Copyright © 1999, American Society for Microbiology. All Rights Reserved.
The Presence of Human Immunodeficiency Virus Type 1 Vpr
Correlates with a Decrease in the Frequency of Mutations
in a Plasmid Shuttle Vector
JEREMY B. JOWETT,1,2YI-MING XIE,1ANDIRVIN S. Y. CHEN1*
Departments of Microbiology & Immunology and Medicine, University of California—Los Angeles School of Medicine,
Los Angeles, California 90095-1678,1and International Diabetes Institute, Caulfield, Victoria 3162, Australia2
Received 21 January 1999/Accepted 3 May 1999
The human immunodeficiency virus type 1 (HIV-1) Vpr protein induces cell cycle arrest at the border of G2
and M similar to the arrest caused by agents which damage DNA. We determined whether the presence of Vpr would affect the ability of cells to repair DNA. We developed a shuttle vector system to analyze the effect of Vpr upon the repair of UV-damaged DNA. Our results demonstrated that the presence of Vpr decreased the rate of deletions in this system. Of note, cells arrested in G2by other genotoxic agents also increased the frequency
of DNA repair of UV-damaged shuttle vectors. We did not observe any direct effect of Vpr upon the rate of double-strand break repair and/or nucleotide excision repair of genomic DNA in cells. Our results suggest a role for HIV-1 Vpr in altering the frequency of DNA repair, a property which may have importance for HIV-1 replication and pathogenesis.
The genome of human immunodeficiency virus (HIV), the etiological agent of AIDS, contains nine genes, five of which have been determined to be essential for replication in vitro. The remaining four genes,vpr, vpu,vif, andnef, have conse-quently been dubbed nonessential accessory genes, and they play various roles, either directly, in enhancing the efficiency of the replication cycle of HIV, or indirectly, in enhancing sur-vival in the presence of the immune system in vivo and poten-tially in the causation of disease (10, 11, 27).
Vpr encodes an 11-kDa protein that is incorporated into the virion via an interaction with the carboxy-terminal portion of the Gag precursor protein (17). Vpr has been proposed to promote nuclear localization of the viral preintegration com-plex following infection (5, 15), although this role has been disputed (13). This function of Vpr has been suggested to increase the efficiency of replication of HIV in nondividing cells (3, 7). Furthermore, Vpr has been ascribed additional roles in enhancement of transcriptional activity on various promoters, including the HIV long terminal repeat (6), and in the rescue of viral expression from latently infected cell lines (20), although the roles of these effects on the viral life cycle have yet to be determined.
We and others have reported that the HIV type 1 (HIV-1)
vprgene product causes cell cycle arrest in HIV-infected target cells (14, 16, 32, 33). We further proposed that cell cycle arrest induced by Vpr may play a role in pathogenesis by inhibiting the clonal expansion and effective immune response of T cells (16, 31).
Phenotypic characterization of the Vpr-mediated arrest re-vealed that the cells arrested in the second gap (G2) phase of the cell cycle, prior to initiation of mitosis (M). The Cdc2 kinase, a major component controlling the transition of cells from G2to M, was found to be predominantly in the inactive state (16, 30). This point of arrest can also be triggered in a cell following certain types of DNA damage. Alkylating agents and
ionizing radiation cause cells to arrest or delay in G2 and prevent or postpone the onset of mitosis, depending upon the severity of the damage. This pause in cell cycle progression has been suggested as a period in which repair to the DNA is carried out, after which the cell resumes cell cycle progression into mitosis (21, 24, 26). We identified a phenotypic similarity between Vpr-mediated cell cycle arrest and arrest caused by DNA-damaging agents (30). The attributes identified included growth arrest in the G2phase of the cell cycle with hyperphos-phorylation of Cdc2 kinase and the ability of methylxanthines to reverse cell cycle arrest by both Vpr and nitrogen mustard. We proposed, therefore, that Vpr may mediate growth arrest either through (i) directly causing DNA damage, (ii) interfer-ing with DNA damage repair processes, or (iii) modulatinterfer-ing a DNA damage detection pathway (checkpoint). In this study we determined whether the presence of Vpr influences the rate of DNA damage and repair in cells. We examined the effect of Vpr on both cellular DNA and exogenously introduced non-viral templates.
Our results indicate that the presence of Vpr results in a reduction in the frequency of deletions in a shuttle vector system. Vpr, however, did not detectably affect nucleotide ex-cision repair or double-strand break repair on cellular DNA.
