0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.24.15007–15015.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Induction of Innate Immune Response Genes by Sin Nombre
Hantavirus Does Not Require Viral Replication
Joseph Prescott,
1Chunyan Ye,
1Ganes Sen,
2and Brian Hjelle
1,3*
Infectious Diseases and Inflammation Program, Department of Pathology,1and Departments of Biology and Molecular Genetics
and Microbiology,3University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131, and Department of
Molecular Biology, The Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, Ohio 441952
Received 19 July 2005/Accepted 23 September 2005
Maladaptive immune responses are considered to be important factors in the pathogenesis of the two diseases caused by hantaviruses, hemorrhagic fever with renal syndrome and hantavirus cardiopulmonary syndrome (HCPS). While the intensity of adaptive antiviral T-cell responses seems to correlate with the severity of HCPS, there is increasing evidence that innate antiviral responses by endothelial cells, the native targets for hantavirus infection in vivo, are induced within hours of exposure to infectious hantaviruses. To investigate early events in the innate response to Sin Nombre virus (SNV), the principal etiologic agent of HCPS in North America, we treated human endothelial cells with live virus, or virus subjected to inactivation by UV irradiation at minimal doses required to inhibit replication, and assayed host expression of interferon-stimulated genes (ISG) by microarray and reverse transcription-PCR. We show herein that a variety of ISG are induced between 4 and 24 h after exposure to both live and killed virus. The levels of such induction at early time points (before 24 h) were generally higher in cells treated with SNV particles that had been killed by exposure to UV irradiation. Additionally, SNV exposed to increasing doses of UV irradiation induced ISG better than live virus despite increased disruption of viral RNA integrity. However, SNV replication was required for continued ISG overexpression by 3 days posttreatment. These results suggest that hantavirus particles may themselves be capable of early induction of ISG and that ongoing production of viral particles during infection could contribute to the pathogenic process.
Sin Nombre virus(SNV) is a New World hantavirus (
Bunya-viridae: Hantavirus) that is an important etiological agent of
hantavirus cardiopulmonary syndrome (HCPS), a recently rec-ognized human disease (36). Old World hantaviruses cause a related illness known as hemorrhagic fever with renal syn-drome. Hantaviruses are rodent-borne viruses with a tri-mented negative-sense genome comprised of L, M, and S seg-ments encoding the viral polymerase, envelope glycoproteins G1 and G2, and the nucleocapsid protein, respectively (23).
The natural reservoir host of SNV is the deer mouse (
Peromy-scus maniculatus), a common North American mammal with a
large habitation range (9, 33). Deer mice that have been in-fected by SNV show no apparent pathogenic affects and main-tain the virus in a persistent infection, although the deer mouse does develop a specific immune response to SNV infection, as exemplified by the presence of high titers of neutralizing anti-bodies (8). Although there are several other hantaviruses that cause HCPS in the New World, SNV accounts for the over-whelming majority of cases reported in North America (13, 47). SNV is primarily transmitted to humans via accidental inha-lation of virus-contaminated rodent urine, feces, or saliva (37). HCPS is characterized by an initial onset of flu-like symptoms followed by acute pulmonary edema resulting from increased vascular permeability, but death is usually attributable to hy-potension and cardiogenic shock, leading some investigators to use the term “hantavirus cardiopulmonary syndrome” rather
than “hantavirus pulmonary syndrome” (46). Approximately 380 cases of HCPS have been reported in the United States as of early 2005, with a 38% mortality rate (www.cdc.gov/ncidod /diseases/hanta/hps/index.htm).
Vascular endothelial cells are the primary targets of SNV infection in humans, which the virus infects without causing any apparent cytopathic effects (34, 47). A subsequent increase in pulmonary vascular permeability contributes significantly to the disease process, which is consistent with the known role of endothelial cells in regulation of vascular permeability (35, 43). Additionally, it is observed that patients who succumb to HCPS have an abundance of cytokine-producing cells in lung tissues. Recently, it was further suggested that virus-specific CD8⫹T cells may contribute to the severity of disease (14, 25, 31, 34, 47). These observations suggest that immune-mediated mechanisms induced by virally infected endothelial cells are important in the pathogenesis of SNV, but it remains unclear whether innate or acquired immune mechanisms or both are important in the pathogenesis of HCPS.
To understand the initial events that lead to disease, it would be helpful to examine viral interactions with host endothelial cells and to characterize the innate antiviral responses associ-ated with infection. Alpha/beta interferons (IFN-␣/) have long been known to be induced by viral infections to produce an antiviral state in cells in an autocrine and paracrine fashion (19, 38). One of the best-characterized antiviral pathways for the induction of IFN-␣/is mediated through double-stranded RNA (dsRNA) acting on toll-like receptor 3 (TLR3) (1, 42). This interaction induces the transcription of the chemokines RANTES and IP-10 and of interferon-stimulated genes (ISG) 15, 54, 56, and 60 and 2⬘-5⬘-oligoadenylate synthetase (OAS)
* Corresponding author. Mailing address: Dept. of Pathology, Uni-versity of New Mexico, 329CRF, MSC08 4640, 1 UniUni-versity of New Mexico, Albuquerque, NM 87131-0001. Phone: (505) 272-0624. Fax: (505) 272-4401. E-mail: [email protected].
15007
on November 8, 2019 by guest
http://jvi.asm.org/
(18, 28). Newly synthesized IFN-then binds cell surface re-ceptors, activating the Jak/STAT pathway, which in turn results in the transcription of additional ISG and additional IFN-␣/ via IRF3-, -7-, and -9-related pathways. The above events are known to be initiated via dsRNA, which is an intermediate product of active replication by single-stranded RNA (ssRNA) viruses. Viral dsRNA can induce the transcription of certain ISG through an IFN-independent mechanism. However, re-cent investigations have shown that some viruses can induce ISG activation through an uncharacterized dsRNA-indepen-dent, replication-independent pathway, through the RNA he-licase RIG-I and, in other cases, ssRNA has been implicated in ISG signaling through TLR7 or TLR8 (6, 11, 40).
To assess host events that are involved in SNV recognition and subsequent responses, we used microarrays to identify genes that are differentially regulated after exposure to either live SNV or replication-defective, UV-inactivated SNV, under the presumption that genes that are regulated in response to replicating virus but not by UV-inactivated virus are involved in antiviral responses that require the ongoing expression of viral gene products and/or the presence of the replication in-termediate, dsRNA. By contrast, genes induced by the UV-killed SNV are responses to recognition of one or more com-ponents of the viral particle, possibly in the context of its internalization by the cell.
We found that SNV and UV-inactivated SNV elicit a similar gene expression profile at 1 day postexposure in human pri-mary endothelial cells; the response to killed virus often ex-ceeded that of the live virus at early time points, and the response to live virus exceeded that of killed virus at later time points. Many of the up-regulated genes are involved in char-acterized ISG pathways. We show that there is a rapid induc-tion of ISG even by particles that have sustained substantial damage to the RNA genome, a finding that all but excludes active viral transcription in induction of the signaling cascade and that calls into question whether dsRNA or ssRNA is acting as the proximate ligand that elicits expression of ISG. Further-more, IFN-␣/are not transcriptionally induced by exposure to SNV particles in our assays.
