0022-538X/83/010206-09$02.00/0
Copyright © 1983,AmericanSociety for Microbiology
Effect
of
Intracellular
Vesicular Stomatitis
Virus mRNA
Concentration on the Inhibition of Host Cell Protein Synthesis
W.M. SCHNITZLEIN, M. KERRY O'BANION, MARY K. POIROT, AND M. E. REICHMANN*
Departmentof Microbiology, University of Illinois, Urbana, Illinois 61801
Received 13 August1982/Accepted 1 October 1982
Inhibition of host cellular proteinsynthesis by vesicular stomatitis virus (VSV) has been suggested to be primarily the result of competition for ribosomes between cellularand viral mRNAs (H. F.Lodishand M. Porter, J.Virol., 36:719-733, 1980; Lodish and Porter, J. Virol. 38:504-517, 1981). This hypothesis was investigated byregulating theextentof VSV mRNAsynthesis through theuseof defectiveinterferingparticles.Althoughintracellular VSV mRNA concentrations decreased by as much as a factor of 14 at high multiplicities of infection of defectiveinterfering particles, theinhibitionof host cellprotein synthesisby VSV decreased byamaximum of only 10%. The dataalsoindicated that under these conditions the protein-synthesizingcapacity of the cells was notexhausted. We concluded thatcompetition for cellular ribosomes couldnothavebeen the major factor in the inhibition of host cellproteinsynthesisby VSV. This conclusionwas further supported by inhibition data obtained with VSV mutants. The ts G22 mutant, defective in replication butnot inprimary transcription, inhibited host proteinsynthesis atthe nonpermissivetemperature (39°C)tothesame extent as didwild-type virus,eventhough it generated only 30to50% of theamountof viral mRNA as did wild-type virus. Conversely, in infections with the Rl mutant, which did not inhibit host cell protein synthesis, the amount of total and polysome-bound viral mRNA was indistinguishable from that obtained in infec-tionsbywild-type virus.
Infection of mammalian cells with vesicular stomatitisvirus(VSV) results in the inhibition of cellular macromolecularsynthesis(1, 12-14, 28, 29).Although it has become clear that transcrip-tion ofatleast some viral functions is required for this inhibition (12,29), neither thenatureof thesefunctions northeir cellulartargetsites are
known. Mostoftheinhibitionofcellularprotein synthesis byVSV takesplaceatthetranslational level (10). Stannersetal.(26) have isolated VSV mutantswhichareunabletoinhibit hostprotein synthesis; however, the natureofthe mutation was not established. Nuss et al. (16) have sug-gested that VSV mRNA might initiate protein synthesis more efficiently than does cellular mRNA and thus take overthe protein-synthesiz-ing components ofthe cell. Lodish and Porter (10) have determined the sizes of polysomes translating various cellularmRNAs in uninfect-ed and VSV-infected cells and concluded that theinhibition of cellularproteinsynthesisisdue primarily to competition for ribosomes by a
largeexcessofviral mRNAs. Since thenumber of ribosomes in polysomes ofviral and cellular mRNAsofcomparable sizeswasabout the same in infected cells, these authors concluded that
the
efficiency
of viralmRNAinitiation couldnotsignificantly differ from that of cellular host mRNAs. The competitive nature of viral and host mRNA translation was also reported in a laterpublication by Lodish and Porter (11). A correlation between the intracellular viral mRNAconcentration and theextentof cellular protein synthesis inhibition in infections with various VSVwild-type andmutant isolateswas demonstrated.
Intracellular VSV mRNA concentrations are basically determined by two events, primary transcription by the incoming virion inoculum and amplifiedtranscription during which newly synthesizedviralnucleocapsidsareutilized. Pri-mary transcription usually contributes up to
10%, depending onthe multiplicity of infection (MOI), of the fully amplified level of VSV mRNA(27). By inhibiting virion RNA replica-tion to various degrees, intracellular VSV mRNAconcentrations couldberegulatedin the rangeof10 to100% oftheamplifiedvalue. This should make itpossibleto measure directly the inhibition of host cell protein synthesis as a
function of VSV mRNA concentrations. Since chemical means ofinhibitingvirion RNA repli-cationmightaffect cellularprocesses,regulation
wasaccomplished bytheuseofdefective
inter-206
on November 10, 2019 by guest
http://jvi.asm.org/
VSV INHIBITION OF PROTEIN SYNTHESIS 207
fering (DI) particles. These particles inhibit viri-on replication without interfering with primary transcription (4, 17). Therefore, mixed infec-tions of VSV and various MOIs of DI particles shouldbe suitableforanexamination of thedose effect of VSV mRNA onhostprotein synthesis inhibition. The results presented here show that inhibition of BHK host cellproteinsynthesis by VSV wasessentially independent of DI particle concentration evenwhen the intracellular VSV mRNA concentration dropped to 7.4% of its fully amplified value. We also found that the ts G22 mutant of VSV inhibited BHK cellular protein synthesisto adegreecomparabletothat by the wild-type virion even when the mutant genomewas notreplicated. Finally,we showed that the intracellular and polysomal concentra-tions of viral mRNAs during infection with the Rl mutant(26)wereapproximately thesame as those during infection with thewild-type virion eventhoughthe mutant virus did notinhibit host cell protein synthesis during the first 4 h after infection.
MATERIALSANDMETHODS
Cellsandviruses.Babyhamsterkidneycells
(BHK-21, clone13)weregrown inmonolayersaspreviously
described(7, 21).
The following isolates of the Indiana serotype of
VSV were used in this study: MS (21); ts G22,
obtained from C. R.Pringle (18);andRl,provided by
C. P. Stanners (26). Viralinocula, freed of DI
parti-cles, werepreparedbyafivefoldclonal selection and
enrichment in BHK cellmonolayersat 39°C (MSand
R1) or31°C (tsG22) by the method ofStampferetal.