MATERIALS AND METHODS
Cells and viral stocks.COS cells (African green monkey kidney) were grown
in Dulbecco’s modified Eagle’s medium with 10% calf serum (Gemini). Stocks of HIV-1NL4-3-thyenv(⫺)/vesicular stomatitis virus G protein (VSV-G) and HIV-1NL4-3-thyvprXenv(⫺)/VSV-G were prepared and used to infect target cells as previously described (30). The measurement of levels of infection was per-formed with anti-Thy 1.2 antibodies and flow cytometry as described previously (16). In the case where Vpr was provided in the absence of all other HIV proteins, the vector NLthy⌬Bgl was replaced with both pHRcmvypyvpr and pCMVdR8.2DVPR, which made up the viral stock pHRVpr. The former was derived from pHRcmvluciferase (25) by removal of the luciferase open reading frame and substitution with the epitope-taggedvpropen reading frame. As a control for this experiment, an additional vector was constructed, pHRcmvthy, which was identical to pHRcmvypyvpr except that the murinethy1.2gene (29) replaced the vpr open reading frame (giving the viral stock pHRThy). The latter plasmid, pCMVdR8.2DVPR, was used as a packaging plasmid based on pCMVdR8.2D (25), but with thevpropen reading frame deleted. The R80A mutant of Vpr, prepared in the NLthy⌬Bgl vector, consists of a single-amino-acid change at codon 80 from arginine to alanine and is identical to the mutant previously described (8).
* Corresponding author. Mailing address: Departments of Microbi-ology & ImmunMicrobi-ology and Medicine, UCLA School of Medicine, Los Angeles, CA 90095-1678. Phone: (310) 825-4793. Fax: (310) 794-7682. E-mail: [email protected].
7132
on November 9, 2019 by guest
http://jvi.asm.org/
Shuttle vector.The shuttle vector used in these DNA damage detection ex-periments was the pBK-CMV (pBK) phagemid vector obtained from Stratagene. Where stated, the plasmid was irradiated in Tris-EDTA buffer (pH 7.5) on parafilm in a Stratalinker UV light irradiator at a rate of approximately 6 J/s. At 16 h postinfection, to allow reverse transcription, integration, and expression of viral proteins, the cells were transfected with the pBK shuttle vector by using Lipofection (Life Technologies) according to the manufacturer’s protocol. The cells were harvested after a further 48 h (allowing maximal episomal replication of the plasmid) and lysed, and a Hirt extraction was performed to recover low-molecular-weight DNA. The recovered DNA was transformed into Esche-richia coliand plated on nutrient medium. Colonies were scored after overnight incubation at 37°C and a further 4- to 6-h incubation at 4°C to enhance the blue color. Linearized shuttle vector was prepared by digestion withHindIII, which cuts within the LacZ␣peptide coding region.
CHEF pulsed-field gel electrophoresis.The method and reagents used for
contour-clamped homogeneous electric field (CHEF) pulsed-field gel electro-phoresis were derived from the CHEF genomic DNA plug kit (Bio-Rad). Briefly, cells were harvested with trypsin, washed, and set in a 2% soft agar plug at 2⫻ 106to 3⫻106/ml. The plugs were digested overnight at 50°C with proteinase K (1 mg/ml) and then washed four times in wash buffer. The agarose plugs were then set in the electrophoresis gel (1%) and subjected to CHEF pulsed-field electrophoresis in 0.25⫻Tris-borate-EDTA buffer as follows: initial pulse time, 60 s; final pulse time, 120 s; field strength, 5.2 V/cm; duration, 22.5 h. These settings resolve fragments less than approximately 2 Mbp; the larger fragments will migrate in a “compression” zone at the same point, while unbroken genomic DNA remains at the origin (4, 18).
Unscheduled DNA synthesis (UDS) assay.Cells were infected as described
above. A strong inhibitor of replicative DNA synthesis (hydroxyurea [10 mM]) was added 1 h prior to UV irradiation (30 J/m2). The use of this inhibitor is required because of the potential for high levels of background [3H]thymidine incorporation as the cells progress through DNA synthesis (S) phase. Immedi-ately following UV irradiation, [3H]thymidine was added to the tissue culture medium (50Ci/ml) and pulsed for a further 2 h in the presence of hydroxyurea. The cells were harvested and lysed on a filter prior to being immersed in scintillation fluid and counted. The values reported are the means of triplicate samples.
Statistical analysis.Statistical significance (where indicated) was calculated by
either the chi-square test or Fisher’s exact test (two-tailed method).
RESULTS
Vpr decreases DNA mutation frequency in a transfected shuttle vector.To investigate whether Vpr affects the repair of damaged DNA, we utilized a shuttle vector (pBK) containing thelacZ␣gene under the control of a bacterial promoter as a reporter gene for mutation. When transformed intoE. coli, the colonies turn blue in the presence of inducer and substrate. A white colony would indicate a mutation in either the LacZ␣ peptide reporter gene or in its expression control regions. The vector also contained a eukaryotic origin of replication derived from simian virus 40 (SV40) allowing episomal replication in COS cells to sufficiently high levels for detection after extrac-tion and transformaextrac-tion into bacteria. We first determined conditions which would provide a sufficiently high rate of mu-tation for us to monitor the effects of Vpr. We found that UV irradiation of plasmid DNA at a dose of 2,500 J/m2followed by DNA transfection and recovery after 48 h was sufficient to achieve a mutation frequency of approximately 3%.
