0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Enhanced Transcriptional Activation by E2 Proteins from
the Oncogenic Human Papillomaviruses
ROBERT KOVELMAN,* GRAHAM K. BILTER, EMILIA GLEZER, AMY Y. TSOU,
ANDMIGUEL S. BARBOSA
Department of Virology, Signal Pharmaceuticals, Inc., San Diego, California 92121
Received 5 April 1996/Accepted 29 July 1996
A systematic comparison of transcriptional activation by papillomavirus E2 proteins revealed that the E2 proteins from high-risk human papillomaviruses (human papillomavirus type 16 [HPV-16] and HPV-18) are much more active than are the E2 proteins from low-risk HPVs (HPV-6b and HPV-11). Despite the tropism of HPVs for particular epithelial cell types, this difference in transcriptional activation was observed in a number of different epithelial and nonepithelial cells. The enhanced activities of the E2 proteins from high-risk HPVs did not result from higher steady-state levels of protein in vivo, and in vitro DNA-binding assays revealed similar binding properties for these two classes of E2 proteins. These results demonstrate that the E2 proteins from high-risk HPVs have an intrinsically enhanced potential to activate transcription from promoters with E2-responsive elements. We found that there are also substantial differences between the activation properties of the bovine papillomavirus type 1 E2 protein and those of either of the two classes of HPV E2 proteins, especially with regard to requirements for particular configurations of E2 binding sites in the target promoter. Our results indicate that there are at least three distinct functional classes of E2 proteins and that these classes of E2 proteins may perform different roles during the respective viral life cycles.
The papillomaviruses are a large family of double-stranded DNA viruses that infect a wide variety of epithelial cells. Their genomes are composed of 8 to 10 open reading frames (ORFs) which encode the regulatory proteins essential for completion of the viral life cycle and the structural components of the virion. The proteins encoded by the E2 ORF are regulatory factors which control transcription by RNA polymerase II and, in conjunction with the viral E1 protein, play a role in repli-cation of the viral genome (reviewed in references 23 and 36). The E2 proteins control transcription by binding to specific cognate DNA sites, multiple copies of which are found in the papillomavirus upstream regulatory regions (URRs).
The proteins encoded by the E2 ORF of bovine papilloma-virus type 1 (BPV-1) have been studied more extensively than have those encoded by the E2 ORFs of the human papilloma-viruses (HPVs). Mutational analysis of the BPV-1 E2 protein has shown that the C-terminal 100 amino acid residues are sufficient for both DNA binding and dimerization, while the N-terminal 210 amino acid residues are required for transcrip-tional activation (17, 26, 37, 39). This structure consisting of an N-terminal activation domain and a C-terminal DNA-binding domain is highly conserved in E2 proteins from different pap-illomaviruses, and experiments involving mutagenesis of other E2 proteins or domain swaps between different E2 proteins indicate that the functions of these domains are also conserved (13, 17, 18). Truncated BPV-1 E2 polypeptides containing the C-terminal DNA-binding domain but not the N-terminal acti-vation domain can act as transcriptional repressors (10, 13, 17, 34, 62), and such polypeptides have been observed in BPV-1-transformed cells (10, 31, 33). Whether such truncated E2 polypeptides are produced in vivo after HPV infection is not known.
Depending on the E2-dependent reporter construct used,
the full-length BPV-1 E2 protein can itself act as either a transcriptional activator or a transcriptional repressor (13, 15, 17, 25, 28, 41, 54, 60). Similarly, HPV E2 proteins have been reported to be either activators or repressors of transcription (5, 9, 13, 17, 29, 30, 40, 59). The context surrounding the E2 binding sites is of particular importance in determining the resultant effects of the expression of E2 proteins, with the
configuration found at the 39end of the URRs of genital HPVs
typically resulting in E2-dependent repression of transcription of the nearby E6 and E7 coding regions (5, 15, 44, 59, 60). However, because standard monolayer tissue culture systems do not support the entire life cycle of any HPV, the relevant in vivo functions of full-length or truncated HPV E2 proteins in a productive viral infection are not known.
Indeed, there is no clear consensus from previous studies on whether expression of the BPV-1 and HPV E2 proteins in standard tissue culture systems results in a similar modulation of E2-dependent transcription. Early studies indicated that there might be differences between the activities of the BPV-1 and HPV E2 proteins (29, 59, 60), but these differences might have arisen solely because the expression vectors employed for the BPV-1 and HPV E2 proteins were not equivalent or be-cause subcloning of the E2 ORFs into these vectors introduced variable noncoding sequences which affected expression levels. More recent studies (6, 61) have employed equivalently con-structed expression plasmids containing the BPV-1 and HPV type 16 (HPV-16) E2 coding regions. Whereas Ushikai et al. (61) came to the conclusion that these two E2 proteins are similar in their activation properties, the experiments of Bou-vard et al. (6) indicated that their comparative activities are dependent upon the reporter system used. In any case, neither these studies nor the earlier studies included a determination of protein expression levels, so it is unclear whether any dif-ferences in E2 activity were due to difdif-ferences in steady-state protein levels or to an actual divergence between their intrinsic transcriptional regulatory properties.