MATERIALS AND METHODS
Cells and virus.We purchased pooled primary human umbilical vein endo-thelial cells (HUVEC) from Clonetics (San Diego, CA) and propagated them in EBM-2 medium supplemented with the bullet kit (Clonetics), which supplies growth factors, including vascular endothelial growth factor. We previously de-scribed the isolation of SNV strain SN77734 from a New Mexican deer mouse (7); SNV was propagated and the titer was determined in Vero E6 cells under strict standard operating procedures using biosafety level 3 facilities and prac-tices (CDC registration number C20041018-0267). For preparation of UV-inac-tivated SNV, we placed 100l of virus stock (typically 1.5⫻106
to 2⫻106
focus-forming units/ml) in each well of a 96-well plate and subjected the virus to UV irradiation at 254 nm for various time intervals (⬃5 mW/cm2
). We estab-lished that virus inactivation was complete at 10 s of exposure using a focus assay (3). We also determined the effects of UV irradiation on the viral genome and protein constituents G1, G2, and N with real-time reverse transcription-PCR (RT-PCR) and Western blotting, respectively (unpublished data).
HUVEC infections.We plated HUVEC at passage 1 to 3 onto 12-mm glass coverslips in 12-well tissue culture plates or in plastic tissue culture flasks (Corn-ing) at a density of 10,000 cells/cm2or 2,500 cells/cm2, respectively. Cells were
incubated at 37°C in the presence of 5% CO2, and medium was changed every
2 days. When the cells reached 85 to 90% confluence, the medium was removed and we then added either live SNV or UV-inactivated SNV at a multiplicity of infection (MOI) of 1 to the culture vessel. We incubated the virus or killed virus
for 1 h at 37°C in the presence of 5% CO2with periodic rocking. We then
removed the virus-containing medium, washed the cells once in phosphate-buffered saline (PBS), and replaced it with fresh medium.
IFA.For immunofluorescence assay (IFA) studies, we grew HUVEC on glass coverslips and infected or mock infected them for 1 or 3 days with SNV and then washed them in PBS and fixed them onto the slides with 4% paraformaldehyde for 30 min. After another wash in PBS, we permeabilized them by treatment with 0.2% Triton X-100 for 7 min and then washed and preblocked with 4% PBS-bovine serum albumin (BSA) for 10 min. Primary rabbit antinucleocapsid im-mune sera or control preimim-mune sera at a 1:10,000 dilution in PBS-BSA were then incubated with the cells for 1 hour at room temperature. After the cells were again washed, we applied a secondary fluorescein isothiocyanate (FITC)-labeled anti-rabbit immunoglobulin G (IgG) antibody (Boehringer Mannheim) at a 1:1,000 dilution in PBS-BSA to the cells for 1 h at room temperature. We then washed the coverslips in distilled water and mounted them onto glass slides. We observed and photographed the stained cells using a Zeiss Axioskop fluorescent microscope equipped with a Zeiss Axiocam digital camera (Carl Zeiss, Germany). The fraction of cells that were infected was estimated through review of multiple random fields from two separate infection experiments.
RNA preparation and microarrays.We exposed 80% confluent HUVEC at passage 2 in T75 flasks to either live or UV-inactivated SNV (MOI of 1.0) or mock-infected cultures treated identically with HUVEC medium in duplicate as described above. At 1 and 3 days posttreatment, total RNA was isolated using the RNeasy kit (QIAGEN). We conducted replicate target synthesis and replicate microarray experiments on different days from replicate cell cultures. A poly(A)⫹spike in control (Affymetrix) was added to the total RNA as a control for the efficiency of target synthesis, followed by using the GeneChip Expression cDNA synthesis kit (Affymetrix) to produce cDNA. We generated biotinylated cRNA targets using the in vitro transcription labeling kit (Affymetrix). To assess the quality of the RNA, we examined it visually to determine whether higher-molecular-weight forms were in abundance with the RNA Nano LabChip kit with a Bioanalyzer 2100 instrument (Agilent Technologies, Palo Alto, CA). After determining that the RNA was of good quality, we hybridized 15g of fraction-ated cRNA (Affymetrix Clean-up module) to Affymetrix HG-U133 PLUS 2.0 microarray slides for 16 h and then washed and stained the arrays using the fluidics FS 450 station (Affymetrix). Fluorescence images were captured using the GeneChip scanner, and all chips were scaled to 500 using the Affymetrix GeneChip operating software. The fluorescence data were analyzed using Gene-Spring version 6.1 software (Silicon Genetics, Redwood City, CA). We statisti-cally analyzed replicate chips for each gene using a Studentttest. One-day and 3-day gene expression values for duplicate treated samples were normalized relative to the mean values of mock-infected samples from 1-day and 3-day cultures, respectively. We statistically compared the intensity of hybridization for individual genes to controls using a one-way analysis of variance, with signifi-cance set toPⱕ0.05. Regulated transcripts were filtered by having an expression fluorescence intensity of⬎250, a twofold or more change in expression, and genes with a present signal, indicated by specific target hybridization to perfectly matched probes but not mismatched probes differing by a single base, in at least one treatment sample for up-regulated genes and at least one control sample for down-regulated genes.
Real-time SYBR Green and TaqMan RT-PCR.For SYBR Green RT-PCR, we performed reverse transcription using 2g of total RNA and random hexamer primers in 50-l reaction mixtures using the Applied Biosystems (ABI) TaqMan reverse transcription reagents kit (ABI, Foster City, CA). For PCR, we used the Applied Biosystems SYBR Green core reagents kit to perform reactions in triplicate using 3l of cDNA and a 0.4M final concentration of each primer in a 25-l total reaction volume. The primers we used were the following: GAPDH, sense (5⬘-GGAAGCTCACTGGCATGGC-3⬘) and antisense (5⬘-TAG ACGGCAGGTCAGGTCCA-3⬘); RANTES, sense (5⬘-TGCCTCCCCATATTC CTCGG-3⬘) and antisense (5⬘-TGGGTTGGAGCACTTGCCAC-3⬘); MxA, sense (5⬘-TGATCCAGCTGCTGCATCCC-3⬘) and antisense (5⬘-GGCGCACCT TCTCCTCATAC-3⬘); ISG56, sense (5⬘-TCTCAGAGGAGCCTGGCTAAG-3⬘) and antisense (5⬘-CCACACTGTATTTGGTGTCTAGG-3⬘); ISG15, sense (5⬘ -TGGTGGACAAATGCGACGAA-3⬘) and antisense (5⬘-CAGGCGCAGATT CATGAAC-3⬘); IFI35, sense (5⬘-TCCCCCTGGTATTCCGAGGA-3⬘) and an-tisense (5⬘-TGTTGCAGCACCTGCTCAGC-3⬘); and TLR3, sense (5⬘-ACC TGAATTTGAAACGGTCTTTTACT-3⬘) and antisense (5⬘-CTTCCATGTTAA GGTGCTCCAAAC-3⬘). We incubated the reaction mixtures at 50°C for 2 min and then 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 min using an ABI Prism 7000 sequence detection system. We subjected the reactions to dissociation curve analysis to exclude the possibility of nonspecific amplification, and we then calculated the change for each gene using the mean
on November 8, 2019 by guest
http://jvi.asm.org/
of the change inCtvalues (⌬Ct) normalized to theCtvalues of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for each sample (2⫺⌬⌬Ct
).