We determined the effect of Vpr on the mutation frequency by expressing Vpr in cells prior to transfection with the re-porter plasmid. COS cells were infected at a high multiplicity of infection with a pseudotyped virus based on the HIV-1NL4-3 strain carrying either a wild-type Vpr [HIV-1NL4-3-thyenv(⫺)/ VSV-G] or a frame-shifted mutant of Vpr, termed VprX [HIV-1NL4-3-thyvprXenv(⫺)/VSV-G] that results in truncation of approximately one-third of the protein (16, 29). A sufficient innoculum of virus was used to ensure that more than 90% of the cells were infected, which was verified by Thy 1.2 expres-sion and flow cytometry (data not shown). The mutant protein does not induce G2cell cycle arrest. At 16 h after infection, the cells were transfected with the pBK shuttle vector and har-vested after a further 48 h. The recovered episomal DNA was transformed intoE. coli, and the blue and white colonies were
counted. The presence of Vpr reduced the mutation frequency occurring at the lacZ gene locus by approximately 10-fold compared to that of mock- or VprX-infected cells (Table 1). Although the magnitude varied in different experiments, a 10-fold or greater level of reduction of the mutation frequency of the shuttle vector was consistently observed with Vpr.
We confirmed that the effect was due specifically to Vpr by using a point mutant of Vpr (R80A) that is expressed stably yet fails to cause cell cycle arrest (8). We found that VprR80A resulted in a mutation frequency in our shuttle vector (2.6%) similar to that of mock-infected COS cells (2.9%), while wild-type Vpr resulted in a 0.16% mutation frequency (Table 2).
We verified that Vpr alone was the cause of the observed reduction in mutation frequency and that it was not a result of other viral proteins present in the NL-Thy vector. We prepared a retroviral vector capable of only Vpr production following infection (pHRVpr). The level of suppression was approxi-mately 10-fold that of the control (pHRThy) and was similar to that obtained with virus carrying the wild-typevprgene (Table 3).
Vpr reduces the frequency of deletion mutants. Plasmids
were recovered from randomly selected white colonies and analyzed by nucleotide sequence analysis for the type of
mu-TABLE 1. Vpr decreases mutation frequency in UV-damaged shuttle vectora
Virus stock
Expt 1 Expt 2 Expt 3
No. of white/blue
colonies % White colonies
No. of white/blue
colonies % White colonies
No. of white/blue
colonies % White colonies
Mock 18/527 3.30 8/1,318 0.60
Vpr 0/2,415 ⬍0.04 0/1,557 ⬍0.06 0/142 ⬍0.7 VprX 17/1,708 0.98 6/888 0.67 25/80 24.0
Pvalue ⬍0.0001 0.0021 ⬍0.0001
[image:2.612.312.551.91.180.2]aCOS cells (2⫻105) were either mock infected or infected with HIV-1NL4-3-thyenv(⫺)/VSV-G or HIV-1NL4-3-thyvprXenv(⫺)/VSV-G. At 16 h postin-fection, the cells were transfected with 4g of pBK-CMV plasmid (UV irradi-ated at 2,500 J/m2 by using Lipofectin. After a further 48 h, the cells were harvested and low-molecular-weight DNA was recovered by Hirt extraction and transformed intoE. coli. Blue and white colonies were scored to give mutation frequencies. The observed number of colonies for Vpr-infected cells was found to differ significantly from those for mock- (experiments 1 and 2) and VprX (experiment 3)-infected cells, with thePvalues given in the bottom row of the table. Experiments 1, 2, and 3 are independent repetitions of the same protocol.
TABLE 2. Decreased mutation frequency correlates with G2
arrest phenotypea
Virus stock No. of white/bluecolonies White%
colonies
Mock 36/1,191 2.90
Vpr 2/1,184 0.16
VprX 10/832 1.20
VprR80A 55/2,055 2.60
aCOS cells (2⫻105) were either mock infected or infected with HIV- 1 NL4-3-thy env(⫺)/VSV-G (Vpr), HIV-1NL4-3-thyvprXenv(⫺)/VSV-G (VprX), or HIV-1 NL4-3-thyenv(⫺)/VSV-G containing an arginine-to-alanine mutation in Vpr at codon 80 (VprR80A). At 16 h postinfection, the cells were transfected with 4g of pBK-CMV plasmid (UV irradiated at 2,500 J/m2) by using Lipofectin. After a further 48 h, the cells were harvested and low-molecular-weight DNA was recovered by Hirt extrac-tion and transformed intoE. coli. Blue and white colonies were scored to give mutation frequencies. The results observed for Vpr were statistically significant (P⬍ 0.0001) when compared to mock infection. Conversely, those observed for R80A were not significant (P⫽0.58). This experiment was repeated twice with similar results.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.312.551.566.639.2]tation that abolishes LacZ␣peptide function. All of the mu-tations were found to be deletions in thelacZ␣peptide open reading frame and/or its promoter region (data not shown).