(25). After plaque purification, the ts G22 isolate
exhibited a temperature-sensitive dependency of
greater thantwoordersofmagnitude. The Rl isolate
hasbeenpreviously reportedtoexhibitanattenuated
cytopathic effect (26).However,after clonalselection,
we observed the samedegree ofcytopathiceffect with
either Rloritswild-typeparent in BHK cells. Inspite
of thisdiscrepancy, theclonally enriched Rl didnot
inhibit cellularproteinsynthesisaspreviously
report-ed(26). The MS isolatewasalso usedforasourceof
DIparticles,VSIMS DI 0.23(5',5%)(seereference 19 fornomenclature), whichwereobtainedbyundiluted
passageof the viral inoculum.
Preparationandpurificationofvirions, DIparticles,
andparticle-boundRNAs.Theisolation of virions and
DIparticles has been reportedpreviously(2).
Concen-trations of DI particles were determined by
absor-bance measurements at 260 nm
(A260)
with a Cary 14spectrophotometer (Applied Physics Corp.,
Monro-via, Calif.). EffectiveDIparticleMOIswere
calculat-edasfollows. Virionplaque assays indicated that one
A260unitcorrespondedto 3x 109PFU.Assumingthat
the same percentage of virion and DI particles is
biologically active and correcting for the one-third
difference in their sizes (20),wetook oneA260unit to
beequivalenttoapproximately1010active particles.
To radioactively label virion proteins, MS
virus-infected cells wereoverlaid withserum-and
methio-nine-free mediumcontaining4pCiof[35S]methionine
(1,000Ci/mmol; NewEngland Nuclear Corp., Boston,
Mass.) perml.After 18 to 24 h ofincubationat39°C,
virions were isolated as described above and then
pelletedat35,000 rpm and 4°C for 90 min inaBeckman
R40 rotor. Viral pellets were lysed and prepared for
electrophoresis as described below for cellularpellets.
MS virion RNA,radioactivelylabeledwith tritium in
all fourbases, was prepared as previously described
(22).
Interferenceassays.Interferenceby DI particles was
quantitated as previously described (2) except that
progenyvirus was measured at 4h instead of 18 h after
infection.
Assay for intraceilular protein synthesis. BHK
con-fluent monolayers in 60-mm (5 x 106cells) or 100-mm
(1 x 107cells)tissue culture plates were either virus
infected at an MOI of 5 or mockinfected at39°Cwith
occasionalagitation. Forexperiments using DI
parti-cles, MS virions andappropriate dilutions of DI
parti-cles were allowedtoadsorbsimultaneously. After 30
min,the cells were overlaid with prewarmedmedium
andmaintained at 39°C.
At various times after infection, the medium was
replaced with methionine- and serum-free medium
containing 0.5 to 1.0 ,uCi of[355]methionine. After 30
min (viralinfections) or 45 min (DI particle infections),
the cells were washed twice with cold
phosphate-buffered saline and then overlaid with 5 ml of 10o
trichloroacetic acid (TCA). After 5 min, the TCA was
replaced with5ml offresh 10%o TCA foranadditional
5 min. Intracellular pools of radioactive precursors
were measured bydissolving 0.9 ml of the first TCA
rinse in 6 ml ofAquasol (New England Nuclear) and
counting ina Beckman LS-250 scintillation counter.
The cells were thendehydrated with two 5-min washes
ofethanol-ether (3:1), air dried, solubilized in 1 ml of
freshly made 0.2% NaOH, added to 6 ml of Aquasol, and counted.
Preparation and gel electrophoresis of intracellular
proteins. Infected and mock-infected cells in 60-mm
plates were pulse-labeled for 30 min at 3 h
postinfec-tion with 4,uCi of[35S]methionine per ml. The cells
wererinsed twice with coldphosphate-buffered saline
and then overlaid with 2 mlof cell resuspension buffer
(25 mMTris-hydrochloride [pH 7.9],1mM MgCl2,0.4
mM CaC12, 0.5 mMdithiothreitol) (24). After being
scraped from the plates andpelletedat2,500 rpmfor
10min in aSorvall GLC-1 centrifuge, thecells were
dissolved in 90 ,ul oflysisbuffer (10 mM HEPES
[N-2-hydroxyethylpiperazine-N'-2-ethanesulfonicacid] [pH
7.5],10 mMmagnesiumacetate,0.2%sodiumdodecyl sulfate, 0.25 ,ugofphenylmethylsulfonylfluoride per
ml)(10)towhich 90 ,ul of the samebuffer containing 20
,ugof DNase I (Worthington Diagnostics, Freehold,
N.J.) per ml was added. After1 hat37°C, 90 p.1 ofa
protein-denaturing solution (30 mM Na2PO4, 7.5%
sodiumdodecylsulfate, 15%[vol/vol]
2-mercaptoeth-anol,30%o [vollvoly glycerol) (10)wasadded, and the
samples were boiled for 3 min immediately before
being loaded on gels. Separation of proteins was
accomplished in10%opolyacrylamide slab gels by the
method of Laemmli(6). Gels wereeither(i) fixed for
15 min in10%oaceticacid-5% glycerol, dried inagel
drier (Bio-Rad Laboratories, Richmond, Calif.), and
autoradiographed with Kodak XAR-5 X-ray film
(Eastman KodakCo., Rochester, N.Y.) for48to96 h
at -70°Cor(ii) divided into individual lanes andcut
VOL. 45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
into 1-mm sections withaMickle Gel Slicer (Brink-mannInstruments Inc.,Westbury, N.Y.). The slices
were dissolved overnight in 5 ml of 3% Protosol in
Econofluor (New EnglandNuclear)at37°Cand
count-ed.
Isolationof intracellular and polysome-bound RNAs.