We performed TaqMan RT-PCR for viral S-segment RNA as previously described (8). Briefly, we extracted viral RNA from 140l of HUVEC culture supernatant using the QIAamp viral RNA mini kit (QIAGEN) or from cells using the RNeasy kit as above. We then reverse transcribed 3l of RNA using an S-segment-specific sense primer (5⬘-AGCACATTACAGAGCAGACGGGC-3⬘) and subjected 5 l of the resulting cDNA to PCR with S-segment-specific primers, sense (5⬘-GCAGACGGGCAGCTGTG-3⬘) and antisense (5⬘-AGATCA GCCAGTTCCCGCT-3⬘), and a fluorescently labeled TaqMan probe (5⬘ -[car-boxyfluorescein]- TGCATTGGAGACCAAACTCGGAGAACTT-[tetramethyl carboxyrhodamine]-3⬘) in triplicate PCRs using an ABI Prism 7000 sequence detection system. We quantitated the viral RNA using a standard curve gener-ated using S-segment templates of known copy number.
Western blotting.We cultured HUVEC in six-well plates and treated them with SNV or UV-killed SNV as above. At 12, 24, or 36 hours posttreatment, we trypsinized and washed the cells once with ice-cold PBS. We made whole-cell
extracts using 200l of a buffer containing 1% Triton X-100, 0.5 M Tris, 0.1 M EDTA, 2% glycerol, 10%-mercaptoethanol, 2% sodium dodecyl sulfate (SDS), and Complete Mini protease inhibitor (Roche). We then separated equal vol-umes of lysates on 12.5% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gels and electrophoretically transferred the proteins to nitrocellulose membranes for 1 h at 100 V. We blocked the membranes in 5% milk–PBS for 1 h and then probed them with a primary rabbit anti-ISG56 antibody for 2 h at a dilution of 1:1,000 or with an anti-actin antibody (Santa Cruz SC-1616-R) at a 1:2,000 dilution. We washed the membranes three times for 5 min each in Western wash and incubated them with a peroxidase-labeled rabbit IgG secondary anti-body for 1 h. To visualize the proteins, we washed the membranes again and developed them using enhanced chemiluminescence and Kodak BioMax MR film. Blocking by neutralizing antisera against SNV. Using a heat-inactivated (56°C, 30 min) serum with a known focus reduction neutralization titer of 1:800/ml, we neutralized an aliquot of live SNV by adding 5 or 2.5l of the plasma to 100l of SNV (1.5⫻106
FFU/ml) and incubated the mixture for 30 min at room temperature with periodic mixing. We then treated HUVEC with this mixture or with SNV alone for 1 hour, removed the supernatant, and added fresh medium. After 6 hours of incubation, we harvested total cellular RNA and quantitated the ISG response using real-time RT-PCR as described above. As controls we also examined the plasma of an SNV-seronegative individual as well as the serum of a rabbit with antibodies raised against recombinant SNV N antigen (titer, 1:106
/ml by Western blotting), a serum which is known to lack any neutralizing activity (data not shown).
Statistical analysis.Statistical analyses were performed on replicate samples using an unpaired Student’sttest. The mean⫾standard error of the mean is represented, and significance (P⬍0.05) is reported where appropriate.
RESULTS
[image:3.585.43.284.68.240.2]Sin Nombre virus, but not UV-killed SNV, infects and rep-licates in human endothelial cells. It has previously been shown that SNV is able to infect human endothelial cells in vitro (16, 41). We cultured primary HUVEC on glass cover-slips and exposed them to live or UV-killed SN77734 SNV at an MOI of 1.0. At 1 and 3 days postinfection (p.i.), we assessed the expression of viral N antigen using an indirect IFA with rabbit anti-N antibody and FITC-conjugated anti-rabbit IgG. Fluorescence microscopy showed that at 1 day p.i., approxi-mately 30 to 40% of the cells displayed visible staining for nucleocapsid antigen, while at 3 days p.i., approximately 70 to 80% of the cells showed such staining (Fig. 1). Infected sam-ples stained with preimmune sera showed staining character-istics similar to mock-infected controls, easily distinguished from specific staining (data not shown).
FIG. 1. Indirect immunofluorescence of SNV-infected HUVEC. HUVEC grown on 12-mm glass slides were mock infected or infected for 1 hour with SNV at an MOI of 1.0 and incubated for 24 (top) or 72 (bottom) hours. Cells were fixed in 4% paraformaldehyde and stained with rabbit N serum (1:10,000) followed by a FITC-labeled anti-rabbit antibody. The numbers of positive cells were quantitated by counting the number of fluorescing cells compared to the total number of cells in multiple random fields. Approximately 30 to 40% of cells showed nucleocapsid staining at 24 h p.i., and 70 to 80% of cells showed staining at 72 h p.i. Experiments were performed in triplicate.
FIG. 2. Live SNV replicates in HUVEC, whereas UV-killed SNV does not. HUVEC were treated for 1 hour with SNV at an MOI of 1.0 (squares) or an identical amount of SNV that had previously been exposed to UV irradiation at 254-nm for 15 seconds (triangles). Cells were washed and incubated for the indicated amount of time while medium was changed every other day. Total cellular RNA from confluent T25s (A) or from 140l of culture supernatant (B) was extracted at the indicated time points and reverse transcribed using an SNV S-segment forward primer (8). Viral RNA was quantitated by TaqMan using an S-segment primer and probe set and standards of known copy number. PCR was performed in triplicate for duplicate experiments. Results are reported as the mean⫾standard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.585.134.456.542.667.2]To confirm that the virus is actively replicating in these cells, we isolated RNA from SNV-infected and UV-inactivated SNV-treated cells and cell culture supernatants exposed to the same amount of virus. RNA was harvested at 1 hour, 1 day, and every other day until 9 days posttreatment, and real-time PCR was used to quantitate cDNA generated with a primer de-signed to transcribe the S-segment negative-sense RNA (8) to determine viral-sense RNA (vRNA) load in these samples (Fig. 2). Immediately following UV treatment, there was a 5.8-fold decrease in the titer of S-segment RNA in the killed virus preparation. Productivity of vRNA in the SNV-infected HUVEC peaked at 24 h p.i., as manifested by a 4.7-fold in-crease in titer compared to cells harvested 1 hour p.i. (Fig. 2A). By contrast, the productivity of S-segment vRNA in superna-tant was highest at 3 days p.i., increasing 7.4-fold from 1 hour p.i. (Fig. 2B). The UV-killed SNV samples showed small in-creases in vRNA at 1 day p.i. for both cell and supernatant vRNA isolations with a 1.2-fold increase in each case, possibly
due to incomplete release of viral particles from the cell sur-faces during the wash step. By 3 days p.i., there was a sharp decrease in vRNA to almost undetectable levels out to 9 days p.i. The SNV-infected and UV-killed SNV-treated HUVEC cul-tures showed similar growth characteristics to control samples throughout the 9-day period of observation, indicating that neither virus treatment interferes significantly with normal cell division and growth in culture (not shown).