Deletions in DNA may arise when a double-strand DNA break occurs, followed by exonuclease digestion and end re-joining, with the size of the deletion determined by the level of exonuclease activity and the rate of end ligation. Alternatively, a deletion may occur when recombination takes place between two homologous regions of DNA. The size of this deletion is determined by the size of the intervening DNA segment be-tween the homologous regions. Deletion junctions in a random selection of recovered mutant shuttle vectors derived from mock, Vpr-, and VprX-containing cultures were examined by nucleotide sequence analysis for evidence of homologous re-combination. We found that the sizes of the deletions varied from one plasmid to the next and that there were no obvious stretches of homology at or near the deletion junctions that would be indicative of homologous recombination (data not shown). This observation is consistent with published work with similar shuttle vector systems (9).
Vpr reduces mutation frequency in the repair of linearized shuttle vector DNA. The above results indicate that deletion mutants could result from cleavage of DNA followed by nu-clease activity. We therefore tested whether Vpr could affect mutation frequency in the shuttle vector cleaved by a restric-tion endonuclease. The shuttle vector was digested to comple-tion with an enzyme that cuts once within thelacZ␣peptide coding region. We observed overall a higher frequency of white colonies, as might be expected following transfection of linear-ized plasmid DNA. As expected, nucleotide sequence analysis of white colonies demonstrated evidence for deletions extend-ing from the cleavage site. We found that the presence of Vpr in this experiment decreased the frequency of mutation by approximately two- to threefold compared to that of either mock-infected or VprX virus-infected cells (Table 4). This result suggested that the presence of Vpr reduces the mutation frequency during the repair and recircularization of linear DNA. One interpretation is that the presence of Vpr facilitates accurate end rejoining of double-strand breaks in DNA. Al-ternatively, the presence of Vpr may inhibit double-strand break rejoining. Since plasmids with double-strand breaks would not amplify in COS cells, any plasmids bearing muta-tions originating from a double-strand break would not be detected.
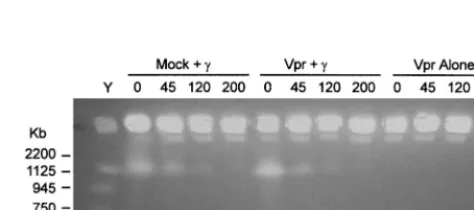
Vpr does not directly affect endogenous cellular DNA repair processes.The above observations are consistent with the idea that the presence of Vpr acts to increase the rate of ligation, thereby preventing the action of nucleases on the free DNA ends; to inhibit cellular exonuclease digestion of free ends; or to inhibit rejoining of double-strand breaks. We tested the possibility that Vpr influences the ability of cellular DNA to repair itself. We measured the rate of endogenous DNA repair either in the presence or absence of Vpr following the gener-ation of double-strand breaks by␥-irradiation. Pulsed-field gel electrophoresis was used to monitor strand breakage and its rate of repair (2, 18). Vpr did not itself cause detectable levels of double-strand DNA breaks (Fig. 1). ␥-Irradiation (80 Gy) caused double-strand breakage as measured by the appearance of a lower-molecular-weight band on a pulsed-field gel (Fig. 1). The DNA was repaired completely by 240 min following irra-diation, as evidenced by the diminishing intensity of the faster-migrating band over time, as has been previously described (18). In the presence of Vpr, the faster-migrating band dimin-ished at a similar rate, indicating that Vpr did not alter the rate of repair of the double-strand DNA breaks induced by irradi-ation. Thus, within the limits of this assay, we conclude that Vpr does not detectably enhance or inhibit the rate of repair of double-strand breaks generated by␥-irradiation. Furthermore, the presence of Vpr itself does not induce double-strand breaks in DNA. However, we could not determine from this
[image:3.612.53.295.93.175.2]FIG. 1. Vpr does not affect rate of endogenous DNA double-strand break repair induced by␥-irradiation. COS cells (6⫻105) were infected with HIV-1NL4-3-thyenv(⫺)/VSV-G (Vpr) to give a⬎90% infection rate as measured by Thy expression at 48 h postinfection. At 18 h postinfection, the cells were exposed to 80 Gy of␥-irradiation and harvested at the indicated time points. The cells were embedded in 1% soft agarose, gently lysed, and subjected to pulsed-field electrophoresis at 5.1 V/cm and 60- to 120-s ramp time for 22 h. Lane Y, Saccharomyces cerevisiae chromosomes were used as markers, with the sizes indicated.
TABLE 3. Vpr causes decreased mutation frequency in the absence of all other viral proteinsa
Virus stock No. of white/bluecolonies White%
colonies
Mock 105/9,225 1.10
VprX 44/7,061 0.62
Vpr 7/4,770 0.15
pHRThy 88/5,954 1.40
pHRVpr 2/2,665 0.07
aCOS cells (2⫻105) were either mock infected or infected with HIV-1NL4-3-thyenv(⫺)/VSV-G (Vpr) or HIV-1NL4-3-thyvprXenv(⫺)/VSV-G (VprX). At the same time, two additional cultures were infected with a virus capable of producing only Vpr (pHRVpr) or a control producing Thy (pHRThy) and no other viral proteins. At 16 h postinfection, the cells were transfected with 4g of pBK-CMV plasmid (UV irradiated at 2,500 J/m2) by using Lipofectin. After a further 48 h, the cells were harvested and low-molecular-weight DNA was re-covered by Hirt extraction and transformed intoE. coli. Blue and white colonies were scored to give mutation frequencies. The observed counts for Vpr com-pared to those for Mock infection and those of pHRVpr comcom-pared to those for pHRThy achieved significance, with bothPvalues less than 0.0001.