In fact, the focus of these studies was on the differences between the BPV-1 E2 protein and HPV E2 proteins, and a
* Corresponding author. Mailing address: Signal Pharmaceuticals, Inc., 5555 Oberlin Dr., San Diego, CA 92121. Phone: (619) 558-7500. Fax: (619) 558-7513. Electronic mail address: rkovelma@signalpharm .com.
7549
on November 9, 2019 by guest
http://jvi.asm.org/
systematic comparison of the activities of E2 proteins from different HPVs has not been reported. There is a precedent for such physiologically relevant activity differences, since it has been demonstrated that the E6 and E7 proteins from the genital HPVs associated with cervical cancer (high-risk HPVs) have a much greater ability to cause cellular immortalization and transformation than do the E6 and E7 proteins from other, low-risk HPVs (2, 3, 47, 56). In the absence of any information on whether the functions of viral proteins other than E6 and E7 from different HPVs are similar or divergent, we decided to undertake a systematic comparison of E2-dependent transcrip-tional activation. We found that the E2 proteins from high-risk HPVs are in fact much more active than are the E2 proteins from low-risk HPVs in both keratinocytes and other, unrelated cell types. The enhanced activation properties of the E2 teins from high-risk HPVs did not result from increased pro-tein levels in vivo, nor did we observe differences in the in vitro DNA-binding properties of the E2 proteins from high-risk and low-risk viruses. We also found that while the BPV-1 E2 pro-tein could be a very potent transcriptional activator in these cells, high-level expression of this E2 protein resulted in the squelching of transcriptional activation. Moreover, we ob-served a striking difference in the ability of HPV and BPV-1 E2 proteins to activate transcription when the configuration of E2 binding sites in the reporter was changed, further underlining the existence of mechanistic differences in the activation prop-erties of these proteins.
MATERIALS AND METHODS
Materials.Unless noted otherwise, chemicals were obtained from Sigma or Fisher Scientific, enzymes were obtained from New England Biolabs, Promega, or Perkin-Elmer, and custom oligonucleotides were obtained from Ransom Hill Bioscience.
Plasmids.Oligonucleotide primers designed to amplify by PCR the coding sequences of the E2 ORFs of HPV-6b, HPV-11, HPV-16, HPV-18, and BPV-1 contained the sequences 59ATTCTAGATGCGGCCGCCATGN1239(upstream
primer) and 59 GAGCTCGAGCGGCCGCTYAN1239 (downstream primer),
where Y5C or T according to the appropriate termination codon and N12is the
stretch of relevant bases for each E2 sequence. The sequences in italics corre-spond to the initiation and termination codons for the E2 ORFs, and the underlined sequences include theXbaI,NotI, andXhoI sites used for subsequent cloning. PCR products of the expected length were digested withXbaI andXhoI and cloned into pGEX-KG (21) cut withXbaI andXhoI. While sequencing the resulting plasmids to determine that PCR amplification did not introduce any mutations into the E2 coding regions, we found that the clones of HPV-16 and HPV-18 used as templates for PCR contained differences relative to the pub-lished sequences (12, 48). For HPV-16, we found a T at position 655 of the E2 ORF in place of a C, resulting in the replacement of Pro-219 with Ser, along with a G at position 171 which does not result in a change in the encoded amino acid. For HPV-18, we found a sequence of GCGT in place of TGCG starting at position 40 of the E2 ORF, resulting in an encoded Ala-Leu instead of Cys-Val at positions 14 and 15; GC instead of CG at position 268, resulting in Ala instead of Arg at position 90; and G instead of C at position 459, resulting in no change in the protein sequence. We also constructed plasmids containing the published HPV-16 E2 ORF sequence and found that the presence of serine in place of proline 219 did not affect E2 activity (data not shown).
In order to generate the pRSV-E2 series of expression vectors, we clonedNotI fragments of the pGEX-E2 series containing E2 ORFs into theNotI-cleaved pOPRSVICAT (Stratagene) vector fragment. In order to facilitate the cloning of E2 ORFs into the other vectors used, we first recloned thisNotI fragment containing the E2 ORFs back into theNotI-cleaved pGEX-E2 vector fragment in the reverse orientation to generate the pGEX-E2-R series of plasmids.Xho I-EcoRI fragments of the pGEX-E2-R series containing E2 ORFs were then cloned intoXhoI-EcoRI-cleaved pRSET C (Invitrogen) to generate the pRSET-E2 series of plasmids for His-pRSET-E2 protein expression in bacteria. In order to generate the pCMV-E2 plasmids used for most of the expression studies re-ported, we subcloned the HindIII-XbaI fragment of the pGEX-E2-R series containing the E2 ORFs into pCB6 (7) cut withHindIII andXbaI. TheBgl II-XbaI fragment of the pCB6-E2 series of plasmids containing the E2 ORFs was then subcloned into the vector fragment resulting from the digestion of pCGE2B (20) withBamHI andSpeI; this contains the cytomegalovirus (CMV) major immediate-early promoter and enhancer driving the E2 proteins and is what we have labeled pCMV-E2.
