800 <
600
u
400 a
200 r,
z~~~~~~~~~~~~~~~~~~~~~~~~~~~~I"
5 10 15 20 25 5 10 15 20 25
FRACTION
FIG. 8. Analysis of pulse-labeledviralDNA inalkalinesucrosegradients. DNArecovered from the HH poolsshown inFig. 6wassedimentedin alkalinesucrosegradientsat17,000rpmfor 16h inanSW27.1
rotor at20°C. Two-thirds ofthenuclearpoolandone-twentiethofthecytoplasmic poolwereanalyzed. A
parallel gradient contained3H-labeled plasmid DNA (pML21) as asedimentation marker. (pML-21 isa
bacterialplasmid that exists as a form IDNA molecule of6.7x 106 daltons andhas a sedimentation
coefficient of70S in alkaline sucrose.) Sedimentation wasfrom right to left. ViralDNApresent in each gradient fractionwasdetectedby annealing with32P-labeled cDNAasdescribed in Materials and Methods.
B Il/ll
4hr. Nuclei I
A.
I
-IIrl
8 hr. Nuclei
11/111
qD
I
0id
'
4-+8 hr. 11/111 Sytoplasmf
i
ll
0
-
.!I
I
-_-J.
VOL. 25,1978 ill
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.505.133.374.401.580.2]0
" 800
z
z 600 400
u
Z 200
l.
10 20 30 10 20 30 10 20 30
-S
0I
a 800 6
w
z 600 Z 4:
400 Q-200 Z a
U1
FRACTION
FIG. 9. Analysisof both strands ofpulse-labeledviral DNA inalkaline sucrose gradients. HH viral DNA
wasrecoveredfromCsCldensitygradients ofthe nucleus andcytoplasmof QT-6cellsinfectedanddensity
pulselabeled as described inFig. 6, except that the chase wasextended until 12 h after infected to increase
the amountofHH DNAtransportedintothe nucleus. The HH DNA samplesfromthecytoplasmofIx 10, cells andfromtheSDS-NaClsupernatantof the nucleifrom5 x 108 cells were sedimented in an alkaline sucrosegradient for16hat17,000rpm in an SW27.1 rotor at20°C.Minus-strand DNA was detected by
annealing to125I-labeledviralRNA, and plus-strand DNA was detected byannealing to 32P-labeled cDNA. To reduce the numberof analyses (sinceanyrapidlysedimenting[form I]DNAmustcontain both strands ofviralDNA), fractions1 to 20wereanalyzedwith '32P]cDNA, whereasfractions21 to 40 weredivided in
halfandanalyzedwith each reagent. Aparallel gradientcontained the3H-labeledpML21DNA (formI)as
asedimentation marker. Sedimentation wasfrom righttoleft.
detected by annealing with [32P]cDNA, sedi-mentedat 4 to8S in both cytoplasmic samples (Fig.9Aand B). Furthermore, the plus strands present inthe nucleus 12hafter infection sedi-mentedat4 to 8Sunlessthey were present in form I molecules (Fig.
90).
Therefore we are unable to detect plus-strand intermediates be-tweenthe shortplusstrands in thelinear mole-cule and thefull-length plus strands present in form I molecules.DISCUSSION
We wish to drawonecentralconclusion from the experiments presented in this report: the linear form of viral DNA, synthesized in the cytoplasm ofcellsinfected withASV, is a
pre-cursor tothe closed circular form found in the
nucleus afew hours later. We haveestablished this relationship by the use of the traditional pulse-chase protocol, which has been used in
many contexts to illustrate precursor-product
relationships.Although pulse-chaseexperiments
most commonlyuse labeling ofprecursor can-didates with radioactive isotopes, the small quantities of viral DNA inRNA tumor virus-infected cellsobligedus touse densitylabeling with BUdRto identifyprecursor DNA, which
wasthen detected by molecularhybridization. Usingthisprotocol, wehave shown that a sig-nificantproportionofthedensity-labeledlinear viralDNA canbe chasedinto formImolecules (Fig. 5-9) and that thisDNA isalsotransported from thenucleus into the
cytoplasn
during the chaseperiod (Fig.6).We encounteredsignificant variabilityin the recovery of the small amounts of viral DNA from the preparative CsCl gradients, thereby impairing our ability to account for all of the viralDNA insubsequentanalyses.On the other hand,since therewasfive times more HH DNA in thenucleus after the chase (Fig. 6) and over 50% of the HH DNA in the nucleus after the chasewasinthe form Iconfiguration (Fig.