TABLE 4. Vpr decreases mutation frequency in transfected linearized shuttle vectora
Virus stock No. of White/bluecolonies White%
colonies
Mock 119/181 40
Vpr 24/80 23
VprX 103/81 56
aCOS cells (2⫻105) were either mock infected or infected with HIV-1NL4-3-thyenv(⫺)/VSV-G (Vpr) or HIV-1NL4-3-thyvprXenv(⫺)/VSV-G (VprX). At 16 h postinfection, the cells were transfected with 4g of pBK-CMV plasmid that was linearized withHindIII. After a further 48 h, the cells were harvested and low-molecular-weight DNA was recovered by Hirt extraction and trans-formed into E. coli. Blue and white colonies were scored to give mutation frequencies. The values observed for Vpr compared to those for VprX and mock infection were found to be statistically significant, with associatedPvalues of ⬍0.0001 and 0.0023, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.315.552.545.650.2]experiment whether the presence of Vpr affects the rate of mutation at or around the breakpoint junctions.
Different repair pathways are utilized by the cell to repair DNA damage, depending upon the nature of the damage to the DNA. Nucleotide excision repair (NER) is the primary mechanism active in repair of cyclobutane pyrimidine dimers, the primary form of damage caused by UV light irradiation. Vpr has been shown to bind to HHR23A (36), a protein that can bind xeroderma pigmentosum-complementing protein group C (XPC). XPC likely plays a role in NER, although its function has not yet been determined (1, 23). A part of normal NER activity is in the resynthesis of the damaged stretch of DNA by using the complementary strand as a template. This has been called UDS and can be measured in cells following UV irradiation by using radioactively labeled nucleotides (12, 34). To test whether Vpr could affect NER, we measured UDS in UV-irradiated cells in the presence and absence of Vpr. UDS was seen as an increase in [3H]thymidine incorporation following UV irradiation. Mock-infected and VprX-infected cells showed approximately a twofold increase in [3 H]thymi-dine incorporation following UV irradiation (Table 5). No significant difference was observed in cells expressing Vpr. Thus, these results indicate that Vpr does not grossly affect the extent of repair synthesis; however, the fidelity of the repair synthesis cannot be assessed by these experiments.
Agents that damage cellular DNA can also decrease shuttle vector mutation frequency.The similarities between Vpr-me-diated G2cell cycle arrest and the cell cycle arrest induced by DNA-damaging agents suggest that Vpr triggers a normal cel-lular checkpoint activated in response to damage of the DNA. These checkpoints are normally activated in order to allow cells to repair damaged DNA prior to further progression through the cell cycle (21, 24, 26). It is conceivable that acti-vation of the checkpoint would also activate DNA repair pro-cesses which have high fidelity for repair of damaged DNA. Such a scenario has been observed following the activation of p53, which, among other effects, induces cell cycle arrest at the G1/S phase and simultaneously activates GADD45, a protein involved in NER (19, 35). An increase in resistance to UV light damage in G2versus G1has been studied in a synchronized cell population. This difference, however, was attributed to an in-creased time for repair in G2until the beginning of the next S phase rather than a specific activation of DNA repair enzymes (28).
Since Vpr alters the frequency of mutation, we determined whether the arrest of cells by other genotoxic agents might have similar effects on the frequency of mutation. COS cells
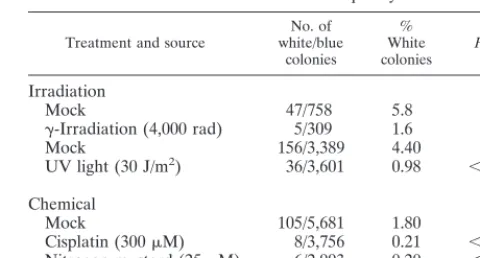
were treated with agents known to cause double-strand DNA breaks and cell cycle arrest in the G2phase of the cell cycle. These were␥-irradiation (40 Gy), cisplatin (300M; 1 h), and nitrogen mustard (25M; 30 min). Treatment with UV irra-diation (30 J/m2) was also included, as it generates a different set of lesions, the majority of which are cyclobutane pyrimidine dimers. Compared to mock-treated samples, all treatments caused a decrease in mutation frequency in the transfected shuttle vector ranging from fourfold to ninefold (Table 6). This data suggests that a common mechanism of action may exist for Vpr and other genotoxic agents in the reduction of muta-tion frequency. One possibility is that both Vpr and DNA damage activate common checkpoint pathways which alter the frequency of DNA repair.
