0022-538X/01/$04.00⫹0 DOI: 10.1128/JVI.75.19.9018–9028.2001
Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Three Herpes Simplex Virus Type 1 Latency-Associated Transcript
Mutants with Distinct and Asymmetric Effects on Virulence
in Mice Compared with Rabbits
GUEY-CHUEN PERNG,1DANIEL ESMAILI,1SUSAN M. SLANINA,1ADA YUKHT,1HOMAYON GHIASI,1,2
NELSON OSORIO,1KEVIN R. MOTT,1BARAK MAGUEN,1LING JIN,1
ANTHONY B. NESBURN,1,2ANDSTEVEN L. WECHSLER1,2*
Ophthalmology Research Laboratories, Cedars-Sinai Medical Center Burns & Allen Research Institute, Los Angeles, California 90048,1and Department of Ophthalmology, UCLA School of Medicine, Los Angeles, California 900242
Received 9 April 2001/Accepted 14 June 2001
Herpes simplex virus type 1 latency-associated transcript (LAT)-null mutants have decreased reactivation
but normal virulence in rabbits and mice. We report here ondLAT1.5, a mutant with LAT nucleotides 76 to
1667 deleted. Following ocular infection of rabbits,dLAT1.5 reactivated at a lower rate than its wild-type parent
McKrae (6.1 versus 11.8%;P ⴝ0.0025 [chi-square test]). Reactivation was restored in the marker-rescued
virus dLAT1.5R (12.6%; P ⴝ 0.53 versus wild type), confirming the importance of the deleted region in
spontaneous reactivation. Compared with wild-type or marker-rescued virus,dLAT1.5 had similar or slightly
reduced virulence in rabbits (based on survival following ocular infection). In contrast, in mice,dLAT1.5 had
increased virulence (P< 0.0001). Thus, deletion of LAT nucleotides 76 to 1667 increased viral virulence in mice
but not in rabbits. In contrast, we also report here that LAT2.9A, a LAT mutant that we previously reported to have increased virulence in rabbits (G. C. Perng, S. M. Slanina, A. Yuhkt, B. S. Drolet, W. J. Keleher, H.
Ghiasi, A. B. Nesburn, and S. L. Wechsler, J. Virol. 73:920–929, 1999), had decreased virulence in mice (Pⴝ
0.03). In addition, we also found thatdLAT371, a LAT mutant that we previously reported to have wild-type
virulence in rabbits (G. C. Perng, S. M. Slanina, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler, J. Virol.
70:2014–2018, 1996), had decreased virulence in mice (P< 0.05). Thus, these three mutants, each of which
encodes a different LAT RNA, have different virulence phenotypes.dLAT1.5 had wild-type virulence in rabbits
but increased virulence in mice. In contrast, LAT2.9A had increased virulence in rabbits but decreased
virulence in mice, and dLAT371 had wild-type virulence in rabbits but decreased virulence in mice. Taken
together, these results suggest that (i) the 5ⴕ end of LAT and/or a gene that overlaps part of this region is
involved in viral virulence, (ii) this virulence appears to have species-specific effects, and (iii) regulation of this virulence may be complex.
Herpes simplex virus type 1 (HSV-1) is a double-stranded neurotropic DNA virus. It is ubiquitous in the general popu-lation and establishes lifelong latent infections in host sensory neurons. Following ocular infection, the virus travels up nerves and establishes latent infection in neurons of the trigeminal ganglia (TG). Via a mechanism that remains to be fully eluci-dated, the latent virus can reactivate at various times through-out the life of the individual and produce recurrent disease. Recurrent HSV-1 infection in the eye can result in corneal scarring, leading to loss of vision. As a result, HSV-1 is one of the most common infectious causes of corneal blindness in the developed world.
During neuronal latency, the latency-associated transcript (LAT) is the only viral gene that is abundantly transcribed (4, 11, 27, 30, 32, 33). The primary LAT transcript is 8.3 kb long (35, 40) and overlaps two viral genes, ICP0 and ICP34.5, in an antisense direction (27, 33). A very stable intron, the 2-kb LAT, is spliced from the primary transcript (9) and is the major LAT RNA detected during latency (6, 30, 31, 36, 37, 39).
LAT is essential for wild-type levels of spontaneous and induced reactivation in the rabbit ocular model (12, 20). This function maps to within the first 1.5 kb of the primary LAT (22). This 1.5-kb region does not overlap ICP0 or ICP34.5, encompasses only the first 837 nucleotides of the stable 2-kb LAT, and does not retain the 2-kb LAT stability (22). Thus, LAT appears to enhance spontaneous reactivation in the rab-bit by a mechanism that does not involve antisense regulation of ICP0, does not require production of the 2-kb LAT, and does not require the presence of a highly stable LAT RNA. We have recently shown that LAT can promote cell survival in vitro and block apoptosis in rabbit TG during acute infection (13, 19). This may increase the number of neurons that become latently infected (25, 28, 34), thereby increasing spontaneous reactivation by increasing the pool of latently infected neurons available for subsequent reactivation. In some situations, it might also affect viral virulence.
LAT-null mutants in which the LAT promoter has been deleted and no LAT RNA is detected appear to have wild-type virulence in animal models (1, 2, 12, 20, 29). This suggests that LAT does not play a role in viral virulence. However, we recently showed that deletion of LAT nucleotides 76 to 447 from a virus that could transcribe only the first 1.5 kb of LAT (producing a mutant designated LAT2.9A), significantly in-creases the death rate in the rabbit ocular model (24). This * Corresponding author. Mailing address: Ophthalmology Research
Laboratories, Cedars-Sinai Medical Center Burns & Allen Research Institute, Davis Bldg., Room 5072, 8700 Beverly Blvd., Los Angeles, CA 90048. Phone: (310) 423-6457. Fax: (310) 423-0225. E-mail: Wechsler @csmc.edu.
9018
on November 9, 2019 by guest
http://jvi.asm.org/
suggests that the first 1.5 kb of LAT is involved in virulence as well as latency. However, the potential role of the remainder of the primary 8.3-kb LAT transcript (the region of LAT from 1.5 to 8.3 kb) has not been addressed.
We describe here a LAT mutant designateddLAT1.5. This mutant has LAT nucleotides 76 to 1667 deleted but contains the entire LAT promoter and expresses the primary 8.3-kb LAT with the exception of the deleted region (i.e., it expresses the region of LAT from 1.67 to 8.3 kb).dLAT1.5 was wild type for ocular replication and eye disease. As expected,dLAT1.5 had reduced spontaneous reactivation compared with the parental McKrae wild-type virus or marker-rescued virus (dLAT1.5R). In rabbitsdLAT1.5 had wild-type or slightly re-duced virulence (as measured by survival). In contrast, in mice, dLAT1.5 was significantly more virulent than dLAT1.5R or wild-type virus.