In order to construct pCMV-6/16E2, which contains the N-terminal activation
domain of HPV-6b and the C-terminal DNA-binding domain of HPV-16, we amplified by PCR a DNA fragment encoding the N-terminal 211 amino acids of the HPV-6b E2 protein by using pCMV-6E2 as the template, a primer to upstream sequences in the vector, and 59GGTGGTTGGCCAAAGATTCAG GAATGGATACTTC 39as the downstream primer. The underlined sequence is anMscI site. We digested the resultant PCR product withMscI andClaI (which cleaves within the upstream sequence) and inserted the fragment intoMscI-Cla I-cleaved pCMV-16E2, which provided the HPV-16 E2 C-terminal domain. Sim-ilarly, in order to construct pCMV-6/16E2, we amplified by PCR a fragment encoding the N-terminal 238 amino acids of the HPV-16 E2 protein by using pCMV-16E2 as the template, a primer to upstream sequences in the vector, and 59TCGTTTCCTAGGGATAGTCGTCTGTGTTTCTTCG 39as the down-stream primer. In this case, the underlined sequence is anAvrII site. The result-ant PCR product was digested withAvrII andHindIII (which also cleaves within the upstream sequence), and we inserted the fragment intoAvrII-Hin dIII-cleaved pCMV-6E2.
In order to create p2xE2BS-luc and p2x2xE2BS-luc, oligonucleotides were synthesized so that after annealing the double-stranded structure represented below resulted:
59CCGGGACCGAAAACGGTTCAACCGAAAACGGTTG 39 39 CTGGCTTTTGCCAAGTTGGCTTTTGCCAACGATC59
The underlined stretches are the E2 binding sites, and the entire double-stranded region is identical to the sequence found in the HPV-6b URR; the homologous regions in the URRs of HPV-11, HPV-16, and HPV-18 are very highly con-served. p2xE2BS-luc was constructed by cloning this double-stranded oligonu-cleotide intoXmaI-NheI-cleaved pGL2-promoter (Promega), which contains a minimal simian virus 40 (SV40) early promoter upstream of the luciferase coding region. We also obtained p2x2xE2BS-luc by the same method; it contains two copies of the oligonucleotide shown above separated by the polylinker sequences between theXmaI andNheI sites of pGL2-promoter, resulting in a spacing of 35 bp from the end of the second E2 binding site to the beginning of the third E2 binding site. p4xE2BS-luc was constructed by ligating an excess of oligonucleo-tides containing consensus E2 binding sites (ACCGN4CGGT, where N45A4or
T4) flanked by overhangingBglII cores (GATC) intoBglII-cleaved
pGL2-pro-moter. The sequence of the insert replacing the originalBglII site of pGL2-promoter is 59AGATCCACCGTTTTCGGTAGATCCACCGTTTTCGGTAG ATCCACCGTTTTCGGTACCGAAAACGGTGGATCT 39, with the E2 bind-ing sites underlined.
In order to construct reporter plasmids containing URR sequences, PCRs were performed with (i) pBR-HPV-6b as the template and 59TCCCCCGGGT AATATATGTGTATATGTACTG 39and 59CTAGCTAGCTCGTTTGCTAA ATTTTAGGGCTGG 39as primers and (ii) pUC-HPV-16 as the template and 59TCCCCCGGGTAAGTATTGTATGTATGTTGAA 39 and 59 GTAGCTA GCTCTTTTGGTGCATAAAATGTCTGC 39as primers. The resultant PCR products were digested withXmaI andNheI (sites in primers are underlined) and cloned intoXmaI-NheI-cleaved pGL2-promoter (Promega) containing an SV40 early promoter upstream of the luciferase gene to generate p6bURR(enhancer) and p16URR(enhancer) or intoXmaI-NheI-cleaved pGL2-basic (Promega) con-taining the luciferase gene to generate p6bURR(promoter) and p16URR(pro-moter). When we sequenced the PCR products generated, we found that there were also differences between the HPV-16 URR sequence in the template we had used and the previously published sequence.