DISCUSSION
Our results demonstrate that the presence of HIV-1 Vpr acts to decrease the frequency of deletion mutations which occur following introduction of a UV-damaged plasmid into cells. Since Vpr induces cell cycle arrest at a G2checkpoint, we determined whether Vpr directly affects the repair of damage to endogenous cell DNA. However, we observed no effect of Vpr upon the rate of either double-strand-break repair or NER of cellular DNA. The effects of Vpr on mutation rates were similar to that observed with other agents which induce cell cycle arrest, such as␥radiation and DNA-damaging chem-icals.
It is noteworthy that a previous study (22) reported that Vpr could reduce the mutation rate of an HIV-1 shuttle vector by approximately fourfold. The design of the experiments sug-gested that the mutations that were observed were a result of errors that occurred during reverse transcription. The muta-tions which we monitored in our experiments were a result of cellular DNA damage repair processes in which reverse tran-scription is not implicated. The majority of the mutations we observed, both in the presence and absence of Vpr, were de-letions, consistent with previous reports of the major types of mutations occurring following transfection of DNA into cells. The most likely mechanism for deletion formation is a double-TABLE 5. Vpr does not affect UDS following UV irradiation of
COS cellsa
Virus stock
[3H]-thymidine incorporated (mean cpm) No UV
irradiation irradiatedUV
Mock 4,508 8,219
Vpr 4,260 8,933
VprX 4,299 9,675
[image:4.612.311.551.91.220.2]aCOS cells (2⫻105) were either mock infected or infected with HIV-1NL4-3-thyenv(⫺)/VSV-G (Vpr) or HIV-1NL4-3-thyvprXenv(⫺)/VSV-G (VprX). At 16 h postinfection, the cells were exposed to UV light (30 J/m2), after which [3H]thymidine (50Ci/ml) was added for 2 h prior to harvest. Hydroxyurea (10 mM) was added to the culture medium from 1 h prior to UV irradiation until the time of harvest. The harvested cells were lysed, placed on a filter, and counted for levels of [3H]thymidine incorporation. This experiment was repeated twice with similar results.
TABLE 6. Agents that damage cellular DNA can also decrease shuttle vector mutation frequencya
Treatment and source white/blueNo. of colonies
% White
colonies Pvalue
Irradiation
Mock 47/758 5.8
␥-Irradiation (4,000 rad) 5/309 1.6 0.0024
Mock 156/3,389 4.40
UV light (30 J/m2) 36/3,601 0.98 ⬍0.0001
Chemical
Mock 105/5,681 1.80
Cisplatin (300M) 8/3,756 0.21 ⬍0.0001 Nitrogen mustard (25M) 6/2,993 0.20 ⬍0.0001
aCOS cells (2⫻105) were exposed to various DNA-damaging treatments. The chemical treatments were cisplatin (300M; 1 h) and nitrogen mustard (25M; 30 min). At 19 h postinfection, the cells were transfected with 4g of pBK-CMV plasmid (UV irradiated at 2,500 J/m2) by using Lipofectin. After a further 48 h, the cells were harvested and low-molecular-weight DNA was recovered by Hirt extraction and transformed intoE. coli. Blue and white colonies were scored to give mutation frequencies. The values for the different agents were compared with those from the mock experiments, and all were found to vary significantly from the control; the associatedPvalues are given in the final column of the table. This experiment was repeated twice with similar results.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.53.293.92.164.2]strand break in the DNA followed by nuclease activity and religation of the free DNA ends. Vpr could decrease the fre-quency of mutations by (i) increasing the rate of accurate rejoining of DNA following breakage, (ii) decreasing the rate of rejoining of ends resulting from double-strand breaks (re-sulting in failure to amplify in COS cells), (iii) increasing the fidelity of nucleotide excision repair of UV-damaged plasmids such that double-strand breaks are less likely to occur, (iv) inhibiting the action of nucleases on the broken free ends of DNA, and (v) enhancing the activity of error-free as opposed to error-prone mechanisms of DNA repair. From our experi-ments, we cannot distinguish among the above possibilities; however, altering the rate of rejoining of double-strand breaks (hypotheses i and ii above) appears less likely, since we did not observe an effect of Vpr on repair of double-strand breaks induced by␥-irradiation of endogenous cellular DNA. If one assumes that the increase in fidelity in our studies is due to mechanisms similar to those in the studies by Mansky (22), as-yet-unknown processes involved in DNA repair common to cellular DNA repair and reverse transcription may be impli-cated.
We hypothesize that Vpr acts along pathways normally used by cells to induce cell cycle arrest at a G2checkpoint. These checkpoints are ordinarily activated in cells following damage by genotoxic agents, such as␥radiation, which induces double-strand breaks in DNA. Failure to arrest at G2and entry into mitosis without repair of double-strand breaks would have catastrophic consequences for the ability of the cell to faith-fully duplicate its genetic material in daughter cells. Since it is so critical for a cell to faithfully replicate and segregate DNA, it is conceivable that activation of DNA repair processes which are more efficient or act with greater fidelity occurs at the G2 checkpoint. In this scenario, the mechanism by which Vpr decreases the frequency of mutations is not a direct effect of Vpr but rather a consequence of the cell cycle arrest check-point induced by Vpr. A similar decrease in the frequency of mutations observed in cells arrested by other genotoxic agents is consistent with this idea.