We previously reported that LAT2.9A has increased viru-lence in the rabbit (24). In contrast, we found here that LAT2.9A had reduced virulence in mice. We also previously reported that a third LAT mutant, dLAT371, has wild-type virulence in rabbits.dLAT371 contains the same 371-nucleo-tide deletion in the 5⬘end of LAT as does LAT2.9A. However, in contrast to LAT2.9A,dLAT371 expresses the remainder of both copies of LAT. Thus,dLAT371 is structurally similar to dLAT1.5 in that both mutants express both copies of LAT with the exception of a deletion in the 5⬘end. Furthermore, both deletions begin at the same StyI restriction enzyme site. In contrast to the case fordLAT1.5, we found thatdLAT371 has decreased (rather than increased) virulence in mice. Thus, compared with wild-type virus and their corresponding mark-er-rescued viruses, each of these three LAT mutants affected virulence differently in rabbits compared with mice. There were also differences among the mutants regarding in which animal model they were more virulent. These results suggest that LAT and/or a gene overlapping part of the 5⬘end of LAT can affect virulence and that this effect can differ in rabbits compared with mice depending on the exact portion of LAT that is deleted.
MATERIALS AND METHODS
Cells and virus.Rabbit skin (RS) cells were grown in Eagle’s minimal essential medium (MEM) supplemented with 5% fetal calf serum (FCS). CV-1 cells were grown in MEM supplemented with 10% FCS. CV-1 cells were used for growth kinetic studies. RS cells were used for all other tissue culture procedures, in-cluding preparation of virus stocks. All mutants were derived from HSV-1 strain McKrae. The parental McKrae virus and all mutants were triple plaque purified and passaged only one or two times prior to use. The construction and properties of dLAT2903, dLAT371, LAT3.3A, and LAT2.9A have been previously de-scribed (20, 22, 24). LAT3.3A was originally designated LAT1.5a (22), but its designation was changed (7, 8, 24) to maintain nomenclature consistency with our other mutants (the number prior to the A indicates the size of the LAT region added between UL37 and UL38).
Construction of the mutantdLAT1.5.The previously clonedBamHI B frag-ment from HSV-1 strain McKrae (21) was digested withMluI, the overhang was filled in using the Klenow fragment and digested withSwaI, andPacI linker was added. The fragment was then digested withPacI, and the products were sepa-rated by agarose gel electrophoresis. A resulting 3.6-kb band (SwaI toMluI; LAT nucleotides⫺798 to⫹2850) which includes the LAT promoter and the entire 2-kb LAT was isolated by electroelution and cloned into thePacI site of plasmid pNEB193 to produce the plasmid SMpNEB193. SMpNEB193 was digested with StyI andHpaI, theStyI overhang was filled in using the Klenow fragment, and the blunt ends were self-ligated to produce the plasmid pLAT1.5dl, which contains a deletion from LAT nucleotide 76 to 1667. pLAT1.5dl was amplified by
trans-formation intoEscherichia colistrain RR1CI857 according to standard proto-cols (21).
dLAT1.5 was generated by homologous recombination of pLAT1.5dl with dLAT2903 as we previously described (20, 22, 23, 26).dLAT2903 is a mutant of HSV-1 strain McKrae containing a 1.8-kb (EcoRV-HpaI) deletion in both copies of LAT that removed 0.2 kb of the LAT promoter and 1.6 kb of the 5⬘end of the primary 8.3-kb LAT transcript (20). Briefly, pLAT1.5dl was cotransfected with infectiousdLAT2903 DNA by the calcium phosphate method. Viruses from the cotransfection were plated, and isolated plaques were picked and screened by Southern analysis. Selected plaques were triple plaque purified and reanalyzed by restriction digestion and Southern analysis to ensure that the LAT promoter had been restored and that the region from LAT nucleotide 76 to 1667 was deleted from both copies of LAT (one in each long repeat). A final plaque was purified and designated dLAT1.5. The marker-rescued virus,dLAT1.5R, was generated as described above by homologous recombination ofdLAT1.5 DNA with the plasmid SMpNEB193. This restored the deletion indLAT1.5 back to wild type.
Replication of virus in tissue culture.CV-1 cell monolayers at approximately 70 to 80% confluency were infected with virus at 0.01 PFU/cell, and all mono-layers were refed with exactly the same amount of MEM containing 10% FCS. Virus was harvested for titration at various times by two cycles of freeze-thawing of the monolayers plus medium (⫺80°C to room temperature). PFU per milli-liter were determined by standard plaque assays on RS cells.
Animals.Rabbits were 8- to 10-week-old female New Zealand White from Irish Farms. Mice were 6-week-old female Swiss Webster from Harlan. Rabbits and mice were treated in accordance with Association for Research in Vision and Ophthalmology, American Association for Laboratory Animal Care, and Na-tional Institutes of Health guidelines.
Rabbit model of ocular HSV-1 infection, latency, and spontaneous reactiva-tion.The McKrae strain of HSV-1 does not require corneal scarification in rabbits or mice for efficient ocular infection (20). Rabbits were bilaterally in-fected without scarification or anesthesia by placing 2⫻105PFU of HSV-1 per
eye into the conjunctival cul-de-sac, closing the eye, and rubbing the lid gently against the eye for 30 s (20). At this dose of HSV-1 McKrae, virtually all of the surviving rabbits harbor a bilateral latent HSV infection in both TG, resulting in a high group rate of spontaneous reactivation with the McKrae strain of HSV-1. In each animal group, tear films were collected from five eyes, each from a different rabbit, on days 3, 5, 7, and 10 postinfection (p.i.) for acute ocular replication. Virulence (animal death) was monitored daily for up to 21 days p.i. Beginning on day 31 p.i., tear film specimens were collected daily from each eye for 26 days as previously described (20, 23, 26), using a nylon-tipped swab. The swab was then placed in 0.5 ml of tissue culture medium and squeezed, and the inoculated medium was used to infect RS cell monolayers. These cell monolayers were observed in a masked fashion by phase light microscopy for up to 5 days for HSV-1 cytopathic effects (CPE). All positive monolayers were blind passaged onto fresh cells to confirm the presence of virus. DNA was purified from positive cultures and analyzed by restriction enzyme digestion and Southern blotting to confirm that the CPE was due to reactivated HSV-1 and that the reactivated virus was identical to the input virus.
Ocular infection of mice.Swiss Webster mice were bilaterally infected without scarification or anesthesia by placing 106PFU of HSV-1 per eye into the
con-junctival cul-de-sac as described for rabbits. Tear films were collected on various days p.i. from one eye per animal. The amount of virus in each tear film was determined by standard plaque assays on RS cells. Virulence was determined by survival at 21 days after ocular infection.
Virus replication in mouse TG and brains.Mice were infected as described above and euthanized at various times p.i. The brain and TG were removed and individually homogenized, debris was pelleted by low-speed centrifugation, and the amount of infectious virus in the supernatant was determined by standard plaque assays on RS cells.
Statistical analyses. Statistical analyses were performed using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, Calif.). Results were considered statistically significant when thePvalue was⬍0.05.