Totalintracellular RNAwasisolatedatvarious times
afterviral infection. Cell monolayersin 100-mmplates
werewashed three times withphosphate-buffered
sa-lineand then lysedwith 5ml of E buffer(40mM
Tris-acetate[pH7.2],20 mM sodiumacetate,1mMEDTA)
containing1% sodiumdodecylsulfate. Thelysedcells
wereextracted twice withanequal volumeof
phenol-CHC13 (1:1)andoncewithanequalvolume ofCHCl3.
The RNA species were thenprecipitated with 3
vol-umesofice-cold ethanolin thepresence of 300 mM
sodiumacetate at-20°C.Afteratleast 8h,the RNA
wascollected bycentrifugationat9,000rpmat4°Cfor
20 min in a Sorvall RC2-B centrifuge, dried under
nitrogen, anddissolved inwater. Theoptical density ofthe RNAsampleswasdeterminedat260nm.The
RNAswerethenreprecipitatedwith ethanol dissolved
in the appropriate volume of water and stored at
-70°Cuntil used.
Polysomes were isolated according to the
proce-duresofLodishand Froshauer(9),with thefollowing modifications: emetine treatment (to inhibit
elonga-tion)wasomitted, cell monolayerswereused instead
ofcells insuspension, andpolysomes werenot
frac-tionatedonsucrose gradients.
At 3.5 h postinfection, the cell monolayers were
washed,scraped off the plates, and poured into chilled
Douncehomogenizers.Thecellswereallowedtoswell for 10 min and then were homogenized 40 times as
describedpreviously (9). Thedetergent-treated
post-nuclearfractionwaslayeredon a0.5-to1.0-mlpad of
40mMHEPES (pH7.5)-80mMNaCl-2.5mM
magne-siumacetatecontaining15%sucroseinSW50.1
nitro-cellulosetubes. Polysomeswerepelleted by
centrifu-gationat45,000rpmin aBeckman SW50.1 rotorfor
1.5h,thetimerequiredtosediment80Sribosomesat
5°C. Aftercentrifugation, thepelletsweresuspended
in 1 ml ofwater(forTCA precipitations) or3 ml of extractionbuffer(10mMTris-hydrochloride [pH 7.5], 100 mMNaCl,10mMEDTA)containing 1%sodium
dodecyl sulfate(forextraction ofRNA).
Quantitation of viral intracellular and
polysome-bound mRNAs. Viral intracellular mRNAswere quan-titated by annealing to a 3H-labeled virion RNA as
describedpreviously (22). When lessthan40% of the virion RNAwasrendered RNase resistant, therewas a
direct relationship between the amount ofannealed virion RNA and theamountofcomplementarymRNA
species.The results(expressedascountsperminute)
were then extrapolated to the total amount of viral
mRNA per infected cell sample and normalized by
correcting for variations in the total cellular RNA
content as determined by A260 readings.
Polysome-bound viral mRNA species werealsocompared ina
similar manner, except that the A260 of RNAs was
usedasthecorrection factor.
Polysome-boundviralmRNAswerealso
quantitat-edby labelingRNA in thepresenceofactinomycinD
asdescribed below. Cellmonolayersweretreatedfor 30minbeforeinfectionwith mediumcontaining7.5,ug
ofactinomycin D (Sigma Chemical Co., St. Louis,
Mo.) per ml. The cells were then mock infected or
infected with MSorRlvirions at anMOI of 5 for 30
min at39°C. Mediumcontaining7.5 ,ugofactinomycin
Dperml was thenadded,andincubationwas
contin-ued at39°C for1h.After thistime,mediumcontaining
7.5 Fg of actinomycin D and 5 ,uCi each of
[5,6-3H]uridine(38 Cilmmol)(New EnglandNuclear) and
[2,8-3H]adenosine (37 Ci/mmol) (ICN Chemical and
RadioisotopeDivision,Irvine, Calif.)permlwas
add-ed. Incubationwascontinued foranadditional 2.5h,
atwhich time polysomes were isolated as described
above.Thepolysomeswerethensuspendedin1mlof
water,precipitated with1 mlof10%oTCA and 100 ,ug
of yeast RNA, collectedon2.8-cmglassfiberfilters,
washed,dried, andcounted.
RESULTS
[image:3.489.255.448.442.540.2]Effect of DI particles on the inhibition of host proteinsynthesis by VSV. WhenBHK cellswere infected withahigh MOIofDIparticles, cellular proteinsynthesiswaspartially inhibited evenin the absence of VSV (Table1, DIparticle MOIs of 28 and 56). This inhibition was not due to virion contamination since cells infected with thenumberofPFUcorrespondingtothelevelof contamination in these inocula did not differ from mock-infected controls in their protein-synthesizing activity. It is more likely that the highMOI ofDIparticlescausedextensive dam-age to the cell membrane, primarily through interactions with the G protein ofthe particle spikes (15, 20). The interfering ability of these DIparticle concentrationswith VSVyields from
TABLE 1. Effect of DI particles of various
interfering ability on BHK host cell protein synthesis
DI Methionine Protein Logreduction particle incorporation synthesis(% ofinfectivity
Mola (cpm) ofcontrol) (PFU/ml)b
0 164,000 100
1.8 160,000 98 1.76
3.5 157,000 96 2.16
7 165,000 101 2.76
14 151,000 92 NDC
28 135,000 82 3.39
56 110,000 67 3.48
a
MOIs
were determined by A260 measurements.Virion plaque assays indicated that one A260 unit
correspondedto3x 109 PFU.Assumingthatthesame
fraction of virions and DI particles was biologically
active and correcting for the difference in size, the
MOI isbased on the valueof1010 particles perA260
unit.
bResults are expressed as log of the difference
between the number ofinfectious particles produced
byBHKcellscoinfected with virions (5PFUpercell)
and DI particles and the number produced by cells
infected with virions only. In the absence of DI
particles, the yield of virionswas1.3x 107PFU/mlat
4hpostinfection.
cND,Notdetermined.
on November 10, 2019 by guest
http://jvi.asm.org/
SYNTHESIS
doubly infected cells (5 PFU of VSVpercell) is also shown in Table 1. The decrease in viral yields ranged from 1.8 to 3.5 logs, which was approximately equivalent to a 98.3 to 99.97% inhibition.