We have proposed previously that one important conse-quence of Vpr action is the crippling of an effective immune response through arresting division of antigen-activated T cells. The results discussed here suggest the hypothesis that enhancing the fidelity of DNA repair may be another function of Vpr important for HIV-1 replication or pathogenesis. As described by Mansky (22), Vpr may act to increase the fidelity of reverse transcription. We demonstrated previously that Vpr packaged within virions is sufficient to induce cell cycle arrest during the initial infection (31). Thus, another point in the viral life cycle where enhanced fidelity of DNA repair could play a role is during integration. Based upon our results, we hypoth-esize that Vpr may act to facilitate integration by creating an environment within the cell or in the context of the preinte-gration complex which facilitates intepreinte-gration. One possibility may be to protect the free ends of unintegrated viral DNA from nucleases, akin to our proposed mechanism for reducing deletions in our transfected shuttle vector. Another possibility may be to enhance the rate of ligation of free viral DNA ends to endonuclease-cleaved cellular DNA. Vpr may therefore promote HIV evasion of possible intracellular defenses against exogenous DNA sequences entering the cell. We can test these possibilities by examining the effect of Vpr on the rate of integration and the structures of preintegration viral DNA intermediates.
ACKNOWLEDGMENTS
We thank Elizabeth Withers-Ward and Kathie Grovit-Ferbas for critical reading of the manuscript and Liz Duarte and Rosie Taweesup for preparation of the manuscript. We are grateful to Dong Sung An for preparation of the pCMVdR8.2DVPR and the pHRcmvthy vectors and to Sheila Stewart for preparation of the pHRcmvypyvpr vector and R80A point mutant of Vpr used in this study.
This work is supported by NIH grant CA70018, The Center for AIDS Research (CFAR), and Amgen. J.B.J. was supported by an American Cancer Society Fellowship.
REFERENCES
1.Aboussekhra, A., and R. D. Wood.1994. Repair of UV-damaged DNA by
mammalian cells and Saccharomyces cerevisiae. Curr. Opin. Genet. Dev.
4:212–220.
2.Ager, D. D., and W. C. Dewey.1990. Calibration of pulsed field gel
electro-phoresis for measurement of DNA double-strand breaks. Int. J. Radiat. Biol.
58:249–259.
3.Balliet, J. W., D. L. Kolson, G. Eiger, F. M. Kim, K. A. McGann, A.
Srini-vasan, and R. Collman.1994. Distinct effects in primary macrophages and
lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: mutational analysis of a primary HIV-1 isolate. Virology
200:623–631.
4.Ble`ocher, D., M. Einspenner, and J. Zajackowski.1989. CHEF
electrophore-sis, a sensitive technique for the determination of DNA double-strand breaks. Int. J. Radiat. Biol.56:437–448.
5.Bukrinsky, M. I., S. Haggerty, M. P. Dempsey, N. Sharova, A. Adzhubel, L.
Spitz, P. Lewis, D. Goldfarb, M. Emerman, and M. Stevenson.1993. A
nuclear localization signal within HIV-1 matrix protein that governs infec-tion of non-dividing cells. Nature365:666–669.
6.Cohen, E. A., E. F. Terwilliger, Y. Jalinoos, J. Proulx, J. G. Sodroski, and
W. A. Haseltine.1990. Identification of HIV-1 vpr product and function. J.
Acquired Immune Defic. Syndr.3:11–18.
7.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau.1995. Vpr is required
for efficient replication of human immunodeficiency virus type-1 in mono-nuclear phagocytes. Virology206:935–944.
8.Di Marzio, P., S. Choe, M. Ebright, R. Knoblauch, and N. R. Landau.1995.
Mutational analysis of cell cycle arrest, nuclear localization, and virion pack-aging of human immunodeficiency virus type 1 Vpr. J. Virol.69:7909–7916.
9.DuBridge, R. B., and M. P. Calos.1988. Recombinant shuttle vectors for the
study of mutation in mammalian cells. Mutagenesis3:1–9.
10. Fauci, A. S.1988. The human immunodeficiency virus: infectivity and
mech-anisms of pathogenesis. Science239:617–622.
11. Fauci, A. S.1993. Multifactorial nature of human immunodeficiency virus
disease: implications for therapy. Science262:1011–1018.
12. Fautz, R., B. Husein, E. Efstathiou, and C. Hechenberger-Freudl.1993.
Assessment of the relation between the initial viability and the attachment of freshly isolated rat hepatocytes used for the in vivo/in vitro DNA repair assay (UDS). Mutat. Res.291:21–27.
13. Freed, E. O., G. Englund, and M. A. Martin.1995. Role of the basic domain
of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol.69:3949–3954.
14. He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau.
1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol.
69:6705–6711.
15.Heinzinger, N. K., M. I. Bukrinsky, S. A. Haggerty, A. M. Ragland, V.
Kewalramani, M. A. Lee, H. E. Gendelman, L. Ratner, M. Stevenson, and M.