RESULTS
Genomic structures of viruses.dLAT1.5 was constructed as
described in Materials and Methods. This mutant contains a deletion from LAT nucleotide 76 (aStyI restriction enzyme site) to 1667 (aHpaI restriction enzyme site) in both copies of the LAT gene (one in each long repeat) (Fig. 1D). This
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 1. Genomic structures of the viruses used in this study. (A) Prototypic genomic structure of parental wild-type McKrae and marker-rescued virusesdLAT1.5R,dLAT2903R, anddLAT371R. TRL and IRL, terminal and internal long repeats, respectively; TRS and IRS, terminal and internal short repeats, respectively; UL and US, long and short unique regions, respectively. The LAT gene is located in the viral long repeats and is thus present in two copies. The primary 8.3-kb LAT transcript is shown expanded below the genome. The stable 2-kb LAT is indicated by a black rectangle. The relative locations of the ICP0 and ICP34.5 transcripts are shown for reference. (B)dLAT2903 (20) has anEcoRV-to-HpaI restriction fragment deletion from LAT nucleotide⫺161 to⫹1667 relative to the start of LAT transcription at⫹1 (XXXX). This deletion removes the core LAT promoter, including the TATA box, and a second proposed promoter near the 5⬘end of the 2-kb LAT. No LAT is transcribed, as indicated by the dashed lines. (C)dLAT371 has a 371-nucleotideStyI-StyI restriction fragment deletion from LAT nucleotide 76 to 447 (X) in both copies of LAT (23). The primary LAT promoter is intact, and the remaining portion of the primary 8.3-kb LAT is transcribed. (D)dLAT1.5 has aStyI-to-HpaI restriction fragment deletion from LAT nucleotide 76 to 1667 (XXX). The primary LAT promoter is intact, and the remaining portion of the primary 8.3-kb LAT is transcribed. (E) LAT3.3A (22) contains the LAT promoter driving the first 1,499 nucleotides of LAT inserted into an ectopic location between the UL37 and UL38 viral genes ofdLAT2903. This virus transcribes only the first 1.5 kb of LAT yet has wild-type spontaneous reactivation. LAT2.9AR (24) was marker rescued from LAT2.9A and has the same genomic structure as LAT3.3A. (F) LAT2.9A (24) has the same genomic structure as LAT3.3A except for aStyI-StyI restriction fragment deletion from LAT nucleotide 76 to 447 in the ectopic LAT insert.
on November 9, 2019 by guest
http://jvi.asm.org/
tion does not alter the primary LAT promoter. Thus,dLAT1.5 transcribes the primary LAT with the exception of the deleted region. The marker-rescued virus,dLAT1.5R, is identical to the parental wild-type strain McKrae (Fig. 1A). Also employed in this study were the following previously described mutants. (i) dLAT2903 (20) (Fig. 1B) has LAT nucleotides ⫺161 to 1667 deleted (a region including the primary LAT promoter and a putative second LAT promoter sometimes called LAP2). (ii)dLAT371 (17, 23) (Fig. 1C) contains aStyI-StyI restriction site fragment deletion in both copies of LAT (LAT nucleotides 76 to 447). TheStyI-StyI deletion in dLAT371 and the StyI-HpaI deletion indLAT1.5 begin at the sameStyI site. Thus, the genomic structure ofdLAT371 is identical to that ofdLAT1.5 except for the size of the deletion in both copies of LAT. (iii) LAT2.9A (24) (Fig. 1F) contains the same LAT deletion as dLAT2903 (Fig. 1B) and therefore makes no LAT from the normal LAT genomic location. LAT2.9A contains an ectopic insert between UL37 and UL38 consisting of the entire LAT promoter and the first 1.5 kb of the primary LAT transcript from which the sameStyI-StyI region deleted indLAT371 (Fig. 1C) has been deleted. This virus therefore expresses only LAT nucleotides 1 to 76 plus 447 to 1499. (iv) LAT3.3A (22) (Fig. 1E), from which LAT2.9A was derived, is included here for clarity. This mutant, originally designated LAT1.5a, was re-named to maintain consistency with some of our other mu-tants. The 3.3 in the designation refers to the approximate size, in kilobases, of the ectopic insert.
Spontaneous reactivation. Rabbit eyes were infected with
2⫻105PFU ofdLAT1.5,dLAT1.5R, or wild-type McKrae per
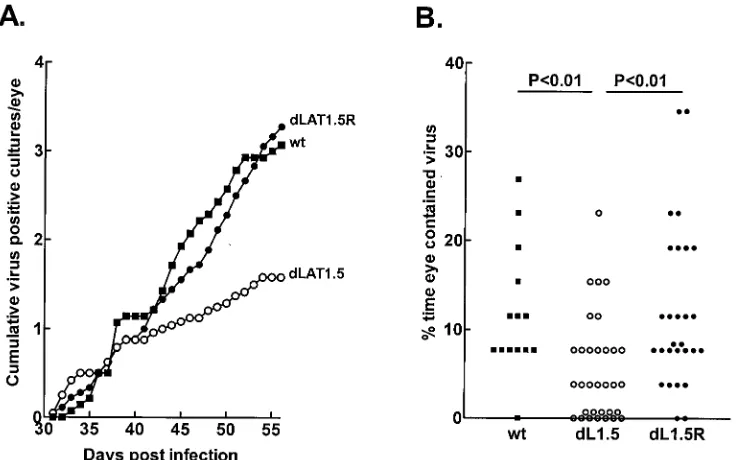
eye as described in Materials and Methods. Starting at 31 days p.i., at which time latency had already been established, tear films were collected daily from all eyes and individually plated on indicator cells (RS cells) to detect spontaneously reacti-vated virus. Southern analysis of spontaneously reactireacti-vated viruses grown from tear films (not shown) confirmed that all cultures which scored positive based on the presence of viral CPE contained reactivated virus and that the spontaneously reactivateddLAT1.5 virus retained the deletion in both copies of LAT. The cumulative number of virus-positive tear film cultures per eye during 26 days for each virus are shown in Fig. 2A. Wild-type McKrae anddLAT1.5R appeared to have sim-ilar average cumulative spontaneous reactivation rates of ap-proximately three positive cultures per eye on day 26 of col-lection. In contrast,dLAT1.5 appeared to have a lower average spontaneous reactivation rate of approximately 1.6 positive cultures per eye.
[image:4.612.120.488.79.309.2]A statistical analysis of positive (spontaneously reactivated) tear film cultures versus total cultures is shown in Table 1. Approximately 6.1% of the tear films from rabbits latently infected with dLAT1.5 contained spontaneously reactivated virus. This was significantly less than the results for wild-type McKrae (11.8%) anddLAT1.5R (12.6%). Because the above analyses do not take into account the number of eyes in each group, we calculated the percentage of time that each eye in each group was positive for spontaneous reactivation (i.e., the percentage of virus- positive cultures) and plotted the results as scattergrams (Fig. 2B). This analysis is considered more strin-gent because it takes into account both the number of eyes and FIG. 2. Spontaneous reactivation ofdLAT1.5. Rabbits were ocularly infected withdLAT1.5 (dL1.5), marker-rescueddLAT1.5R (dL1.5R), or parental wild-type (wt) McKrae. Beginning on day 31 p.i., at which time latency had been established, tear films were collected daily for 26 days, plated on RS cells, and observed for the presence of CPE. Randomly selected positive cultures were confirmed by passage and Southern analysis. (A) Theyaxis represents the cumulative number of HSV-1-positive cultures for each virus group divided by the number of eyes in the group. (B) The percentage of tear films containing spontaneously reactivated virus was calculated for each individual eye. The results are shown as a scattergram. In thedLAT1.5 column, some of the zero points are plotted at 0.7 so that all of the zero points can be visualized. Similarly, in the
dLAT1.5R column, two points with an actual value of 7.7 are plotted at 8.4. ThePvalues shown were determined by one-way ANOVA and Dunnet’s posttest. Results for the wild type anddLAT1.5R were similar (P⬎0.05).
on November 9, 2019 by guest
http://jvi.asm.org/
the period of time in a single calculation (22–24). By this analysis spontaneous reactivation ofdLAT1.5 was also signif-icantly reduced compared with those of the wild-type and marker-rescued viruses (P ⬍0.01). The above analyses indi-cated that spontaneous reactivation ofdLAT1.5 was reduced compared with those of its marker-rescued virus and wild-type virus.