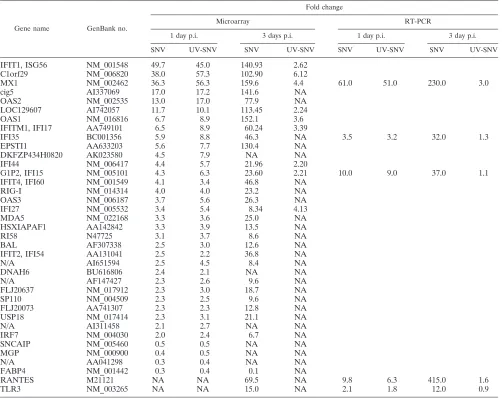
[image:4.585.43.545.81.480.2]SNV and UV-inactivated SNV induced similar responses at 1 day but different responses by 3 days postexposure. We compared the list of differentially regulated genes from HUVEC that had been exposed to live SNV with that of UV-killed SNV to determine which genes were differentially regulated by replicating SNV and which genes could also be induced by the mere presence of the viral particle, respectively. Of the 64 genes that were differentially regulated by SNV and the 70 genes regulated by UV-killed SNV at 24 h p.i., 35 genes appear on both lists (Table 1). Thirty-one of the shared genes
TABLE 1. Microarray and real-time RT-PCR resultsa
Gene name GenBank no.
Fold change
Microarray RT-PCR
1 day p.i. 3 days p.i. 1 day p.i. 3 day p.i.
SNV UV-SNV SNV UV-SNV SNV UV-SNV SNV UV-SNV
IFIT1, ISG56 NM_001548 49.7 45.0 140.93 2.62
C1orf29 NM_006820 38.0 57.3 102.90 6.12
MX1 NM_002462 36.3 56.3 159.6 4.4 61.0 51.0 230.0 3.0
cig5 AI337069 17.0 17.2 141.6 NA
OAS2 NM_002535 13.0 17.0 77.9 NA
LOC129607 AI742057 11.7 10.1 113.45 2.24
OAS1 NM_016816 6.7 8.9 152.1 3.6
IFITM1, IFI17 AA749101 6.5 8.9 60.24 3.39
IFI35 BC001356 5.9 8.8 46.3 NA 3.5 3.2 32.0 1.3
EPSTI1 AA633203 5.6 7.7 130.4 NA
DKFZP434H0820 AK023580 4.5 7.9 NA NA
IFI44 NM_006417 4.4 5.7 21.96 2.20
G1P2, IFI15 NM_005101 4.3 6.3 23.60 2.21 10.0 9.0 37.0 1.1
IFIT4, IFI60 NM_001549 4.1 3.4 46.8 NA
RIG-I NM_014314 4.0 4.0 23.2 NA
OAS3 NM_006187 3.7 5.6 26.3 NA
IFI27 NM_005532 3.4 5.4 8.34 4.13
MDA5 NM_022168 3.3 3.6 25.0 NA
HSXIAPAF1 AA142842 3.3 3.9 13.5 NA
RI58 N47725 3.1 3.7 8.6 NA
BAL AF307338 2.5 3.0 12.6 NA
IFIT2, IFI54 AA131041 2.5 2.2 36.8 NA
N/A AI651594 2.5 4.5 8.4 NA
DNAH6 BU616806 2.4 2.1 NA NA
N/A AF147427 2.3 2.6 9.6 NA
FLJ20637 NM_017912 2.3 3.0 18.7 NA
SP110 NM_004509 2.3 2.5 9.6 NA
FLJ20073 AA741307 2.3 2.3 12.8 NA
USP18 NM_017414 2.3 3.1 21.1 NA
N/A AI311458 2.1 2.7 NA NA
IRF7 NM_004030 2.0 2.4 6.7 NA
SNCAIP NM_005460 0.5 0.5 NA NA
MGP NM_000900 0.4 0.5 NA NA
N/A AA041298 0.3 0.4 NA NA
FABP4 NM_001442 0.3 0.4 0.1 NA
RANTES M21121 NA NA 69.5 NA 9.8 6.3 415.0 1.6
TLR3 NM_003265 NA NA 15.0 NA 2.1 1.8 12.0 0.9
a
Microarray and real-time RT-PCR fold changes of genes regulated in common at 1 day by both treatments and their respective regulations at 3 days posttreatment. The RANTES and TLR3 genes were not regulated at day 1 by microarray but were used for RT-PCR analysis. NA, genes that were not shown to be regulated by microarray analysis.
on November 8, 2019 by guest
http://jvi.asm.org/
were up-regulated, and four were down-regulated. The ratio of fold increase for SNV-infected cells compared to those that received UV-killed SNV for the up-regulated transcripts ranged from 0.62 to 1.18 (average, 0.83), indicating that the UV-inactivated virus regulated these common genes to a sim-ilar or greater extent as the replication-competent virus at 24 h p.i. The average SNV/UV-killed SNV ratio for the down-reg-ulated genes ranged from 0.62 to 1.06 (average, 0.79), again indicating that the virus down-regulated these genes to a sim-ilar extent. Those genes that were up-regulated only by live or only by killed SNV were generally only slightly up-regulated or down-regulated by the treatment, relative to controls that re-ceived medium only. Of the 24 genes up-regulated threefold or more by the live virus, only 5 were not represented on the inactivated virus list. In every case, it is possible that the genes were regulated to a similar extent by the two treatments but were removed from one of the two lists because of statistical stringency or because of target binding to mismatch probes (see Materials and Methods). Similarly, of the 27 genes up-regulated threefold or more by the UV-killed SNV, only 4 were not represented on the SNV-infected lists, and these genes were marked absent although regulated to a similar extent.
Almost half of the genes that were regulated in common at 1 day p.i. are already known to be interferon-inducible or -related genes, including IRF7, ISG15, -17, -27, -35, -44, -54, -56, and -60, Mx1, and OAS1, -2, and -3. All of these genes were regulated to a similar extent by exposure to live or UV-killed SNV. Despite the induction of ISG, no IFN-␣ or - genes were up-regulated, indicating that transcription of the above genes may be induced through an IFN-independent pathway as described previously (44, 48).
By 72 h p.i., we found 17 genes to be up-regulated in com-mon from the SNV (576 genes) and UV-SNV (38 genes) lists. Thirteen were up-regulated and four were down-regulated. The SNV/UV-killed SNV fold change ratio for up-regulated genes ranged from 2.02 to 53.7 (average, 32.1), indicating that in all cases, genes were more strongly induced by replication-competent, live SNV at this later time point. The comparable ratio for down-regulated genes ranged from 0.52 to 1.16 (av-erage, 0.87), indicating that live SNV regulated these genes to a slightly lesser extent. As with the 1-day samples, many of these common genes are known to be regulated by IFN, but by 3 days p.i., these transcripts were greatly decreased in abun-dance in the UV-killed SNV sample compared to the SNV sample. As with the 1-day p.i. samples, no increase in IFN-␣/ transcription by either treatment was observed, and no in-crease in IFN-B was measurable by enzyme-linked immu-nosorbent assay (data not shown) using supernatants of cells exposed to SNV or killed SNV.