Emerman.1994. The Vpr protein of human immunodeficiency virus type 1
influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. USA91:7311–7315.
16. Jowett, J. B., V. Planelles, B. Poon, N. P. Shah, M. L. Chen, and I. S. Y. Chen.
1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2⫹M phase of the cell cycle. J. Virol.69:6304–6313.
17.Kondo, E., F. Mammano, E. A. Cohen, and H. G. Go¨ttlinger.1995. The
p6gag domain of human immunodeficiency virus type 1 is sufficient for the incorporation of Vpr into heterologous viral particles. J. Virol.69:2759– 2764.
18.Lee, S. E., C. R. Pulaski, D. M. He, D. M. Benjamin, M. Voss, J. Um, and
E. A. Hendrickson.1995. Isolation of mammalian cell mutants that are X-ray
sensitive, impaired in DNA double-strand break repair and defective for V(D)J recombination. Mutat. Res.336:279–291.
19.Levine, A. J.1997. p53, the cellular gatekeeper for growth and division. Cell
88:323–331.
20.Levy, D. N., Y. Refaeli, R. R. MacGregor, and D. B. Weiner.1994. Serum Vpr
regulates productive infection and latency of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA91:10873–10877.
21.Maity, A., W. G. McKenna, and R. J. Muschel.1994. The molecular basis for
cell cycle delays following ionizing radiation: a review. Radiother. Oncol.
31:1–13.
on November 9, 2019 by guest
http://jvi.asm.org/
22.Mansky, L. M.1996. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology222:391–400.
23. Masutani, C., K. Sugasawa, J. Yanagisawa, T. Sonoyama, M. Ui, T.
Eno-moto, K. Takio, K. Tanaka, P. J. van der Spek, D. Bootsma, et al.1994.
Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. Embo. J.13:1831–1843.
24. Murnane, J. P.1995. Cell cycle regulation in response to DNA damage in
mammalian cells: a historical perspective. Cancer Metastasis Rev.14:17–29.
25. Naldini, L., U. Blo¨mer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M.
Verma, and D. Trono.1996. In vivo gene delivery and stable transduction of
nondividing cells by a lentiviral vector. Science272:263–637.
26. O’Connor, P. M., D. K. Ferris, G. A. White, J. Pines, T. Hunter, D. L. Longo,
and K. W. Kohn.1992. Relationships between cdc2 kinase, DNA
cross-linking, and cell cycle perturbations induced by nitrogen mustard. Cell Growth Differ.3:43–52.
27. Peterlin, B. M., and P. A. Luciw.1988. Molecular biology of HIV. AIDS
2(Suppl. 1):S29–S40.
28. Petersen, L. N., D. K. Orren, and V. A. Bohr. 1995. Gene-specific and
strand-specific DNA repair in the G1and G2phases of the cell cycle. Mol. Cell. Biol.15:3731–3737.
29. Planelles, V., J. B. Jowett, Q. X. Li, Y. Xie, B. Hahn, and I. S. Y. Chen.1996.
Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Vi-rol.70:2516–2524.
30. Poon, B., J. B. Jowett, S. A. Stewart, R. W. Armstrong, G. M. Rishton, and
I. S. Y. Chen.1997. Human immunodeficiency virus type 1 vpr gene induces
phenotypic effects similar to those of the DNA alkylating agent, nitrogen mustard. J. Virol.71:3961–3971.
31. Poon, B., K. Grovit-Ferbas, S. A. Stewart, and I. S. Y. Chen.1998. Cell cycle
arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science281:266–269.
32. Re, F., D. Braaten, E. K. Franke, and J. Luban.1995. Human
immunode-ficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the acti-vation of p34cdc2-cyclin B. J. Virol.69:6859–6864.
33. Rogel, M. E., L. I. Wu, and M. Emerman.1995. The human
immunodefi-ciency virus type 1 vpr gene prevents cell proliferation during chronic infec-tion.69:882–888.
34. Sawada, S., S. Asakura, H. Daimon, and C. Furihata.1995. Comparison of
autoradiography, liquid scintillation counting and immunoenzymatic staining of 5-bromo-2⬘-deoxyuridine for measurement of unscheduled DNA synthesis and replicative DNA synthesis in rat liver. Mutat. Res.344:109–116.
35. Smith, M. L., I. T. Chen, Q. Zhan, I. Bae, C. Y. Chen, T. M. Gilmer, M. B.
Kastan, P. M. O’Connor, and A. J. Fornace, Jr.1994. Interaction of the
p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Sci-ence266:1376–1380.
36. Withers-Ward, E. S., J. B. M. Jowett, S. A. Stewart, Y. M. Xie, A. Garfinkel,
Y. Shibagaki, S. A. Chow, N. Shah, F. Hanaoka, D. G. Sawitz, R. W.
Arm-strong, L. M. Souza, and I. S. Y. Chen.1997. Human immunodeficiency virus
type 1 Vpr interacts with HHR23A, a cellular protein implicated in nucle-otide excision DNA repair. J. Virol.71:9732–9742.