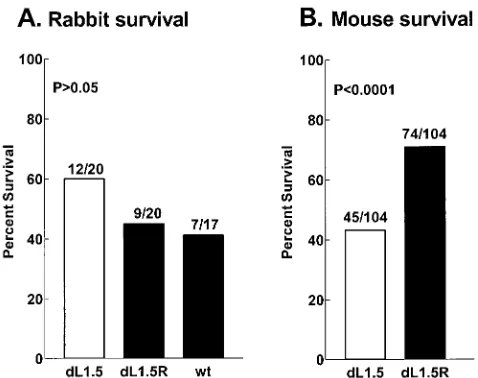
Survival following oculardLAT1.5 infection.Survival in the
rabbits used for the above-described spontaneous reactivation study was quantitated at 21 days p.i. (Fig. 3A). Although not statistically significant, there was a suggestion in this experi-ment of increased survival in the rabbits infected withdLAT1.5 (60%) compared with marker-rescueddLAT1.5R (45%) and wild-type McKrae (41%). Thus, it was of interest to determine if significant differences might be seen with larger numbers of animals. Due to difficulties with using large numbers of rabbits, we decided to examine survival in mice. Groups of 104 mice were infected with 106PFU ofdLAT1.5 ordLAT1.5R per eye.
Surprisingly, instead of the expected decrease in virulence, in micedLAT1.5 was significantly more virulent thandLAT1.5R (Fig. 3B) (43% survival compared with 71% survival; P ⬍
0.0001 [chi-square test]). In a separate experiment (see Fig. 9
below) the virulence ofdLAT1.5R in mice was similar to that of the original wild-type parental virus (P ⫽ 0.39). Thus, as judged by survival of mice following ocular HSV-1 challenge, dLAT1.5 was more virulent than either wild-type virus or its marker-rescued virus.
Replication of dLAT1.5 in tissue culture, eyes, TG, and
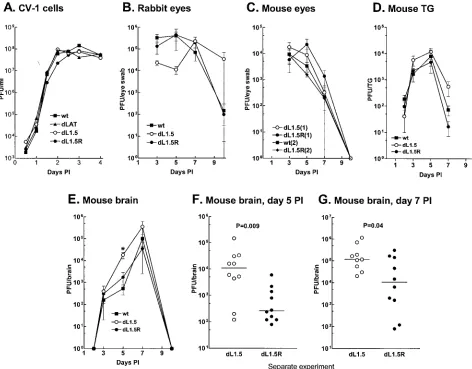
brains.CV-1 cell monolayers were infected at a multiplicity of
infection of 0.01 PFU/cell, and the kinetics of replication were determined as described in Materials and Methods. Replica-tion ofdLAT1.5 was indistinguishable from that ofdLAT1.5R, wild-type virus, or the LAT-null mutantdLAT2903 (Fig. 4A). Rabbits were infected with 2⫻105PFU/eye. Tear films were
collected from five rabbit eyes on days 3, 5, 7, and 10 p.i. Figure 4B shows the average amount of virus detected in the eye swabs at each time point. Although replication of dLAT1.5 appeared to be slightly less than that ofdLAT1.5R or the wild type in rabbit eyes on day 5 p.i., the differences were not significant (P⬎0.05 at all time points by analysis of variance [ANOVA]). In addition, the peak virus titers (day 5 p.i. for the wild type anddLAT1.5R; day 7 fordLAT1.5) appeared to be very similar for all three viruses (P⫽0.8).
Mice were infected with 106PFU/eye, and replication in eyes
was examined. Figure 4C shows the results of two independent experiments. In experiment 1, ocular replication of dLAT1.5 anddLAT1.5R were similar (P⬎0.23 at each time point). In experiment 2, the wild type anddLAT1.5R were similar (P⬎
0.53 at each time point). Thus, replication of dLAT1.5 in mouse eyes was similar to that of its marker-rescued virus, which in turn was similar to that of the original parental wild-type virus. Thus, alterations indLAT1.5 virulence (death rate) did not appear to be due to altered ocular replication.
[image:5.612.52.294.89.146.2]For analysis of replication in mouse TG, five mice per group were ocularly infected for each time point shown (Fig. 4D), and the amount of infectious virus was determined individually for each TG at each time point. There was no significant difference among thedLAT1.5,dLAT1.5R, and wild-type vi-ruses at any time point (P⬎0.1 [ANOVA]). The amounts of virus in the brains of the same mice were similarly determined. On day 5 p.i. (Fig. 4E), the average amount of virus in the brains of mice infected withdLAT1.5 was significantly higher than that of either dLAT1.5R or the wild type (P ⫽ 0.005 [ANOVA]). On day 7 p.i., the day of peak virus titer in the brain, the amount ofdLAT1.5 detected was 3.5- to 10-fold higher than that ofdLAT1.5R or the wild type. However, this difference did not reach statistical significance (P ⫽ 0.1 [ANOVA]), perhaps because only five mice were used per time point. To increase the statistical power, additional experiments were performed using 9 or 10 mice per group. (Fig. 4F and G). On both days 5 and 7 p.i. significantly more virus was detected in the brains of dLAT1.5-infected mice than in those of dLAT1.5R-infected mice (P⫽0.009 andP⫽0.04, respectively [Mann-Whitney rank sum test]). Thus, it appears thatdLAT1.5 grew more efficiently in mouse brains than its marker-rescued virus or wild-type virus. This was consistent with the increased virulence ofdLAT1.5 in mice and suggests that the increased virulence in mice may have been due to increased virus load in the brain. Replication ofdLAT1.5 in rabbit TG or brain was not examined both because this virus did not have significantly altered virulence in rabbits and because these analyses are highly variable in the rabbit.