70),
we conclude that the majority of the form I DNA in the nucleus was synthesized as linear molecules in the cytoplasm. Taken together, these observations support steps ito iii in the proposedscheme for thesynthesisofviral DNA (Fig. 1).The resultspresentedhere donotexclude the possibility that the cytoplasmic form of viral DNA is an open circle rather than a linear molecule. Usingagarose gel electrophoresisand analysiswith restrictionendonucleases,wehave establishedthat the
cytoplasmic
viral DNA mol-ecule is linear (Shank et al., in preparation). Similar results have been observed with theDNA ofmouse mammarytumorvirus (Shank
et al., in preparation); in addition, the open forms ofMoloneymurineleukemiavirus,mouse
sarcoma virus, andspleen necrosis virusDNAs
are in linear rather than open circular
confor-mation (3, 7, 23).
Inview of the evidence for the efficiency of
our chase procedure (Fig. 3 and 4), we were
surprisedtofind thatalarge proportionof viral DNAexhibited the densityofpartially labeled molecules after the chase period (Fig. 5C and
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.505.116.402.71.193.2]CYTOPLASMIC ASV-SPECIFIC DNA 113
D; Fig 6C and D). One obvious interpretation of this result is that many of the viral DNA molecules initiated during the first 4 h after infection were not complete at that time, and that their completion during the chase period conferred an intermediate density upon them. We have recently obtained corroborating evi-dence for this view. Studies of the kinetics of synthesis of viral DNA (underconditions iden-tical to those used in this report) have revealed that minus strandsareslowlyelongated, requir-ing 3 to 6 h for completion, and synthesis of plus strands is retarded about 1 to 2 hrelative
to synthesis of minus strands (Varmus et al., submitted). Thus,atthe time of our chase pro-cedure,onlyaminority of DNA molecules were likely to be completed duplexes which would retain their HH density during the chase pro-cedure. It was, however, impossible for us to lengthenourpulse period, since effective dem-onstrationof the precursor-productrelationship demanded that little or no nuclear DNA be detectableattheconclusion of the BUdR label-ingperiod.
Althoughit is apparent from our results that atleast aportion of the cytoplasmic DNA mi-grates to thenucleus and enters a closed circular form, we do not know whether all of the ob-served cytoplasmic DNA is capable of these transitions. Thelargeamountof HH DNAthat persistedin thecytoplasm duringthe chase(Fig. 6B) might have represented incomplete mole-cules that could not be transported into the nucleus. Previous results from our laboratory and others indicate that viral DNA is synthe-sized inconsiderableexcess overtheintegrated species (12, 17) and that some of this excess
appears to remain in the cytoplasm (36). In addition, weand others have shown thata sig-nificantportion of the unintegrated linear DNA isless than genomelength(12, 25).These short species could represent abortive transcription products,precursorstofull-length forms, and/or precursorstothesmallspeciesofclosed circular viral DNA we have previously described (12). Althoughourexperiments provide direct evi-dence for theprecursor-productrelationship
be-tweenthecytoplasmiclinear and thecovalently
closed nuclear forms of viral DNA,theydonot
address the questionof whether the covalently closed form is the precursor to the integrated provirus (step iv, Fig. 1). In addition, they do notrevealthemolecular natureofthe
circulari-zationprocess. Although thesimplestmodelfor
circularization of theviral DNA wouldinvolve single-stranded regions ("sticky ends") at the
termini of the linearmolecules, suchendshave
notyet been demonstrated, and recombinational
mechanisms of circularization remain equally plausible.
ACKNOWLEDGMENTS
Wethank R. V.Guntaka,L.Levintow,and J. M.Bishop forhelpful discuionof theseexperimentsandmanuscript. ThisworkwassupportedbyPublicHealth Servicegrants CA 12705and CA 19287, awardedbythe National Cancer Institute.H.E.V. istherecipientofResearch Career Devel-opment Award CA 70193 from the National Cancer Institute. P.RS. acknowledges support from Public Health Service
btining grant 5T01 CA 05303 fromthe National Cancer Institute.