Real-time SYBR Green RT-PCR of selected genes confirms the array data.To verify that genes were truly differentially regulated by SNV as assessed by microarrays, we extracted RNA from SNV-infected, UV-killed SNV-treated, and mock-infected HUVEC at 1 and 3 days p.i. and reverse transcribed it into cDNA. Genes that we chose to subject to RT-PCR anal-ysis were either not regulated or up-regulated on the microar-rays. Primers for GAPDH, RANTES, MxA, ISG15, IFI35, and TLR3 were used along with SYBR Green chemistry to quan-titate gene transcripts by real-time PCR. Cycle values (Ct)
were normalized to the optical density of total RNA, and transcript abundance relative to mock-infected samples was calculated (Table 1). In most cases, especially under conditions at which there was substantial induction in expression by treat-ment, real-time PCR showed greater regulation of genes tested. The greatest differential was in the day 3 RANTES sample, with a 5.9-fold difference between array and real-time PCR values.
p56 protein levels are similarly increased by SNV and UV-killed SNV.p56 is an antiviral protein with translation initia-tion inhibitory properties and is known to be strongly induced by many viruses at early time points postinfection (11, 20). To determine whether SNV or UV-killed SNV is able to induce p56 translation, we treated HUVEC with SNV or killed SNV at an MOI of 1.0 for 1 hour and made whole-cell lysates 12, 24, or 36 h posttreatment. We used poly(I-C) as a positive control for ISG56 induction. Equal volumes of lysates were used for SDS-PAGE and Western blotting and probed with anti-ISG56 polyclonal antibodies (see Materials and Methods). Actin lev-els were used as a protein loading control. By 24 h posttreat-ment, there was a strong increase in the expression of ISG56 in both SNV and UV-killed SNV samples. At 36 h posttreatment, there was an additional increase in expression by both treat-ments compared to mock-infected HUVEC at this time point (Fig. 3).
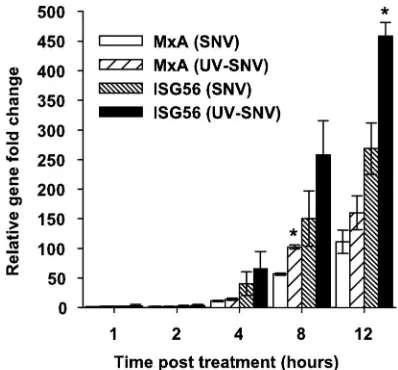
SNV and killed SNV can induce an ISG response by 2 hours posttreatment.To analyze the induction kinetics of the innate response pathway by both virus treatments, we treated cells with live or killed virus for 1 hour and harvested RNA 1, 2, 4, 8, or 12 h posttreatment to be used in real-time PCR experi-ments. Primers for ISG56 and MxA were chosen as markers of an innate response. These genes were chosen because ISG56 was the highest induced gene by live virus at 1 day p.i., and it has been previously demonstrated elsewhere that MxA is in-duced and has antiviral properties in the context of hantavirus infections (24). SNV and UV-killed virus treatment showed increases in transcription of ISG56 by 2 hours posttreatment (2.9-fold and 3.8-fold, respectively) (Fig. 4). By 4 hours post-treatment, MxA was increased 11.3-fold and 14.2-fold in the live and killed virus samples, respectively. At 8 and 12 h post-treatment, levels of ISG56 and MxA continued to increase in both treatment groups, although the UV-killed virus elicited increased transcription all cases.
UV-killed SNV elicits ISG responses independent of vRNA integrity.As described above, UV irradiation significantly re-duces the ability to amplify intact viral RNAs by PCR. To examine the kinetics of ISG gene transcription as a function of viral UV irradiation, we treated HUVEC for 1 hour with SNV exposed to UV for the indicated times (Fig. 5). Six hours postinfection, we performed real-time RT-PCR of cellular RNAs using ISG56- or MxA-specific primers. We also quan-titated S-segment vRNA as a function of UV irradiation im-mediately following exposure (Fig. 5). As a control to exclude the possibility that there is a nonspecific proinduction effect associated with UV irradiation of RNA, we treated HUVEC with poly(I-C) exposed to UV for 0 or 60 seconds and per-formed gene-specific real-time RT-PCR as above. SNV vRNA titers significantly decreased as a function of UV exposure. In contrast, ISG transcription was enhanced by UV exposure and remained above or equal to that of untreated SNV out to 2
on November 8, 2019 by guest
http://jvi.asm.org/
minutes of UV exposure, before finally decreasing to control levels after 8 minutes of exposure. The ability of poly(I-C) to induce an antiviral response was decreased by 2.8-fold after 1 minute of UV exposure (data not shown).
The ISG response is blocked by neutralizing antisera.Vero E6 cells, which we used to propagate the virus, are deficient in IFN-␣/ responses, and so it is unlikely that the results re-ported here are influenced by soluble antiviral mediators like IFN produced by these cells. However, to rule out the
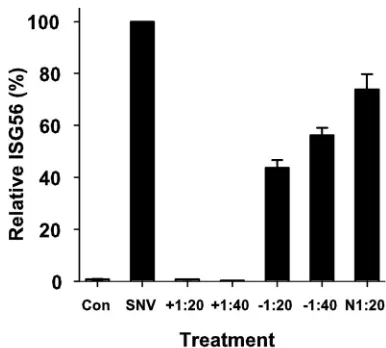
possi-bility that the observed ISG response is induced by nonviral factors, we incubated SNV with various dilutions of heat-inac-tivated plasma obtained from a convalescent HCPS patient or control human sera. We incubated the plasma with a constant amount of virus at ratios of 1:20 or 1:40 (vol/vol) for 30 min at room temperature and then incubated it with HUVEC cul-tures for 1 hour as above. Six hours after treatment, we har-vested total RNA and determined the expression levels of ISG56 by quantitative RT-PCR compared to those of un-treated cells and cells exposed to SNV without prior incubation with antisera or to cells incubated with a 1:20 dilution of rabbit anti-recombinant N sera (Fig. 6). Both dilutions of the conva-lescent-phase sera completely blocked induction of ISG ex-pression, whereas human preimmune and rabbit anti-N sera reduced the ISG response by twofold or less.
DISCUSSION
[image:6.585.317.522.68.237.2]Transcription of ISG can be induced by nucleic acid tem-plates ranging from dsDNA to dsRNA and ssRNA (12, 21, 29). We used UV-killed SNV as a control, in part, to help us determine the role of nucleic acids in induction of ISG tran-scription in the setting of hantavirus infection. Using a 66-nucleotide target from the S genome for TaqMan amplifica-tion, we showed that UV treatment causes a five- to sevenfold diminution of abundance of this very short template which, if extrapolated to consider the entire⬃12.2-kb length of the viral genome, suggests that the RNA genome at large was severely compromised by the UV treatment. The apparent ability of hantavirus particles to elicit ISG is consistent with data re-ported using several other virus systems (11).