FIG. 3. Survival of rabbits (A) and mice (B) infected withdLAT1.5. (A) Survival of the rabbits used in the experiments shown in Fig. 2 was determined on day 21 p.i. The numbers above each bar indicate the number of surviving rabbits/total number of rabbits infected.Pwas determined by the chi-square test for all pairs of rabbits. dL1.5,
dLAT1.5; dL1.5R, dLAT1.5R; wt, parental wild-type McKrae. (B) Mice were bilaterally ocularly infected with 106PFU/eye without
[image:5.612.55.294.466.655.2]scar-ification, and survival was determined at 21 days p.i. TABLE 1. Spontaneous reactivation of dLAT1.5a
Virus No. of positive cultures/total(% positive) PversusdLAT1.5b
dLAT1.5 38/624 (6.1)
dLAT1.5R 59/468 (12.6) 0.0002
McKrae 43/364 (11.8) 0.0025
aNumerical presentation of the spontaneous reactivation data shown in Fig. 2. bBy the chi-square test.
on November 9, 2019 by guest
http://jvi.asm.org/
Virulence of LAT2.9A in mice.LAT3.3A (Fig. 1E) was con-structed by inserting the LAT promoter and the first 1.5 kb of LAT into an ectopic location in the LAT-null mutant dLAT2903. This virus, which expresses just the first 1.5 kb of LAT, has wild-type virulence in rabbits (22) and mice (unpub-lished results). The LAT2.9A genome is structurally identical to that of LAT3.3A, with the exception of a 371-nucleotide
[image:6.612.67.539.79.448.2]deletion between LAT nucleotides 76 and 447 in the ectopic insert. We previously showed that LAT2.9A has increased vir-ulence in rabbits (24). Because of the difference in virvir-ulence found above fordLAT1.5 in mice compared with rabbits, it was of interest to examine the virulence of LAT2.9A in mice, since to our knowledge, this is the only other LAT mutant reported to have altered virulence. Groups of 104 mice were therefore FIG. 4. Replication ofdLAT1.5 in tissue culture, eyes, TG, and brains. (A) Semiconfluent monolayers of CV-1 cells were infected with 0.01 PFU ofdLAT1.5,dLAT1.5R, the LAT-null mutantdLAT2903, or wild-type (wt) McKrae. At the indicated times, the infected-cell monolayers were harvested by freeze-thawing, and the amount of virus was determined by plaque assay on RS cells. The result for each time point is the average of two determinations. (B) Rabbits were ocularly infected with 2⫻105PFU of the indicated virus per eye as described in Materials and Methods.
In each group tear films were collected from five individual eyes (each from a different rabbit) on days 3, 5, 7, and 10, and virus titers were determined by standard plaque assays. There was no statistical difference between the groups on any day (by ANOVA,P⫽0.69 for day 3,P⫽ 0.055 for day 5,P⫽0.76 for day 7, andP⫽0.84 for day 10). (C) Mice were ocularly infected as described above, and virus replication in the eye was determined as for rabbits in panel B. Results from two independent experiments are shown. Experiment 1 (six eyes per group) compared
dLAT1.5 todLAT1.5R. Experiment 2 (five eyes per group) compared the wild type todLAT1.5R. There was no statistical difference between the viruses in each experiment on any day (P⬎0.2 for experiment 1;P⬎0.5 for experiment 2 [Mann-Whitney test]). (D) Mice were infected as described above. Both TG were removed from five mice per group at necropsy on days 2, 3, 5, 7, and 10 p.i., and the amount of infectious virus in each individual TG was determine by plaque assay as described in Materials and Methods. There was no statistical difference on any day (P⬎ 0.1 on each day [ANOVA]). (E) Mice were infected as described above, and brains (five per group per time point) were harvested and virus titers were determined on the days indicated. The asterisk indicates the only day for which statistical significance was obtained (day 5,P⫽0.013 [ANOVA];dLAT1.5 versusdLAT1.5R,P⫽0.02 [Mann-Whitney test];dLAT1.5 versus wild type,P⫽0.008 [Mann-Whitney test]). (F) The experiment in panel E was repeated fordLAT1.5 anddLAT1.5R using 10 mice per group on day 5 p.i. Each data point represents the virus titer from an individual brain on this day. The horizontal lines indicate the medians. ThePvalue was determined by the Mann-Whitney rank sum test. (G) The experiment in panel E was repeated fordLAT1.5 (9 mice) anddLAT1.5R (10 mice) on day 7 p.i., and the results are plotted as in panel F. Error bars indicate standard deviations.
on November 9, 2019 by guest
http://jvi.asm.org/
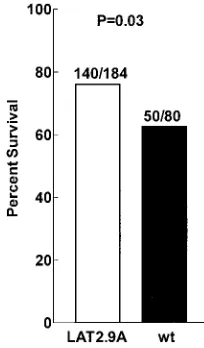
bilaterally ocularly infected with 106PFU/eye, and survival was
determined on day 21 p.i. (Fig 5). LAT2.9A had significantly reduced virulence in mice compared with wild-type virus (P⫽
0.03). As shown below (see Fig. 9), the marker-rescued virus LAT2.9AR (24) and wild-type virus had similar virulence in mice (P⫽0.19). Taken together, these two results indicate that LAT2.9A had reduced virulence in mice compared with both wild-type virus and its marker-rescued virus. Thus, as judged by the ability to kill animals following ocular infection, LAT2.9A had decreased virulence in mice and increased virulence in rabbits, while dLAT1.5 had increased virulence in mice but wild-type or slightly decreased virulence in rabbits.
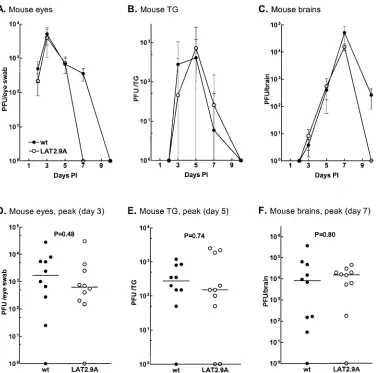
Replication of LAT2.9A.We previously showed that
replica-tion of LAT2.9A in tissue culture and in rabbit eyes was similar to that of wild-type virus, suggesting that the increased viru-lence of LAT2.9A in rabbits is not due to enhanced replication (24). Replication in rabbit TG and brain was not examined because of problems with high variability, as mentioned above. To determine if the decreased virulence of LAT2.9A in mice might be related to decreased replication of the virus in mice, we examined replication in mouse eyes, TG, and brain. Mice were infected and replication was determined as described above for dLAT1.5, using either five eyes, five TG, or five brains per time point per group. The replication kinetics of LAT2.9A were indistinguishable from those of wild-type virus in the eyes, TG, and brains of mice (Fig. 6A, B, and C) (P⬎
0.05 at each time point). For further confirmation, 10 eyes, 10 TG, or 10 brains were similarly analyzed on the day of peak virus titer as determined from these results (Fig. 6D, E, and F). Again, no differences were detected (P ⬎ 0.05). Thus, the decreased virulence of LAT2.9A in mice did not appear to be a result of decreased virus load. Since replication of LAT2.9A was indistinguishable from that of wild-type virus, there was no need to compare replication of LAT2.9A with that of its mark-er-rescued virus.
Virulence ofdLAT371 in mice.dLAT371 contains the same
StyI-StyI restriction fragment deletion (LAT nucleotides 76 to 447) as LAT2.9A. LikedLAT1.5,dLAT371 contains a deletion
starting at LAT nucleotide 76 in both copies of the otherwise unaltered LAT gene. The only difference between the struc-tures of dLAT1.5 and dLAT371 is the size of the deletion (1,591 nucleotides for dLAT1.5 versus 371 nucleotides for dLAT371). Although LAT2.9A contains the same deletion as dLAT371, LAT2.9A contains only one copy of LAT, the LAT is in an ectopic location, and LAT2.9A transcribes LAT only up to LAT nucleotide 1499. Thus, although we have previously shown thatdLAT371 has wild-type virulence in rabbits, it was of interest to examine the virulence ofdLAT371 in mice.