LITERATURE CITED
1.Campbell,A. 1962.Episomes.Adv.Genet. 11:101-145. 2.Canaani, E.,P.Duesberg,and D.Dina.1977.Cleavage mapof linearmouse sarcomavirus DNA. Proc.Natl. Acad.Sci.U.S.A.74:29-33.
3. Coffin, J. M., and W. A. Haseltine. 1977. Terminal redundancyand theoriginofreplicationof Rous sar-coma virus RNA. Proc. Natl. Acad. Sci. U.S.A. 74:1908-1912.
4. Commerford,S. L 1971.Iodinationofnucleicacids in vitro.Biochemistry10:1993-1999.
5. Dahlberg, J.E., R.C.Sawyer,J.M. Taylor, A. J. Faras,W. E.Levinson,H. M.Goodman,and J.M. Bishop. 1974. Transcription of DNA from the 70S RNA of Rous sarcoma virus. I. Identification of a
speciesof 4S RNAwhich serves asprimer.J.Virol. 13:1126-1133.
6. Friedrich, R., H. J.King,B.Baker, H. E.Varmus, H.M.Goodman, andJ. M.Bishop.1977. Character-izationof DNAcomplementarytonucleotidesequences atthe 5'-terminus of the aviansarcomavirus genome. Virology79:198-215.
7.Fritsch, E., and H. M. Temin. 1977. Formation and structureof infectious DNA ofspleennecrosisvirus. J. Virol. 21:119-130.
8. Garapin,A. C., H. E. Varmus, A. J. Faras,W. E. Levinson, and J. M. Bishop. 1973. RNA-directed DNAsynthesis byvirions ofRoussarcoma virus: fur-ther characterization of thetemplatesand theextent of theirtranscription.Virology52:264-274.
9. Gianni,A.M.,D.Smotkin,and R. A.Weinberg.1975. Murineleukemia virus: detection ofunintegrated dou-ble-stranded DNA forms of theprovirus. Proc.Natl. Acad. Sci. U.S.A.76:447-451.
10.Gianni,A. M., and R.A. Weinberg. 1975. Partially single-strandedformof freeMoloneyviral DNA. Na-ture(London)256:646-648.
11.Guntaka,R.V.,B.W.J.Mahy,J. M.Bishop, andH. E.Varmus.1975.Ethidiumbromide inhibits appear-anceofclosed circular viral DNA andintegrationof virus-specific DNA in duckcellsinfected by avian sar-comavirus. Nature(London)253:507-511.
12.Guntakca,R.V.,0. C. Richards,P.R.Shank,H. J. Kung,N.Davidson,E.Fritsch,J. M.Bishop, and H. E.Varmus. 1976.Covalentlyclosedcircular DNA ofaviansarcomavirus:purification from nuclei of in-fectedquailtumorcelLsandmeasurementby electron microscopy and gel electrophoresis. J. Mol. Biol. 106:337-357.
13.Haseline,W.A.,D.G.Kleid,A.Panet, E. Rothen-berg,andD.Baltimore.1976.Orderedtranscription of RNAtumorvirusgenomes. J. Mol. Biol.106:109-131. 14. Haseltine,W.A.,A.Maxam,and W.Gilbert. 1977. Rous sarcomavirusgenome isterminallyredundant: the 5' sequence. Proc. Natl. Acad. Sci. U.S.A. 74:989-993.
15.Hirt,B.1967.Selective extraction ofpolyoma DNA from VOL. 25,1978
on November 10, 2019 by guest
http://jvi.asm.org/
infected mouse cell cultures. J. Mol. Biol. 26:365-369. 16. Hudson, B., W. B. Upholt, J. Devinny, and J. Vino-grad. 1969. The useof an ethidium analogue in the dye-buoyant density procedure for the isolation of closed circular DNA: the variation of the superhelix density ofmitochondrialDNA. Proc. Natl. Acad. Sci. U.S.A.62:813-830.
17. Khoury,A. T.,andH.Hanafusa. 1976. Synthesis and integration ofviral DNA inchickencellsatdifferent times after infection with variousmultiplicities of avian oncornaviruses. J. Virol. 18:383-400.
18. Kung,H. J., J. M. Bailey, N. Davidson, M.0. Nicol-son,and R.M.McAllister.1975.Structure,subunit composition, andmolecular weight of RD-114 RNA. J. Virol. 16:397-411.
19. Levinson,W. E. 1967.Fragmentation of the nucleus in Rous sarcoma virus-infected chickembryo cells. Virol-ogy 32:74-83.