InTable 2, the relationship amongVSV
inhi-bition of host cell protein synthesis, DI particle concentration, and intracellular virus mRNA concentration is shown. The latter was
deter-minedby annealing samples of RNA from infect-edcells witharadioactively labeled virionRNA
probe and then measuring RNase A- and Tl-resistantcounts.Theannealingswereperformed with concentration ranges of cellular RNA
whichgavealinear annealingresponse, i.e., up
to 40% of the saturation level of the probe. These datawereextrapolatedtogive the relative mRNA concentrations, whichare expressed as
countsperminute of VSV RNAprobe annealed by the RNAcontentoftwo60-mmplates of cells after normalization by A260 values (Table 2). These dataarealsoexpressedasapercentageof the VSV mRNA synthesized in the absence of DIparticles. Atveryhigh MOIs ofDIparticles, the mRNA levels droppedto7% ofthe amount
synthesized by virus only. This rate ofmRNA
synthesis is consistent with that of virion pri-marytranscription (27), which isnotinhibitedby DI particles (4, 17). The higher value of 11%
obtained in the presence ofcycloheximide (Ta-ble 2) might be due to incomplete inhibition of protein synthesis (8). Indeed, a slight incorpo-ration of methionine was observed under these conditions (Table 2). In spite of the 14-fold
change in VSV mRNA concentration,the
incor-poration of [35S]methionine into proteins by the cells remained thesame overthe entirerangeof
DI particle concentrations and did not differ appreciably from the incorporation by cells in-fected with virion alone (Table 2).
The 35S incorporation experiments did not
necessarily prove that host protein synthesis wasunaffected by the VSV mRNA levels. Since
the DI particles inhibited VSV replication, viral protein synthesiswasprobably diminished in the
doubly infected cells and could have been
conm-pensated for by increased cellular protein
syn-thesis. This isexactly what would be expected if the ribosomes were in limiting supply. It was,
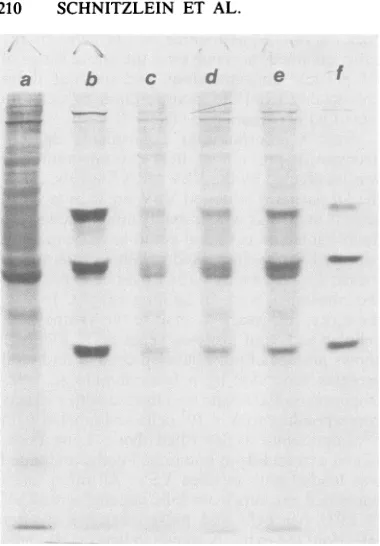
therefore, necessaryto analyze the synthesis of cellular and viral proteins separately. Figure 1
showsprofiles of pulse-labeled cellular and viral proteins separated by polyacrylamide gel elec-trophoresis. Each lanewasloaded withextracts
correspondingto5 x 105cells and labeled with [35S]methionineasdescribed above. Lanea con-tainedextractsfrom uninfected cells, and lane f
was loaded with purified VSV. All other lanes
containedextractsfrom cells infectedwithVSV (5 PFU per cell) and pulse-labeled 3 h after infection; theextractsshown in lanesc,d, ande were isolated from cells also infected with 59, 15, and 3.7 DI particles percell, respectively.
An examination of lanes a and b of Fig. 1 demonstrated the strong inhibition of cellular protein synthesis by VSV in this experiment. Bands corresponding to cellular proteins are barely discernible in lane b. The four strong
[image:4.489.40.438.469.599.2]bands in lane b which corresponded to viral proteins L, G, N, and M (N and NS were not
TABLE 2. Incorporation of methionine in BHK cells infected with VSVa in the presence and absence of DI
particles'
Methionine Protein Viral mRNA
DIparticleMOIC incorporation synthesis(% VSV mRNA synthesis(t
(cpm) ofcontrol) ofcontrol)
0 114,000 69 1.19 x 105 100.0
1.8 102,000 62 3.21 x 104 27.0
3.5 93,000 56 1.68 x 104 14.0
7 102,000 62 1.30 x 104 11.0
14 NDe ND 1.06 x 104 8.9
28 104,000 64 8.86 x 10' 7.4
56 101,000 61 8.33 x 103 7.0
0(NoVSV) 164,000 100 0.00 0.0
0(+ cyclo- 2,900 2 1.30 x 104 11.0
heximidef)
a
VSV
concentration,5 PFUpercell.bAllmeasurements were made4 hafter infection.
c DIparticle concentrations weredeterminedasdescribedforTable 1.
dThe relative concentrationof viralmRNAisexpressedasthe total3Hcountsof virion RNA which could be
rendered RNaseresistant after annealing. This value has been normalized for107cells by measurements of A260
of cellular RNA. The results were corrected fornonspecific annealing by subtractingthe3H countsof virion
RNA made RNase resistant afterannealingwithuninfectedintracellular RNA.
'ND, Notdetermined.
fCycloheximideconcentration,50 ,ug/ml.
VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
a.
b..
3u-.
FIG. 1. Gel anal)
BHKcells, either ml
or doubly infected
described in the te
[35S]methionine for:
tion. Cellextracts fi
were electrophoresei
gel (6). Lanes: a,me
cells infected with
particle MOIs of0,
markerVSVprotein
separated) indicat synthesis had tak particlestothe V' effectonthe inten
spondedtocellular proteins. EvenatanMOI of
d e f was15 (lane d), when the synthesis of viral proteinsseverelyinhibited and the intracellular VSV mRNA concentration was suppressed to
ap-proximately 9% of its normal value (Table 2), the intensity of the bands correspondingto cellular proteinsincreased only marginally.