As done above withdLAT1.5 and LAT2.9A, 104 mice per group were infected with 106PFU ofdLAT371,dLAT2903, or
dLAT2903R per eye. dLAT2903 is a LAT-null mutant, and dLAT2903R is its marker-rescued virus (Fig. 1A and B) (20). This particular combination of mutants was used because this experiment was originally aimed at comparingdLAT371 with the LAT-null mutant.dLAT371 had reduced virulence com-pared with eitherdLAT2903 ordLAT2903R (20) (Fig. 7) (P⫽
0.0004 andP⫽0.008, respectively [chi-square] test). As shown below (see Fig. 9), in mice the virulences ofdLAT2903R and dLAT371R (marker-rescueddLAT371) were both similar to that of the wild type (P ⫽0.19 andP ⫽ 0.68, respectively). Thus, consistent with LAT2.9A, but in contrast todLAT1.5, in mice,dLAT371 was less virulent than wild-type virus or its marker-rescued virus.
Replication ofdLAT371.We previously reported that
repli-cation ofdLAT371 was indistinguishable from that of wild-type virus in tissue culture and in rabbit eyes. The kinetics of rep-lication ofdLAT371 in mouse eyes, TG, and brain were exam-ined, using 10 samples per time point per group (Fig. 8A, B, and C). Scattergrams are also shown for the day of peak rep-lication at each site (Fig. 8D, E, and F). AlthoughdLAT371 appeared to grow slightly less well than wild-type virus in mouse eyes (P⫽0.043 on day 3 p.i. [Mann-Whitney rank sum test]), replication ofdLAT371 in mouse TG and brains was indistinguishable from that of wild-type virus (P⬎0.05 for all times andP⫽0.74 andP⫽0.68, respectively, for peak titers). Thus, it appears unlikely that decreased replication of dLAT371 in mouse brains could account for the decreased virulence of this mutant in mice. Since replication ofdLAT371 in TG and brains was wild type, comparisons withdLAT371R were not made.
Virulence of marker-rescued viruses.Groups of mice were
ocularly infected as described above, and virulence was deter-mined based on survival on day 21 p.i. The virulences of dLAT1.5R, LAT2.9AR,dLAT371R, anddLAT2903R were all indistinguishable from that of wild-type McKrae (Fig. 9) (P⬎
0.05).
DISCUSSION
[image:7.612.119.221.78.252.2]The virulence results presented here can perhaps be most simply summarized by separating the mouse and rabbit data. Thus, in mice, compared with wild-type virus and marker-rescued viruses (all of which had wild-type virulence),dLAT1.5 had increased virulence, while LAT2.9A and dLAT371 had decreased virulence. In rabbits, compared with wild-type and marker-rescued viruses, LAT2.9A had increased virulence, dLAT371 had wild-type virulence, anddLAT1.5 had wild-type or slightly reduced virulence.
FIG. 5. Virulence of LAT2.9A in mice. Mice were ocularly infected as described for Fig. 4, and survival was determined at 21 days p.i. Numbers above each bar indicate the number of surviving mice/total number of mice.Pwas determined by the chi-square test. wt, wild type.
on November 9, 2019 by guest
http://jvi.asm.org/
LAT is essential for wild-type levels of spontaneous reacti-vation in the rabbit ocular model. We previously showed that expression of just the first 1.5 kb of the 8.3-kb LAT, as found in LAT3.3A, is sufficient for wild-type levels of spontaneous reactivation (22). Thus, a major function involved in sponta-neous reactivation in the rabbit maps to within the first 1.5 kb of LAT, a region representing less than 20% of the primary LAT transcript.dLAT1.5 was constructed and studied in this work because it represents a mutant complementary to LAT3.3A. Thus, while LAT3.3A expresses only the first 1.5 kb of LAT,dLAT1.5 expresses all of LAT except for a similar region (LAT nucleotides 76 to 1667). As expected, like the LAT-null mutantdLAT2903,dLAT1.5 had reduced spontane-ous reactivation. This demonstrated that in the rabbit ocular model, not only is the first 1.5 kb of LAT sufficient for wild-type spontaneous reactivation, but part or all of the first 1.5 kb is also required for wild-type levels of spontaneous reactiva-tion. In addition, these results suggest that LAT does not contain a region outside of the first 1.67 kb of LAT that is
[image:8.612.117.493.77.450.2]FIG. 6. Replication of LAT2.9A in mouse eyes, TG, and brains. (A, B, and C) Experimental procedures and analyses were as described for Fig. 4C, D, and E, respectively, except that 10 samples were used per group at each time point. ThePvalue was⬎0.05 at all time points. (D, E, and F) Scatterplots showing the virus titers for individual eyes, TG, and brains on the day of peak virus titer for the samples shown in panels A, B, and C, respectively. The horizontal lines indicate the median (as opposed to the means shown in panels A, B, and C). Error bars indicate standard deviations.
FIG. 7. Virulence ofdLAT371 in mice. Mice were ocularly infected and survival was determined as described for Fig. 4.Pwas determined by the chi-square test.dL371,dLAT371;dLAT,dLAT2903;dLATR,
dLAT2903R.
on November 9, 2019 by guest
http://jvi.asm.org/
independently able to support wild-type levels of spontaneous reactivation
We previously showed that compared with wild-type virus, LAT2.9A (Fig. 1E) has increased virulence in rabbits (i.e., increased death rate following ocular infection) compared with marker-rescued or wild-type virus (24). In contrast, as reported here, in mice LAT2.9A was significantly less virulent than marker-rescued or wild-type virus. Thus, the virulence pheno-type for LAT2.9A not only appeared to be very different in mice compared with rabbits, it appeared to be asymmetric in rabbits compared with mice (i.e., more virulent than the wild type in one species and less virulent than the wild type in another species).
Note that in this report we use the term virulence to mean death following ocular infection. We are not distinguishing between neuroinvasiveness (the ability of the virus to get to the brain following infection at a peripheral site) and neuroviru-lence (the ability of the virus to kill the animal once it reaches the brain).
We also showed in this report that following ocular infection
of rabbits,dLAT1.5 had virulence similar to or slightly less than that of wild-type virus or marker-rescueddLAT1.5R. In contrast, in mice,dLAT1.5 was significantly more virulent than the wild type ordLAT1.5R. Thus, the virulence phenotype for dLAT1.5 also appeared to differ in mice compared with rab-bits. The asymmetric virulence results in mice versus rabbits with LAT2.9A anddLAT1.5 were unexpected. Although mu-tations that alter the virulence of HSV-1 in mice but not rabbits have been reported (38), we are not aware of any previous report of a mutant that is more virulent than its marker-res-cued and parental wild-type viruses in one animal species and less virulent than its marker-rescued and parental wild-type viruses in a second species.
