Interaction between the Cellular Protein eEF1A and the 3′-Terminal Stem-Loop of West Nile Virus Genomic RNA Facilitates Viral Minus-Strand RNA Synthesis
16
0
0
Full text
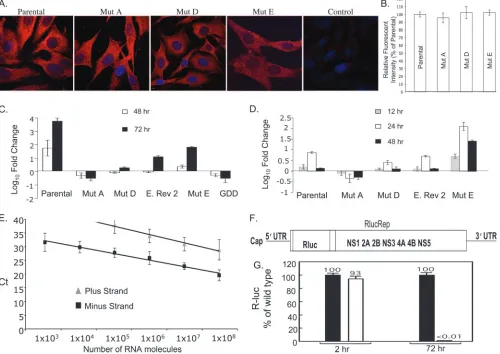
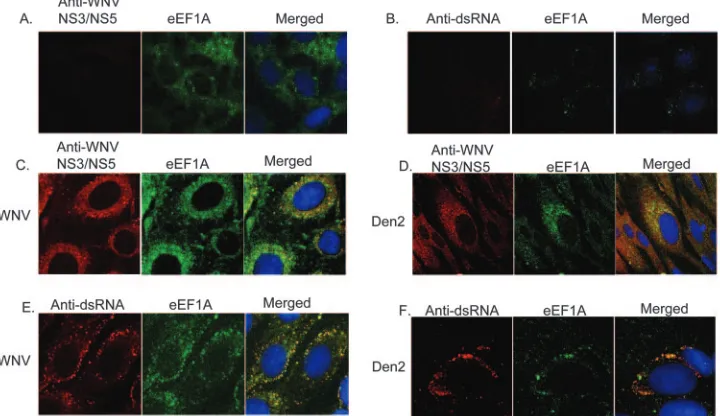
Figure
+3
Related documents