To quantitate the effect of DIparticlesonthe
inhibitionofcellularprotein synthesis by VSV, thegels of each lane weresliced, and the radio-activecontentwasdetermined. Duetothe
pres-enceof viralprotein bands in lanes b throughe
of Fig. 1, the radioactivity corresponding to
cellular and viral proteins was corrected as
follows. Since 30% of the cellular proteins in lane a of Fig. 1 (uninfected cells) were in the
corresponding positions of viral proteins, the radioactivity in lanes b through e of Fig. 1,
representing cellular protein bands only, was
divided by 0.70. These data, whichrepresent a
relativemeasureoftruecellular protein synthe-sis, are shown inTable 3, as aretheamountsof VSV proteins synthesized relativetototal cellu-larproteins under the various conditions of this experiment. It should be noted that the values for theVSVproteins have been corrected for the
ysis of proteins synthesized in presenceofcellularproteins by subtracting 30%
with VSV and DI paricles As of the respective cellular protein radioactivities
xt, the cells were labeled with (Table 3). At DIparticleMOIsof 15and 59, the 30 min beginning 3hafter infec- data illustrate that even though intracellular
rom equivalent numbers of cells VSV mRNA was suppressed to the level of d througha10%polyacrylamide primary transcription (Table 2), cellularprotein
)ck-infected cells;b,c,d, ande, synthesis was enhanced by only 10% over that
5 PFU of VSV per cell and DI takingplacein the absence ofDIparticlesandin
59, 15, and 3.7, respectively; f, the presence of a 10-fold-greater VSV mRNA
Isfrompurifiedvirions. content.This seemstobe themaximum level of
competition thatmay occurbetweencellularand ed thatextensive viralprotein viralmRNAin thisconcentrationrange. ;en place. The addition of DI Inhibition ofproteinsynthesis by VSVmutants.
SV inoculum hadaverysmall Group II temperature-sensitive mutants of the
[image:5.489.58.248.56.329.2]isity of the bands whichcorre- Indiana serotype of VSV are defective at the
TABLE 3. Inhibition of BHK hostcellprotein synthesis byVSVa in the presence and absence ofDI
particles
Methionineincorporation VSVprotein
DIparticle intoncelllaorotin Cellular proteinsynthe- Methionine incorporation synthesis(% MOlb into cellular(cpm)c sis(%ofcontrol) intoVSVproteins(cpm)d of total pro-teinsynthesis)
0 42,500 14 101,000 70
3.5 59,000 19 38,000 39
15 70,000 23 24,000 25
59 74,000 24 26,000 26
0 (NoVSV) 310,000 100
a
VSV
concentration, 5 PFU/cell.b See Table1for MOI determinations.
c Cellswerelabeledatbetween 3 and 3.5hpostinfection.Thecounts wereobtained from slicedgelssuchas
that shown inFig.1.Thirty percent of the control counts (noVSV)werein areascorrespondingtoviralprotein
bands. The numberswereobtained afterappropriatecorrections.
d These numbers were obtained after subtracting 30% ofthe respective cellular protein counts from the
determined values.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.489.57.448.524.615.2]nonpermissive temperatures in virion RNA rep-lication butnotin primary transcription (27). If high concentrations of viral mRNAs are re-quired for inhibition of host protein synthesis, then mutants belonging to this group should not influence host protein synthesis at 39°C but should behave like the wild typeat31°C. Protein synthesisincells infected with mutant ts G22 at 39°C was compared with that in cells infected with wild-type virus bymeasuring the incorpo-ration of [35S]methionine during 30-min pulses. The data, expressed as percent methioine incor-poration as compared with that in uninfected cells, are shown inFig. 2. It is clear that the ts
G22 mutant inhibited protein synthesis as effi-ciently as did thewild-type virus. However, the viral mRNA content of wild-type- and mutant-infected cells was not the same. At 3 and 4 h after infection, the VSV mRNA contentof the mutant-infectedcells was two- to threefold low-erthan that of thewild-type-infectedcells(Table 4). As in the case of cells doubly infected with DIparticles and withwild-type VSV,the lower level of intracellular viral mRNA had no effect ontheability ofthe virustoinhibit hostprotein synthesis.
The effect of the Rl mutant on host protein synthesiswasalso examined. Inagreement with previous reports (26), we found that in cells infected with this virus, protein synthesis was not inhibitedduring the first 3 h after infection.
,00K
90 - \
!;iso ~ OUS Oi NFCTO
°
0\
tL
5o0
uU 0
-2 3 4
HOURSPOS1 INFECT ION
FIG. 2. Inhibition of protein synthesis in BHK
cellsafter infection with MSwild-type(0)andtsG22
mutant (O) VSV at 39°C and 5 PFUper cell. After
infection the cellsweremaintainedat39°C and
pulse-labeled with
[3"S]methionine
(1 ,uCi/ml)for 30 minathourly intervals.TCA-precipitablecounts were
deter-mined as described in the text. The counts were
compared with those obtained from similarlylabeled
mock-infected cells andareexpressedaspercentages
ofincorporation by the uninfected cells. All
determi-nations representanaverage oftwocellplates.