[image:9.612.113.495.77.449.2]Also surprising was thatdLAT1.5 and LAT2.9A had oppo-site phenotypes. That is, compared with wild-type virus and the mutant-specific marker-rescued virus, one mutant had de-creased virulence in mice while the other mutant had inde-creased virulence in mice, and vice versa in rabbits. This is of particular interest, because both of these viruses contain deletions of part of the LAT gene. The most direct conclusion to draw from FIG. 8. Replication ofdLAT371 (d371) in mouse eyes, TG, and brains. Experimental procedures and analyses were done as described in the legend to Fig. 6. ThePvalue was⬎0.05 by the Mann-Whitney rank sum test at all time points except for mouse eyes on day 3. By the Student
ttest, thePvalue was 0.13 for this time point. wt, wild type. Error bars indicate standard deviations.
on November 9, 2019 by guest
http://jvi.asm.org/
these results is that different LAT deletion mutants can affect virulence differently and that the direction of virulence (i.e., increased or decreased compared with the wild type) can differ in mice versus rabbits. This in turn strongly suggests that in addition to being involved in the HSV-1 latency cycle of es-tablishment of and reactivation from latency, LAT is also in-volved in viral virulence.
Because dLAT1.5 and LAT2.9A have significant genomic structural differences in addition to the size of the LAT dele-tion beginning at LAT nucleotide 76, it was of interest to also examine the virulence of a third LAT mutant,dLAT371 (23). All three of these mutants (dLAT1.5, LAT2.9A, and dLAT371) contain a deletion starting at LAT nucleotide 76 (Fig. 1).dLAT371 anddLAT1.5 have identical genomic struc-tures except for the size of the deletion (371 versus 1,591 nucleotides). In contrast, LAT2.9A anddLAT371 have signif-icantly different genomic structures (Fig. 1C and F) but contain identical 371-nucleotide deletions in LAT. Consistent with LAT2.9A, but in contrast todLAT1.5,dLAT371 had reduced virulence in mice. Thus, in mice, the larger 1,591-nucleotide deletion indLAT1.5 resulted in increased virulence, while the smaller 371-nucleotide deletion in dLAT371 and LAT2.9A resulted in decreased virulence.
We previously reported that dLAT371 has wild-type viru-lence and wild-type spontaneous reactivation in rabbits (23). This is in contrast to LAT2.9A, which has increased virulence and decreased spontaneous reactivation in rabbits. Since these mutants have identical deletions in the 5⬘ end of LAT, the phenotypic differences in rabbits appear to be due to either (i) the lack of LAT nucleotides 1500 to 8324 in LAT2.9A, (ii) undetected differences in LAT expression or stability in LAT2.9A compared with dLAT371, (iii) the single copy of
LAT in LAT2.9A, or (iv) the ectopic location of LAT in LAT2.9A. However, we think that the ectopic location and the single LAT copy are unlikely possibilities, since LAT3.3A and LAT2.9AR, which contain the same single-copy ectopic insert but without the 371-nucleotide deletion, both have wild-type virulence and wild-type spontaneous reactivation.
HSV-1 kills rabbits and mice by causing encephalitis. In this study, virulence refers to this encephalitic death following oc-ular infection. In a simple model, the relative level of HSV-1 virulence would be expected to be directly related to the virus load in the brain. That is, if there was more virus in the brain, there would be more encephalitic death. FordLAT1.5 in mice, this situation appeared to hold.dLAT1.5 was more virulent in mice than its marker-rescued virus, and the amount of dLAT1.5 in mouse brains was significantly increased compared with that of its marker-rescued virus. In contrast, this model did not appear to hold for LAT2.9A or dLAT371 in mice. These mutants had reduced virulence in mice, yet the viral load in the brain was the same as that of wild-type virus. This strongly suggests, and supports a previous report (15), that the virus load in the brain is not always a good predictor of neu-rovirulence for HSV-1.
LAT may partially inhibit productive gene expression (3, 10, 18). In addition, it has been proposed that a 0.7-kb transcript beginning and terminating just upstream of the LAT promoter affects viral virulence (38). Both of these findings suggest that deletion of the LAT promoter in LAT-null mutants might alter virulence. However, we are not aware of any reports of a LAT-null mutant with altered virulence (1, 5, 14, 16, 20). We have, however, previously reported that LAT2.9A has in-creased virulence in rabbits (24). Thus, although complete lack of LAT expression (see Fig. 1B) does not appear to alter virulence, expression of just specific portions of LAT, as in LAT2.9A (Fig. 1F),dLAT371 (Fig. 1C), and dLAT1.5 (Fig. 1D), can alter virulence. Thus, we have a situation in which a large deletion (i.e., LAT-null mutants, which have no LAT promoter and which are therefore equivalent to deletion of the entire gene) has no impact on virulence, while smaller dele-tions which are encompassed by the larger deletion (those in LAT2.9A,dLAT371, anddLAT1.5) do alter virulence. As we previously speculated in regard to LAT2.9A (24), this situation is similar to that seen in promoter-mapping studies. Because promoters usually have multiple functional elements that may work either in concert or antagonistically, deletion of increas-ingly larger portions of the 5⬘ end of a promoter often pro-duces multiple increases and decreases in promoter activity. The situation with LAT2.9A,dLAT371, anddLAT1.5 is anal-ogous. These different deletions produced different increases and decreases in virulence, suggesting that LAT may have multiple functional elements. Since LAT may not encode a protein (8), it is possible that the LAT RNA may directly or indirectly regulate the expression or function of viral and/or cellular genes that in turn influence virulence. In this case, the different deletions may alter the LAT RNA structure or sta-bility, which in turn affects the LAT RNA function.
[image:10.612.67.280.74.267.2]Finally, the results presented here strongly suggest that LAT has multiple functions, since it affects both virulence and spon-taneous reactivation.
FIG. 9. Virulence of marker-rescued viruses in mice. Mice were ocularly infected as described for Fig. 4, and survival was determined at 21 days p.i. The numbers above each bar represent the number of surviving mice/total number of mice. TheP value in the box was determined by the chi-square test using a 5-by-2 contingency table. The
Pvalues above individual bars were determined by the chi-square test compared with the wild type. wt, wild-type McKrae;dL15R, marker-rescued dLAT1.5; dLATR, marker-rescued dLAT2903; dL371R, marker-rescueddLAT371; LAT2.9R, marker-rescued LAT2.9A.
on November 9, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants EY07566, EY11629, and EY12823; the Discovery Fund for Eye Research; and the Skirball Program in Molecular Ophthalmology.
We thank Anita Avery for her expert technical support and Steven J. Robbins for productive discussions.
REFERENCES
1.Block, T. M., S. Deshmane, J. Masonis, J. Maggioncalda, T. Valyi-Nagi, and N. W. Fraser.1993. An HSV LAT null mutant reactivates slowly from latent infection and makes small plaques on CV-1 monolayers. Virology192:618– 630.
2.Bloom, D. C., G. B. Devi-Rao, J. M. Hill, J. G. Stevens, and E. K. Wagner. 1994. Molecular analysis of herpes simplex virus type 1 during epinephrine-induced reactivation of latently infected rabbits in vivo. J. Virol.68:1283– 1292.
3.Chen, S. H., M. F. Kramer, P. A. Schaffer, and D. M. Coen.1997. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol.71: 5878–5884.
4.Deatly, A. M., J. G. Spivack, E. Lavi, and N. W. Fraser.1987. RNA from an immediate early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc. Natl. Acad. Sci. USA 84:3204–3208.