FIG. 3. p56 protein expression is up-regulated at 24 h p.i. by live and UV-killed SNV. HUVEC were treated for 1 hour with either conditioned medium (lanes 1 and 9), with SNV (lanes 2, 4, and 6), with SNV that had been exposed to UV for 15 seconds (lanes 3, 5, and 7), or with poly(I-C) (50g/ml) (lane 8). Cells were then washed and incubated for 12 h (lanes 1 to 3), 24 h (lanes 4 and 5), or 36 h (lanes 6 to 9), and whole-cell lysates were prepared. An equal volume of lysate was separated on 12.5% SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were probed with a rabbit anti-p56 antibody or an antiactin antibody, followed by a peroxidase-labeled anti-rabbit antibody. Films were devel-oped using enhanced chemiluminescence.
[image:6.585.48.278.68.183.2]FIG. 4. Either live or UV-killed SNV induces ISG transcription at early times postexposure. HUVEC were exposed to live or UV-killed virus (15 seconds) an an MOI equivalent of 1.0 for 1 hour and washed and incubated in normal medium. At the indicated times, total RNA was extracted from cells and subjected to real-time RT-PCR using MxA- or ISG56-specific primers. Changes in abundance of each mRNA were normalized to GAPDH, and the corresponding changes were calculated using the 2⫺⌬⌬Ct method (see methods). PCR was performed in triplicate for duplicate experiments. Results are reported as the mean⫾standard error of the mean.ⴱ,P⬍0.05 compared to samples that had been treated with live SNV at the same time point.
FIG. 5. SNV exposed to UV irradiation induces ISG transcription despite increased disruption of vRNA. SNV was exposed to UV irra-diation for the indicated amount of time and either immediately as-sayed for S-segment vRNA by TaqMan using a 66-nucleotide target amplimer (7, 8) (open circles and rightyaxis) or used to treat HUVEC. After treatment for 1 hour, HUVEC were washed and incubated for 6 hours, and total cellular RNA was extracted and used for real-time RT-PCR using p56- and MxA-specific primers (left y axis). Gene changes were normalized to GAPDH and calculated using the 2⫺⌬⌬Ct method (see Materials and Methods). PCR was performed in triplicate for duplicate experiments. Results are reported as the mean⫾ stan-dard error of the mean.ⴱ,P⬍0.05 compared to mRNA abundance in cells exposed to nonirradiated SNV (0 seconds).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.62.261.435.620.2]Our results were generally consistent with those reported by other investigators examining host responses of endothelial and epithelial cells to infection with pathogenic hantaviruses (17, 32), but through the use of UV-killed SNV we were able to make several new observations. We confirmed that viral replication occurred in HUVEC that were infected with live SNV and that ISG were induced strongly by 24 h p.i. However, we were also able to demonstrate that killed SNV, even under circumstances in which the ssRNA genome had been nearly completely destroyed, was still able to elicit an ISG response of similar or greater magnitude to that seen with live virus as early as 4 hours p.i. Considering that dsDNA and dsRNA are likely to be present in hantavirus preparations at vanishingly low concentrations if at all, the fact that dramatic reductions in ssRNA integrity induced by UV irradiation did not reduce the intensity of ISG responses at 24 h suggests that the inductions of ISG were triggered by either a non-nucleic acid component or possibly an RNA that is associated with the hantavirus particle but was too small a target to be destroyed by moderate doses of UV light. It is also possible that UV irradiation alters the structural properties or aggregation of the virus, resulting in a particle that better promotes receptor cross-linking and thus enhances ISG production. Focus assays using this inacti-vated virus preparation confirm that the UV-killed virus is replication defective, and Western blotting demonstrates that there is no significant degradation of viral structural antigens (C. Ye and B. Hjelle, unpublished observations).
Many of the ISG regulated here are involved in previously characterized pathways of the host defense. Some RNA vi-ruses, in some contexts, can induce a similar antiviral state as
seen here through pathways, such as the pattern recognition receptor TLR3, via dsRNA intermediates produced as a prod-uct of the replication of a viral ssRNA genome or by cellular responses to viral proteins (1, 42). In those experiments, a strong antiviral response was produced in the absence of either dsRNA intermediates or de novo viral protein synthesis by the UV-killed virus. The fact that cellular gene expression in our experiments at this early time point was similar in the two treatment groups despite active viral replication in the HUVEC infected with live virus indicates that these changes in gene expression are likely induced by virus-cell interactions, such as binding of a receptor or internalization, and not by processes involved in replication, such as the production of dsRNA and viral proteins. The demonstration that increased induction of ISG continued to occur in viral preparations that had been subjected to increasing doses of UV light that were well beyond that needed to completely inactivate the virus strongly suggests, in concert with the results of others, that viral transcription is not necessary to transduce signals leading to induction of ISG (11). The induction of an ISG response through a replication-independent pathway has recently been shown for three negative-sense RNA viruses (11). Treatment of cells with these viruses resulted in the activation of many ISG and downstream effectors. The host recognition mecha-nisms by which inactivated viruses are recognized have yet to be elucidated, but they may involve recognition of viral glyco-proteins and/or signaling by host receptor and internalization machinery through cytoplasmic pathways. For example, repli-cation-defective human cytomegalovirus can elicit ISG expres-sion that is likely mediated by cellular interactions with the envelope glycoprotein gB (6).
While many of these regulated gene pathways are involved in known IFN-␣/-related pathways, transcription of IFNs themselves was not observed with either the live or killed virus in these experiments. It is known that most IFN-␣/expression is transcriptionally regulated (4, 27), and the absence of any IFN gene induction as suggested by the microarray experi-ments indicates that little or no new IFN is being produced by these cells. Transcription of IFN and ISG results in many antiviral cellular responses, including apoptosis, differential regulation of protein synthesis, up-regulated major histocom-patibility complex class I expression, elaboration of cytokines and chemokines, and degradation of RNA. It has been previ-ously demonstrated that ISG induction can proceed through an IFN-independent pathway, and this may account for the inductions seen here (2, 20, 44, 45). That such pathways exist is supported by experiments demonstrating that cytomegalovirus can elicit antiviral responses in IFN null cells (6).
[image:7.585.65.258.67.241.2]By 3 days p.i., many additional IFN-related genes were up-regulated by the live virus, including the chemokines IP-10, IP-9, and RANTES, but few genes continued to be up-regu-lated by the killed virus. These chemokine genes can also be induced via an IFN-independent pathway and are known to be induced by other hantaviruses (17, 41). It is likely that an antiviral dsRNA recognition pathway is induced by the live virus at this time point, because TLR3, MyD88, STAT1 and -2, and protein kinase R are induced, but there is still an absence of IFN transcripts. Other researchers have detected increases in expression of IFN-␣/in response to hantaviruses (17, 26).