TABLE 4. Relative amounts of VSV mRNAs in
cellsinfectedwith wild-type (MS) and mutantviruses
Time Relativeconcnof viral mRNA(cpm)a
after
infection MS tsG22 MS
Rl
(h)
1 2.87 x 103 7.82 x 103 1.59X 104 1.35 x 104
2 2.23 x 104 2.36x 1047.90 x 1047.99 x 104
3 4.64x 104 1.91 X 1041.32 x 105 1.42 x 105
4 7.74x 104 2.42 x 104 1.67 x 105 2.07 x 1O0
aThe relative concentrations of viral mRNA are
expressed as RNase-resistant counts of 3H-labeled
virion RNAafter
annealing
asdescribed for Table 2.One hourlater, protein synthesisincreased con-siderably above the value of the noninfected cells (Fig. 3). This increase could be due to a
significant contribution of viral proteins to the total cellular protein synthesis. On the other hand, the wild-type virus inhibitedtotal protein synthesis at 4 hpostinfection by approximately 45%. Becauseof aprobablecontributionbyviral proteinstototal protein synthesis,this value of
E
a
.C)
cn in
z
0
co
-I
2 3 4
[image:6.489.250.443.93.192.2]HOURS POST INFECTION
FIG. 3. Incorporation of[35S]methionine by BHK
cellsmock infected(0)orinfected with MS wild-type
(U) orRl mutant(0) VSV. Cells wereinfectedat 5
PFU percelland incubated at39°C. Duplicate plates
werepulse-labeledfor 30 min at hourly intervals with
[35S]methionine (1 ,uCi/ml), and TCA-precipitable counts weredetermined asdescribed in thetext. The
average values of[35S]methionine incorporation are
plotted on the left ordinate as a function of time
postinfection. The right ordinate represents percent
inhibition ofprotein synthesis in cells infected with
MS wild-type VSV as compared with that in mock-infected cells.
VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.489.247.443.335.559.2] [image:6.489.43.234.409.572.2]inhibitionis,if anything,toolow. Ifacorrection based on the Rl data was to be made, the inhibition by the wild type would amount to about65%.
Todetermine whether the inability of theRl mutant toinhibit host protein synthesiswasdue to lower intracellular concentrations of viral mRNA than those in the wild-type infection, annealing experiments with a radioactive VSV virionRNAprobewereperformed. The relative concentrations of wild-type and Rl mRNAs isolated from cellsruninparallel with the inhibi-tionmeasurements(Fig. 3) areshownin Table4. Nosignificant differences between viral mRNA concentrations inwild-type andRl mutant infec-tionswereobserved up to 4hafter infection.
The determinations of intracellular VSV mRNAby thehybridizationtechniques (Table4) do not necessarily represent biologically active mRNAs capable of binding to ribosomes. To determine whether the Rl mutant produced mostlyinactive mRNAswhich didnotcompete with cellularmRNAsfor ribosomes,we investi-gatedthepolysome-bound fraction of wild-type and Rl mRNA. These experiments were per-formed in two ways. In the first experiment, actinomycin D-treated cells infected with either wild-typeor Rlviruswerelabeledat 1.5 to3.5h postinfection with [3H]uridine and [3H]adeno-sine, and polysomes wereisolated asdescribed above. The incorporation of radioactive counts from an equivalent number of cells (based on A260 measurements) from wild-type- and Ri virus-infected cellswere23,900 and25,300cpm, respectively. In the secondexperiment, the acti-nomycin D and the radioactive nucleosidepulse wereomittedtoeliminateanypossible effecton polysomecomposition dueto immediate inacti-vation of cellular transcription. The isolated polysomes were phenol-CHCl3 extracted, and the VSV mRNA content was determined by annealing witharadioactive virionRNAprobe. The numberof RNase-resistantcounts (normal-izedto107cells)obtained from wild-type infec-tions was 7.49 x 104 cpm, and that from Rl infections was 8.94 x 104 cpm. Under these conditions,theinhibition of cellular protein syn-thesis asmeasured by
[35S]methionine
incorpo-ration was 53% for wild-type and 6% for Rl virus infections. These data indicated that a comparable quantity of viralmRNAs was pres-entin thepolysome fraction of wild-type-andof Rl virus-infected cells.DISCUSSION
The intracellular regulation of VSV mRNA levels by means ofDIparticles as described in this workeliminated the necessityto use chemi-cal meansofregulation which might have intro-duced unwantedside effectsoncellular
metabo-lism. This procedure also made it possible to follow the correlation between VSV mRNA contentandinhibition of cellularprotein synthe-sis in infections by the same wild-type VSV isolate, rather thantocorrelate theseparameters in different wild-type isolates (11). It was not clear whether theincrease in intracellular VSV mRNAwithdecreasingDIparticle-to-cellratios in the inocula (Table 2) was due to a larger number of cellsnotinfected with DIparticlesat all or to a general increase in VSV mRNA synthesis in all cells. The kinetic interpretation ofDIparticleinterferenceby Bellett and Cooper (3) and morerecently by Sekellick and Marcus (23) would suggest that total inhibition ofviral amplification was accomplished by a single DI particle per cell. Therefore, increasing the DI particle MOI should have hadno furthereffect on the VSV RNA concentration in the cells already doubly infected, and the observed de-creaseinVSV mRNA would beentirelydueto infection of cells whichwerefree of DIparticles at the lower MOIs. Unfortunately, the experi-mental datawerenot accurateenough to distin-guish between the two possibilities. Thus, at a DIparticleMOI of 1.8, the VSV mRNA concen-trationwas 27%of itsamplified value (Table2). The zero term of the Poisson distribution pre-dicts that16.4% ofthe cells would befreeofDI particles while still infected with VSV and that 83.5% would be infected with both particles. The predicted VSV mRNA content based on thisstatistic is (0.164 + 0.835 x 0.07) x 100 =
22.3%. Considering theerror in the determina-tions of MOI and intracellular mRNA content, this value isnot verydifferent from the experi-mentally determined 27%.
In any case,when allcellswereinfectedeither withDIparticles andvirionorwith virion alone, the 7or100%level of VSV mRNA, respective-ly, was a true reflection of the intracellular contents. Under these conditions, even though the VSV mRNAcontent increased 14-fold, the incorporation of
[35S]methionine
into TCA-pre-cipitable countsremained the same. To ensure that the DI particles orvirus did not influence[35S]methionine
uptake by the cells, TCA-solu-ble counts were also measured in washed cells andwerefoundtobeidentical innoninfected,inVSV-infected,
and in VSV- and DIparticle-infectedcells (data not shown).