5.Deshmane, S. L., M. Nicosia, T. Valyi-Nagy, L. T. Feldman, A. Dillner, and N. W. Fraser.1993. An HSV-1 mutant lacking the LAT TATA element reactivates normally in explant cocultivation. Virology196:868–872. 6.Dobson, A. T., F. Sederati, G. Devi-Rao, W. M. Flanagan, M. J. Farrell, J. G.
Stevens, E. K. Wagner, and L. T. Feldman.1989. Identification of the latency-associated transcript promoter by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J. Virol.63:3844–3851.
7.Drolet, B., G. Perng, R. Villosis, S. Slanina, A. Nesburn, and S. Wechsler. 1999. Expression of the first 811 nucleotides of the HSV-1 latency associated transcript (LAT) partially restores wild type spontaneous reactivation to a LAT null mutant. Virology253:96–106.
8.Drolet, B. S., G. C. Perng, J. Cohen, S. M. Slanina, A. Yukht, A. B. Nesburn, and S. L. Wechsler.1998. The region of the herpes simplex virus type 1 LAT gene involved in spontaneous reactivation does not encode a functional protein. Virology242:221–232.
9.Farrell, M. J., A. T. Dobson, and L. T. Feldman.1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA 88:790–794.
10. Garber, D. A., P. A. Schaffer, and D. M. Knipe.1997. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol.71:5885– 5893.
11. Gordon, Y. J., B. Johnson, E. Romanowski, and T. Araullo-Cruz.1988. RNA complementary to herpes simplex virus type 1 ICP0 gene demonstrated in neurons of human trigeminal ganglia. J. Virol.62:1832–1835.
12. Hill, J. M., F. Sedarati, R. T. Javier, E. K. Wagner, and J. G. Stevens.1990. Herpes simplex virus latent phase transcription facilitates in vivo reactiva-tion. Virology174:117–125.
13. Inman, M., G. Perng, G. Henderson, H. Ghiasi, A. Nesburn, S. Wechsler, and C. Jones.2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J. Virol.75:3636–3646.
14. Izumi, K. M., A. M. McKelvey, G. Devi-Rao, E. K. Wagner, and J. G. Stevens. 1989. Molecular and biological characterization of a type 1 herpes simplex virus (HSV-1) specifically deleted for expression of the latency-associated transcript (LAT). Microb. Pathog.7:121–134.
15. Javier, R. T., K. M. Izumi, and J. G. Stevens.1988. Localization of a herpes simplex virus neurovirulence gene dissociated from high-titer virus replica-tion in the brain. J. Virol.62:1381–1387.
16. Leib, D. A., C. L. Bogard, M. Kosz-Vnenchak, K. A. Hicks, D. M. Coen, D. M. Knipe, and P. A. Schaffer.1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol.63:2893–2900.
17. Loutsch, J. M., G. C. Perng, J. M. Hill, X. Zheng, M. E. Marquart, T. M. Block, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.1999. Identical 371-base-pair deletion mutations in the LAT genes of herpes simplex virus type 1 McKrae and 17syn⫹result in different in vivo reactivation phenotypes.
J. Virol.73:767–771.
18. Mador, N., D. Goldenberg, O. Cohen, A. Panet, and I. Steiner.1998. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J. Virol.72:5067–5075.
19. Perng, G., C. Jones, H. Ciacci-Zanella, G. Henderson, A. Yukht, S. Slanina, F. Hofman, H. Ghiasi, A. Nesburn, and S. Wechsler.2000. Virus induced neuronal apoptosis blocked by the herpes simplex virus latency associated transcript (LAT). Science287:1500–1503.
20. Perng, G. C., E. C. Dunkel, P. A. Geary, S. M. Slanina, H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler.1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol.68:8045–8055. 21. Perng, G. C., H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler.1994.
An improved method for cloning portions of the repeat regions of herpes simplex virus type 1. J. Virol Methods46:111–116.
22. Perng, G. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, and S. L. Wechsler. 1996. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J. Virol.70:976–984.
23. Perng, G. C., S. M. Slanina, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler. 1996. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J. Virol.70:2014–2018. 24. Perng, G. C., S. M. Slanina, A. Yuhkt, B. S. Drolet, W. J. Keleher, H. Ghiasi,
A. B. Nesburn, and S. L. Wechsler.1999. A herpes simplex virus type 1 latency-associated transcript mutant with increased virulence and reduced spontaneous reactivation. J. Virol.73:920–929.
25. Perng, G. C., S. M. Slanina, A. Yukht, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.2000. The latency-associated transcript gene enhances establish-ment of herpes simplex virus type 1 latency in rabbits. J. Virol.74:1885–1891. 26. Perng, G. C., R. L. Thompson, N. M. Sawtell, W. E. Taylor, S. M. Slanina, H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler.1995. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol.69:3033–3041.
27. Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J. R. Lokensgard, and S. L. Wechsler.1987. Detection of latency-related viral RNAs in tri-geminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol.61:3820–3826.
28. Sawtell, N. M., and R. L. Thompson.1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol.66:2157–2169. 29. Sedarati, F., K. M. Izumi, E. K. Wagner, and J. G. Stevens.1989. Herpes
simplex virus type 1 latency-associated transcription plays no role in estab-lishment or maintenance of a latent infection in murine sensory neurons. J. Virol.63:4455–4458.
30. Spivack, J. G., and N. W. Fraser.1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol.61:3841–3847. 31. Stevens, J. G.1990. Transcripts associated with herpes simplex virus latency.
Adv. Exp. Med. Biol.278:199–204.
32. Stevens, J. G., L. Haarr, D. D. Porter, M. L. Cook, and E. K. Wagner.1988. Prominence of the herpes simplex virus latency-associated transcript in tri-geminal ganglia from seropositive humans. J. Infect. Dis.158:117–123. 33. Stevens, J. G., E. K. Wagner, G. B. Devi-Rao, M. L. Cook, and L. T. Feldman.
1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science235:1056–1059.
34. Thompson, R. L., and N. M. Sawtell.1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol.71:5432–5440.
35. Wagner, E. K., G. Devi-Rao, L. T. Feldman, A. T. Dobson, Y. F. Zhang, W. M. Flanagan, and J. G. Stevens.1988. Physical characterization of the herpes simplex virus latency-associated transcript in neurons. J. Virol.62: 1194–1202.
36. Wechsler, S. L., A. B. Nesburn, R. Watson, S. M. Slanina, and H. Ghiasi. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol.62:4051–4058.
37. Wechsler, S. L., A. B. Nesburn, J. Zwaagstra, and H. Ghiasi.1989. Sequence of the latency-related gene of herpes simplex virus type 1. Virology168:168– 172.
38. Zhu, J., W. Kang, M. E. Marquart, J. M. Hill, X. Zheng, T. M. Block, and N. W. Fraser.1999. Identification of a novel 0.7-kb polyadenylated transcript in the LAT promoter region of HSV-1 that is strain specific and may con-tribute to virulence. Virology265:296–307.
39. Zwaagstra, J., H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.1989. In vitro promoter activity associated with the latency-associated transcript gene of herpes simplex virus type 1. J. Gen Virol.70:2163–2169.
40. Zwaagstra, J. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, S. C. Wheatley, K. Lillycrop, J. Wood, D. S. Latchman, K. Patel, and S. L. Wechsler.1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J. Virol.64:5019–5028.