FIG. 6. Preexposure of SNV to plasma obtained from a patient convalescing from HCPS that bears neutralizing antibodies abrogates the induction of ISG56 mRNA expression. A control plasma sample from a seronegative patient (⫺), a convalescent-phase plasma sample from an HCPS patient (⫹), or rabbit anti-rec-N antiserum were each heat inactivated for 30 min and then incubated with SNV at either a 1:20 or 1:40 dilution for 30 min at room temperature. We exposed HUVEC to antiserum-treated or untreated SNV for 1 hour, harvested RNA 6 hours after treatment, and then subjected total cellular RNA to quantitative RT-PCR using ISG56-specific primers. Changes were nor-malized by measuring ISG56 mRNA abundance relative to GAPDH mRNA and compared to that observed with untreated HUVEC (Con). PCR assays were performed in triplicate for each of the duplicate exper-iments. Results are reported as the mean⫾standard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
Differences in this observation could be due to the virus used, the MOI, or the time points selected for analysis.
While confirmatory quantitative RT-PCR experiments gen-erally showed very slightly increased expression of ISG with live SNV relative to killed SNV at 24 h (after which time SNV presumably has begun to replicate), at earlier time points (be-tween 4 and 12 h p.i.) the UV-killed virus up-regulates ISG56 and MxA to a greater extent than does the live virus. Addi-tionally, unlike the poly(I-C) control, virus exposed to UV for longer times resulted in an increase in ISG56 and MxA tran-scription out to approximately 2 minutes exposure, while the ability to detect RNAs of viral stocks immediately following UV exposure sharply decreased by 15 seconds of exposure. This could perhaps be the result of viral evasion or antagonism strategies utilized by the live virus. It has been shown that some other members of the familyBunyviridaeencode a nonstruc-tural protein, NSs, which inhibits cellular IFN responses (5, 39). The ISG response observed is unlikely due to a soluble mediator with the ability to induce ISG contained within the virus stock, because the activity that induces ISG copurifies with viral particles in density gradients (data not shown). Fur-thermore, a convalescent-phase human plasma that exhibited a high titer of neutralizing antibodies, unlike the plasma of an uninfected control individual or antibodies against SNV-N an-tigen alone, was able to block induction of ISG56 mRNA when preincubated with the virus.
The substantial induction of the mRNAs of the chemokines RANTES (70-fold), IP-10 (247-fold), and IP-9 (258-fold) by the virus at 3 days p.i. is in accordance with the hypothesis that the disease involves immune mediators, as these molecules have the ability to recruit and activate T cells. Additionally, the cytokine interleukin-15 and the interleukin-15 receptor, which is a known proinflammatory molecule that is a T-cell activator and is expressed in many disorders, including pulmonary in-flammatory diseases, was up-regulated fivefold (30).
Hantaviruses enter cells through receptor-mediated endocy-tosis via3 integrins (15, 16). Integrins mediate many cellular functions, including adhesion and migration, and are able to transduce intracellular signals. In endothelial cells, many of these signals are involved in vascular homeostasis and devel-opment (10, 22). It is possible that binding of integrins or accessory receptors by viral particles results in the activation of the observed antiviral responses. It is important to establish to what extent the activation of innate immune responses might contribute to pathogenesis by hantaviruses in vivo and how the native host species for hantaviruses escape this pathogenic process.
ACKNOWLEDGMENTS
This work was supported by U.S. Public Health Services grants U01 AI 56618 and U01 AI054779. J.P. was supported by National Institute of Allergy and Infectious Diseases grant 1 T32 AI07538.
The microarray experiments were performed using facilities in the Keck-UNM Genomics Resource and the UNM Cancer Research and Treatment Center. Images in this paper were generated in the UNM Cancer Center Fluorescence Microscopy Facility, which received sup-port from NCRR 1 S10 RR14668, NSF MCB9982161, NCRR P20 RR11830, NCI R24 CA88339, NCRR S10 RR19287, NCRR S10 RR016918, the University of New Mexico Health Sciences Center, and the University of New Mexico Cancer Center.
REFERENCES
1.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell.2001. Recog-nition of double-stranded RNA and activation of NF-B by Toll-like recep-tor 3. Nature413:732–738.
2.Bandyopadhyay, S. K., G. T. Leonard, Jr., T. Bandyopadhyay, G. R. Stark, and G. C. Sen.1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem.270:19624–19629. 3.Bharadwaj, M., C. R. Lyons, I. A. Wortman, and B. Hjelle.1999.
Intramus-cular inoculation of Sin Nombre hantavirus cDNAs induces cellular and humoral immune responses in BALB/c mice. Vaccine17:2836–2843. 4.Bhattacharya, S. S., M. Kulka, K. A. Lampel, T. A. Cebula, and B. B.
Goswami.2004. Use of reverse transcription and PCR to discriminate be-tween infectious and non-infectious hepatitis A virus. J. Virol. Methods 116:181–187.
5.Billecocq, A., M. Spiegel, P. Vialat, A. Kohl, F. Weber, M. Bouloy, and O. Haller.2004. NSs protein of Rift Valley fever virus blocks interferon pro-duction by inhibiting host gene transcription. J. Virol.78:9798–9806. 6.Boehme, K. W., J. Singh, S. T. Perry, and T. Compton. 2004. Human
cytomegalovirus elicits a coordinated cellular antiviral response via envelope glycoprotein B. J. Virol.78:1202–1211.
7.Botten, J., K. Mirowsky, D. Kusewitt, M. Bharadwaj, J. Yee, R. Ricci, R. M. Feddersen, and B. Hjelle.2000. Experimental infection model for Sin Nom-bre hantavirus in the deer mouse (Peromyscus maniculatus). Proc. Natl. Acad. Sci. USA97:10578–10583.
8.Botten, J., K. Mirowsky, D. Kusewitt, C. Ye, K. Gottlieb, J. Prescott, and B. Hjelle.2003. Persistent Sin Nombre virus infection in the deer mouse ( Pero-myscus maniculatus) model: sites of replication and strand-specific expres-sion. J. Virol.77:1540–1550.
9.Childs, J. E., T. G. Ksiazek, C. F. Spiropoulou, J. W. Krebs, S. Morzunov, G. O. Maupin, K. L. Gage, P. E. Rollin, J. Sarisky, and R. E. Enscore.1994. Serologic and genetic identification of Peromyscus maniculatus as the pri-mary rodent reservoir for a new hantavirus in the southwestern United States. J. Infect. Dis.169:1271–1280.
10.Clark, E. A., W. G. King, J. S. Brugge, M. Symons, and R. O. Hynes.1998. Integrin-mediated signals regulated by members of the rho family of GTPases. J. Cell Biol.142:573–586.
11.Collins, S. E., R. S. Noyce, and K. L. Mossman.2004. Innate cellular re-sponse to virus particle entry requires IRF3 but not virus replication. J. Virol. 78:1706–1717.
12.Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and R. Sousa.2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science303:1529–1531.
13.Elliott, L. H., T. G. Ksiazek, P. E. Rollin, C. F. Spiropoulou, S. Morzunov, M. Monroe, C. S. Goldsmith, C. D. Humphrey, S. R. Zaki, and J. W. Krebs. 1994. Isolation of the causative agent of hantavirus pulmonary syndrome. Am. J. Trop. Med. Hyg.51:102–108.