The incorporation data did not exclude the possibility that in doubly infected cells host proteinsynthesis actually increasedoverthat in cellsinfected with virus alone but this increase wasexactlycompensated for bythe decrease in synthesis of viral proteins due to interference. The polyacrylamide gel electrophoresis profiles (Fig. 1)visually demonstrated that thiswas not the case. Moreover, from the sliced gels, a
on November 10, 2019 by guest
http://jvi.asm.org/
quantitative determination of the contribution of cellular and viral protein synthesis to total pro-tein synthesis was also made (Table 3). These dataindicated that inhibition of cellular protein synthesis was only slightly dependent on intra-cellular levels of viral mRNA, probably not exceedingabout10% of the total inhibiting activ-ity in the range of mRNA concentrations mea-sured.This 10% may therefore reflect the extent ofcompetition for ribosomesby very high levels of VSV mRNA. Although these data do not prove conclusively that the competition for all available ribosomes had not already taken place when VSV mRNA levels reached 7% of their amplified value, an examination of the poly-acrylamide gel profiles in Fig. 1 is informative in relation to thisquestion. In lanes c, d, and e, it canbeseenthatasthe DIparticleconcentration was decreasing (and VSV mRNA increasing), viralprotein synthesis strongly increased. This clearly indicated that the protein-synthesizing ability ofthe cells represented in lane d (VSV mRNA, 9%) was not exhausted and that all available ribosomes could not have been fully utilized in translation. Thisconclusioncan also bereached byexaminingthedata in Table 3. In no case do theradioactive counts obtained from infectedcells addup to thelevels of radioactiv-ity in theproteinsof noninfected cells.
That concentrations of intracellular VSV mRNAof between 7 and 100% oftheamplified level had onlymarginaleffectson theinhibition ofprotein synthesis was further confirmed by the data obtained with VSVmutants tsG22 and Rl.The Rl mutant,whichin our handsdidnot inhibitincorporationof[35S]methionineup to4h after infection (see also reference26),generated the same amountof mRNA as did the wild type. However, as pointedoutby Lodish and Porter (10, 11),comparisons ofthese kinds should not be basedonhybridizationassays, sincealarge and variable fraction of the VSV mRNAsmight betranslationally inactive and would not partici-pate in any competition for ribosome binding. To examine whether the wild type and the Rl mutantgenerated comparable amounts of active mRNAs, polysomes were isolated from both types of infection under equivalent conditions. Nodifference in the VSV mRNA content of the polysomal fraction in the two infections was observed. This result was also consistent with the observation that the yields of Rl and wild-type VSV virions were comparable (data not shown), whereas alower viral mRNA concen-tration inRl infections might lead to relatively loweryields of this virus.
The polysomal fraction of viral mRNA was not measuredinthe experiments with DI parti-cle- andVSV-infected cells. The criticism of the hybridization assay in those experiments would
only be valid if the DI particles preferentially inhibited the generation of inactive mRNAs while the total content of active RNA remained the same at the 7 and 100% levels of hybridizable mRNA. Such an interpretation is not plausible in view of the severe interference with viral protein synthesis (Fig. 1 and Table 3). A possible effect of DI particles on mRNA sequestration is also eliminated by the data in Table 1. At least up to an MOI of 14, DI particle infections in the absence of VSV had very little effect on host cell protein synthesis.
Recentinvestigations of the inhibition of cel-lular protein synthesis by VSV have led to the conclusionthat VSV mRNAs havenoadvantage in the initiation or elongation process of poly-peptide synthesis. These conclusions were based on the average polysome size of active cellular and viral mRNAs of comparable lengths (10). However, in the infected cell, chain initia-tionrates arereduced equallyfor both types of mRNAs, asreflected in generally reduced sizes of polysomes.This has beeninterpretedasbeing consistent with thehypothesisof alimiting sup-ply of ribosomes in the infected cells when a large excess of viral mRNAs is generated. The lack ofcorrelation between high levels of viral mRNAs andthe extent ofinhibition ofcellular protein synthesis demonstrated in this paper conflicts with that interpretation. Other differ-ences between infected and noninfected cells must serve as abasis for the inhibition. It should benoted that recentinvestigationsof the inhibi-tion ofcellularprotein synthesis by encephalo-myocarditis virus in Lcellshave alsoeliminated competition, inactivation of cellular mRNAs, andinactivationof caprecognitionfunctionsas possible mechanisms of the inhibition. Instead, it was proposed that the reduced activity in infected cells is due to reversible changes (5). The in vitro assays would not register such reversible changes, especially if they were caused by an imbalance of the low-molecular-weightcomponents for which the in vitro trans-lation system has been optimized. Similarly, sequestration of the cellular mRNA could also bereversiblesothat when total RNAis isolated from infected and noninfected cells, an in vitro translational assay using mRNAs from the two types of cells would notrevealany differences. The answer to these problems may therefore not beobtainable through theuseof in vitro systems andmayrequirenew approaches.
ACKNOWLEDGMENTS
We thankJ. J.Holland,C.R. Pringle,andC.P.Stanners for their VSVwild-type andmutantisolates, DelphinePillote for excellent technical assistance, and Marjorie Gillett for typingthismanuscript.
Thisworkwassupported in partbyPublic Health Service research grant Al 12070 from the National Institutes of VOL.
on November 10, 2019 by guest
http://jvi.asm.org/
Health. M.K.P. was the recipient of a U.S. Public Health Service PredoctoralTraineeship from a cellular and molecular biology training grant (GM 7283).
LITERATURE CITED
1. Baxt,B., and R.Bablanian.1976. Mechanism ofvesicular stomatitisvirus-inducedcytopathic effects. II. Inhibition of macromolecular synthesis inducedby infectious and defective-interferingparticles.Virology72:383-392. 2. Bay, P.H. S., and M. E.Reichmann. 1979.UV
inactiva-tion of the biological activity of defective interfering particlesgeneratedby vesicular stomatitis virus. J. Virol. 32:876-884.