20.Mizuuchi, K., and H. A. Nash. 1976. Restriction assay forintegrativerecombinationofbacteriophageXDNA in vitro: requirementforclosedcircular DNA substrate. Proc. Natl. Acad. Sci. U.S.A. 73:3524-3528.
21.Moscovici,C.,M.G. Moscovici,H.Jimenez,M. M. C. Lai,M.J.Hayman, andP. K.Vogt.1977.Continuous tissueculture celllinesderived fromchemically induced tumorsofJapanesequail. Cell11:95-104.
22.Radloff,R., W.Bauer, and J.Vinograd.1967. A dye-buoyant-density methodforthe detection and isolation ofclosedcircularduplexDNA:the closed circular DNA inHeLacells.Proc.Natl.Acad.Sci. U.S.A. 57:1514-1521. 23. Ringold, G.M., K. R. Yamamoto, P. R. Shank, and H. E. Varmus. 1977. Mousemammary tumor virus DNA in infected ratcells:characterization of uninte-gratedforms.Cell10:19-26.
24. Schwartz,D.E.,P.C.Zamecnik, andH. L.Weith. 1977.Rous sarcoma virus genome isterminally redun-dant: the 3' sequence. Proc. Natl. Acad. Sci. U.S.A. 74:994-998.
25. Smotkin, D.,F. K.Yoshimura,and R. A.Weinberg. 1976.Infectious, linear, unintegratedDNA ofMoloney murineleukemiavirus. J.Virol.20:621-629.
26. Studier, F. W. 1965.Sedimentation studies of the size andshapeof DNA. J. Mol. Biol. 11:373-390.
27. Sutton,W. D. 1971. Acrude nuclease preparationsuitable for use inDNA reassociation experiments. Biochim. Biophys. Acta 240:522-531.
28. Svoboda,J. 1960. Presence of chicken tumorvirus in the sarcomaof theadultratincubated after birth with Rous sarcomatissue. Nature(London)186:980-981. 29. Taylor,J.M.,and R.Illmensee. 1975. SiteontheRNA
of aviansarcoma virusatwhichprimeris bound. J. Virol. 16:553-558.
30. Temin,H. M. 1971. Mechanism of celltransformationby RNA tumor viruses. Annu. Rev. Microbiol. 25:609-648. 31. Varmus, H. E., R. V.Guntaka,C. T.Deng,and J. M. Bishop. 1974.Synthesis, structure and function of avian sarcomavirus-specificDNA inpermissive and non-per-missive cells. ColdSpringHarborSymp.Quant. Biol. 39:987-996.
32.Varmus,H.E.,R. V.Guntaka,W.J.Fan, S.Heasley, and J.M. Bishop. 1974. Synthesisof viral DNA in thecytoplasmof duckembryofibroblasts and in enu-cleated cells after infectionby aviansarcoma virus. Proc. Natl. Acad. Sci. U.S.A. 71:3874-3878.
33.Varmus, H.E., S.Heasley,J.Linn, and K. Wheeler. 1976.Use of alkalinesucrosegradientsinazonalrotor todetect integrated andunintegratedavian sarcoma virus-specific DNA in cells. J. Virol. 18:574-585. 34. Varmus, H.E.,and P. R. Shank. 1976. Unintegrated
viral DNA is synthesized in thecytoplasm of avian sarcoma virus-transformed duck cells by viral DNA polymerase.J. Virol. 18:567-573.
35. Varmus, H. E., P. K.Vogt,andJ. M. Bishop.1973. Integrationofdeoxyribonucleicacidspecificfor Rous sarcoma virusafter infection ofpermissiveand non-permissive hosts. Proc. Natl. Acad. Sci. U.S.A. 70:3067-3071.
36. Weil, R.,and J.Vinograd. 1963.Thecyclic helixand cycliccoil formsofpolyoma viralDNA. Proc. Natl. Acad.Sci.U.S.A.50:730-738.
37. Weinberg, R. A 1977. Structure of the intermediates leadingtotheintegratedprovirus.Biochim.Biophys. Acta 473:39-55.
38.Weintraub, H. 1972. Bi-directional initiation ofDNA synthesis in developing chick erythroblasts. Nature (London)New.Biol. 236:195-197.
on November 10, 2019 by guest
http://jvi.asm.org/