14.Ennis, F. A., J. Cruz, C. F. Spiropoulou, D. Waite, C. J. Peters, S. T. Nichol, H. Kariwa, and F. T. Koster.1997. Hantavirus pulmonary syndrome: CD8⫹ and CD4⫹cytotoxic T lymphocytes to epitopes on Sin Nombre virus nucleo-capsid protein isolated during acute illness. Virology238:380–390. 15.Gavrilovskaya, I. N., E. J. Brown, M. H. Ginsberg, and E. R. Mackow.1999.
Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by3 integrins. J. Virol.73:3951–3959.
16.Gavrilovskaya, I. N., M. Shepley, R. Shaw, M. H. Ginsberg, and E. R. Mackow.1998.3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. USA95:7074–7079. 17.Geimonen, E., S. Neff, T. Raymond, S. S. Kocer, I. N. Gavrilovskaya, and
E. R. Mackow.2002. Pathogenic and nonpathogenic hantaviruses differen-tially regulate endothelial cell responses. Proc. Natl. Acad. Sci. USA99: 13837–13842.
18.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott.2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regu-lation of interferon-stimulated genes. J. Virol.76:5532–5539.
19.Grandvaux, N., B. R. tenOever, M. J. Servant, and J. Hiscott.2002. The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis.15:259–267.
20.Guo, J., K. L. Peters, and G. C. Sen.2000. Induction of the human protein p56 by interferon, double-stranded RNA, or virus infection. Virology267: 209–219.
21.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer.2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science303:1526–1529. 22.Hynes, R. O., B. L. Bader, and K. Hodivala-Dilke.1999. Integrins in vascular
development. Braz. J. Med. Biol. Res.32:501–510.
23.Jonsson, C. B., and C. S. Schmaljohn.2001. Replication of hantaviruses. Curr. Top. Microbiol. Immunol.256:15–32.
24.Khaiboullina, S. F., A. A. Rizvanov, V. M. Deyde, and S. C. St. Jeor.2005. Andes virus stimulates interferon-inducible MxA protein expression in en-dothelial cells. J. Med. Virol.75:267–275.
on November 8, 2019 by guest
http://jvi.asm.org/
25.Kilpatrick, E. D., M. Terajima, F. T. Koster, M. D. Catalina, J. Cruz, and F. A. Ennis.2004. Role of specific CD8⫹T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immu-nol.172:3297–3304.
26.Kraus, A. A., M. J. Raftery, T. Giese, R. Ulrich, R. Zawatzky, S. Hippenstiel, N. Suttorp, D. H. Kruger, and G. Schonrich.2004. Differential antiviral response of endothelial cells after infection with pathogenic and nonpatho-genic hantaviruses. J. Virol.78:6143–6150.
27.Lenardo, M. J., C. M. Fan, T. Maniatis, and D. Baltimore. 1989. The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell57:287–294. 28.Lin, R., C. Heylbroeck, P. Genin, P. M. Pitha, and J. Hiscott.1999. Essential
role of interferon regulatory factor 3 in direct activation of RANTES che-mokine transcription. Mol. Cell. Biol.19:959–966.
29.Lund, J. M., L. Alexopoulou, A. Sato, M. Karow, N. C. Adams, N. W. Gale, A. Iwasaki, and R. A. Flavell.2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA101:5598–5603. 30.McInnes, I. B., and J. A. Gracie.2004. Interleukin-15: a new cytokine target
for the treatment of inflammatory diseases. Curr. Opin. Pharmacol.4:392– 397.
31.Mori, M., A. L. Rothman, I. Kurane, J. M. Montoya, K. B. Nolte, J. E. Norman, D. C. Waite, F. T. Koster, and F. A. Ennis.1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis.179:295–302.
32.Nam, J. H., K. A. Hwang, C. H. Yu, T. H. Kang, J. Y. Shin, W. Y. Choi, I. B. Kim, Y. R. Joo, H. W. Cho, and K. Y. Park.2003. Expression of interferon inducible genes following Hantaan virus infection as a mechanism of resis-tance in A549 cells. Virus Genes26:31–38.
33.Nerurkar, V. R., J. W. Song, K. J. Song, J. W. Nagle, B. Hjelle, S. Jenison, and R. Yanagihara.1994. Genetic evidence for a hantavirus enzootic in deer mice (Peromyscus maniculatus) captured a decade before the recognition of hantavirus pulmonary syndrome. Virology204:563–568.
34.Nolte, K. B., R. M. Feddersen, K. Foucar, S. R. Zaki, F. T. Koster, D. Madar, T. L. Merlin, P. J. McFeeley, E. T. Umland, and R. E. Zumwalt.1995. Hantavirus pulmonary syndrome in the United States: a pathological de-scription of a disease caused by a new agent. Hum. Pathol.26:110–120. 35.Pober, J. S., M. S. Kluger, and J. S. Schechner.2001. Human endothelial cell
presentation of antigen and the homing of memory/effector T cells to skin. Ann. N. Y. Acad. Sci.941:12–25.
36.Schmaljohn, C., and B. Hjelle.1997. Hantaviruses: a global disease problem. Emerg. Infect. Dis.3:95–104.
37.Schmaljohn, C. S.1996. Bunyaviridae: the viruses and their replication, p. 1447–1471.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Lippincott-Raven, Philadelphia, Pa.
38.Sen, G. C.2001. Viruses and interferons. Annu. Rev. Microbiol.55:255–281. 39.Soldan, S. S., M. L. Plassmeyer, M. K. Matukonis, and F. Gonzalez-Scarano. 2005. La Crosse virus nonstructural protein NSs counteracts the effects of short interfering RNA. J. Virol.79:234–244.
40.Sumpter, R., Jr., Y. M. Loo, E. Foy, K. Li, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr.2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol.79:2689–2699.
41.Sundstrom, J. B., L. K. McMullan, C. F. Spiropoulou, W. C. Hooper, A. A. Ansari, C. J. Peters, and P. E. Rollin.2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol.75:6070–6085. 42.Takeda, K., and S. Akira.2004. TLR signaling pathways. Semin. Immunol.
16:3–9.
43.Wagner, C. R., R. M. Vetto, and D. R. Burger.1984. The mechanism of antigen presentation by endothelial cells. Immunobiology168:453–469. 44.Wathelet, M. G., P. M. Berr, and G. A. Huez.1992. Regulation of gene
expression by cytokines and virus in human cells lacking the type I interferon locus. Eur. J. Biochem.206:901–910.
45.Weaver, B. K., K. P. Kumar, and N. C. Reich.1998. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell. Biol.18:1359–1368. 46.Young, J. C., G. R. Hansen, T. K. Graves, M. P. Deasy, J. G. Humphreys, C. L. Fritz, K. L. Gorham, A. S. Khan, T. G. Ksiazek, K. B. Metzger, and C. J. Peters.2000. The incubation period of hantavirus pulmonary syndrome. Am. J. Trop. Med. Hyg.62:714–717.
47.Zaki, S. R., P. W. Greer, L. M. Coffield, C. S. Goldsmith, K. B. Nolte, K. Foucar, R. M. Feddersen, R. E. Zumwalt, G. L. Miller, and A. S. Khan.1995. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol.146:552–579.
48.Zhu, H., J. P. Cong, G. Mamtora, T. Gingeras, and T. Shenk.1998. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. USA95:14470–14475.