3. Bellett,A.J. D., and P. D. Cooper. 1959.Someproperties ofthe transmissible interfering component of vesicular stomatitis virus preparations. J. Gen. Microbiol. 21:498-509.
4. Huang,A. S., and E. K. Manders. 1972.Ribonucleicacid synthesisofvesicularstomatitis virus.IV. Transcription bystandardvirus in the presence of defectiveinterfering particles.J.Virol. 9:909-916.
5. Jen, G., and R. E. Thach.1982.Inhibition ofhost transla-tion in encephalomyocarditis virus-infected L cells: a novelmechanism.J.Virol. 43:250-261.
6. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head ofbacteriophage T4. Nature(London) 227:680-685.
7. Leamnson, R. N., and M. E. Reichmann. 1974.The RNA ofdefective vesicularstomatitis virusparticles in relation toviral cistrons. J. Mol. Biol.85:551-568.
8. Lewis, J. B., H.Esche, J. E. Smart, B. W. Stillman, M. L. Harter, and M. B. Mathews. 1979. Organization and expression ofthe left third of the genome of adenovirus. ColdSpringHarborSymp. Quant.Biol. 44:493-508. 9. Lodish, H. F., and S. Froshauer. 1977. Relative rates of
initiation oftranslation of different vesicular stomatitis messenger RNAs. J. Biol. Chem. 252:8804-8811. 10. Lodish,H.F., and M. Porter.1980.Translational control
ofproteinsynthesisafterinfection byvesicular stomatitis virus. J. Virol. 36:719-733.
11. Lodish, H. F., and M. Porter. 1981. Vesicularstomatitis virus mRNA and inhibition of translation of cellular mRNA-Is there a P function in vesicular stomatitis virus? J.Virol.38:504-517.
12. Marvaldi,J., M. J.Sekellick,P.I.Marcus, and J. Lucas-Lenard.1978. Inhibition ofmouseL cellproteinsynthesis
by ultraviolet-irradiated vesicular stomatitis virus re-quirestranscription.Virology84:127-133.
13. McAMister, P.E., and R. R. Wagner. 1976. Differential inhibitionofhostproteinsynthesisin L cellsinfectedwith RNA-temperature-sensitive mutants of vesicular stoma-titis virus. J. Virol. 18:550-558.
14. McGowan, J. J., andR.R. Wagner. 1981. Inhibition of cellular DNAsynthesis by vesicular stomatitis virus. J. Virol. 38:356-367.
15. McSharry, J. J., and Choppin, P. W. 1978. Biological properties of the VSV glycoprotein. I. Effects of the isolatedglycoproteinonhost macromolecularsynthesis. Virology 84:172-182.
16. Nuss, D.L., H.Opperman,and G.Koch. 1975. Selective blockage of initiation of host protein synthesis in RNA virus infected cells. Proc. Natl. Acad. Sci. U.S.A. 72:1258-1262.
17. Perrault, J., and J. J. Holland. 1972. Absence of tran-scriptase activity ortranscription-inhibiting ability in de-fectiveinterfering particles of vesicular stomatitis virus. Virology 50:159-170.
18. Pringle, C. R. 1970.Genetic characteristics of conditional lethal mutants ofvesicular stomatitis virus induced by 5-fluorouracil, 5-azacytidine, and ethyl methane sulfonate. J.Virol. 5:559-567.
19. Reichmsnn, M. E., D. H. L. Bishop, F. Brown, J.Crick, J. J. Holland, C.-Y. Kang, R. Lazzarini, S. Moyer, J. Perrault, L. Prevec, C. R. Pringle, R. R. Wagner, J. S. Younger, and A. S. Huang. 1980. Proposal for a uniform nomenclature for defectiveinterferingviruses of vesicular stomatitis virus. J. Virol. 34:792-794.
20. Relchmann, M. E., and W. M. Schnitzlein. 1979. Defec-tive interfering particles of rhabdoviruses. Curr. Top. Microbiol. Immunol. 86:123-168.
21. Schnitzlein, W. M., and M. E. Reichmann. 1976. The size andcistronicoriginof defective vesicular stomatitis virus particle RNAs in relation tohomotypic and heterotypic interference. J. Mol. Biol. 101:307-325.
22. Schnitzlein, W. M., and M. E. Reichmann. 1980. Inhibi-tion of vesicular stomatitis virusreplication by adenosine. Virology 103:123-137.
23. Sekellick, M. J., and P. I. Marcus. 1980. Viral interfer-enceby defective particles of vesicular stomatitis virus measured in individual cells.Virology 104:247-252. 24.Shmookler, R. J., J. Buss, and M. H. Green. 1974.
Proper-ties ofthe polyoma virus transcription complex obtained from mousenuclei.Virology57:122-127.
25. Stampfer, M., D. Baltimore, and A. S. Huang. 1971. Absence ofinterferenceduringhigh-multiplicityinfection by clonally purified vesicular stomatitisvirus. J. Virol. 7:407-411.
26. Stanners, C. P., A. M. Francoeur, and T. Lam. 1977. Analysisof VSV mutant with attenuated cytopathogenic-ity:mutation in viralfunction,P, for inhibition ofprotein synthesis. Cell11:273-281.
27. Unger, J.T., and M. E. Reichmann. 1973. RNAsynthesis in temperature-sensitive mutants of vesicular stomatitis virus. J. Virol. 12:570-578.
28. Weck, P. K., and R. R.Wagner. 1978.Inhibition ofRNA synthesisin mousemyeloma cells infected with vesicular stomatitis virus. J. Virol. 25:770-780.
29. Wertz, G. W., and J. S. Youngner. 1972. Inhibition of protein synthesisin L cellsinfectedwith vesicular stoma-titis virus. J. Virol. 9:85-89.
on November 10, 2019 by guest
http://jvi.asm.org/