Cells and transfections.C33-A and A431 cells were obtained from the Amer-ican Type Culture Collection, U373 MG cells were provided by Peter Ghazal (The Scripps Research Institute, La Jolla, Calif.), and early-passage primary human keratinocytes were purchased from Clonetics. C33-A, A431, and U373 MG cells were grown in Dulbecco’s modified Eagle medium (Mediatech) plus 10% heat-treated fetal bovine serum (Gemini). Primary keratinocytes were grown in KGM (Clonetics). Calcium phosphate transfections using Profection reagents (Promega) were performed on C33-A and U373 MG cells for 16 h, at which time cells were washed twice with phosphate-buffered saline (PBS) and fed with fresh medium. A431 cells were transfected with Tfx-50 (Promega), and primary keratinocytes were transfected with Lipofectin (Life Technologies), each essentially according to the manufacturer’s instructions. Cells were lysed 40 to 48 h after the beginning of transfection in eitherb-galactosidase reporter lysis buffer (Promega) for C33-A and U373 MG cells or cell lysis buffer (Analytical Luminescence Laboratory) for A431 cells and primary keratinocytes. Theb -ga-lactosidase reporter lysis buffer was compatible with the determination of both luciferase andb-galactosidase activities, so the same lysates could be used in parallel for both assays; however, this buffer was not effective in lysing A431 cells or primary keratinocytes, for which we used the cell lysis buffer, which was not compatible withb-galactosidase activity measurement. Although the expression of E2 proteins caused a small increase inb-galactosidase activity, the results obtained were similar with or without normalization tob-galactosidase activity. All transfection experiments were performed at least three times to ensure the reproducibility of the data.
Antibodies and immunoblot analysis.Antibodies to the BPV-1 E2 protein have been described previously (1). In order to generate antibodies against the HPV-6b and HPV-16 E2 proteins, we produced glutathione S-transferase (GST)–6E2 and GST-16E2 fusion proteins inEscherichia coliJM109 from
on November 9, 2019 by guest
http://jvi.asm.org/
mids pGEX-6E2 and pGEX-16E2 essentially as described by Smith and Johnson (49) and then cleaved the E2 proteins away from the GST moiety with thrombin essentially as described by Guan and Dixon (21). Polyclonal antisera were gen-erated by multiple injections of rabbits with either the HPV-6b E2 protein or the HPV-16 E2 protein (Alpha Diagnostic, Inc.).
For Western (immunoblot) analysis of transfected cell extracts, transfected cells were washed and collected by scraping into PBS, centrifuging at low speed, and resuspending the cell pellet in buffer C (25 mM HEPES [N -2-hydroxyeth-ylpiperazine-N9-2-ethanesulfonic acid]-NaOH [pH 7.9], 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5% Triton X-100, 5 mM dithiothreitol, 1 mM
phen-ylmethylsulfonyl fluoride, 5mg of aprotinin per ml, 5mg of leupeptin per ml, and 5 mg of pepstatin per ml). After rotation for 30 min at 48C, extracts were centrifuged and supernatants were kept for loading onto sodium dodecyl sulfate– 10% polyacrylamide gels. Alternatively, we also lysed cells directly inb -galacto-sidase reporter lysis buffer (Promega) and obtained similar results. After elec-trophoresis, proteins were transferred to nitrocellulose membranes (Hybond ECL; Amersham) which were then sequentially incubated in Tris-buffered saline (TBS)–0.05% Tween 20–5% nonfat milk blocking agent (Bio-Rad), TBS–0.05% Tween 20–the relevant primary antiserum, and TBS–0.05% Tween 20–1% bo-vine serum albumin (BSA)–donkey anti-rabbit immunoglobulin G antibodies conjugated to horseradish peroxidase (Amersham) prior to detection by chemi-luminescence using reagents from Amersham or Alpha Diagnostic, Inc. Gels always included titrations of the relevant bacterial His-E2 proteins as standards. Expression and purification of His-tagged E2 proteins.Plasmids pRSET-6E2 and pRSET-16E2 were transformed intoE. coliBL21(DE3)(pLysS) (Strata-gene), and precultures inoculated with colonies containing these plasmids were incubated overnight at 378C. Cultures inoculated from these precultures were incubated at 378C until the optical density at 600 nm reached 0.6 to 0.8. IPTG (isopropyl-b-D-thiogalactopyranoside) was added to a final concentration of 1 mM, and after an additional 2.5 h, bacteria were collected by centrifugation and pellets were stored at2708C. After being thawed on ice, pellets were resus-pended in buffer A (20 mM Tris-HCl [pH 7.9], 0.5 M NaCl) plus 1 mM phen-ylmethylsulfonyl fluoride, 5mg of aprotinin per ml, 5mg of leupeptin per ml, and 5mg of pepstatin per ml. Triton X-100 was added to 0.1%, and tubes were placed at2708C for 10 min prior to ultracentrifugation at 240,0003gat 48C for 60 min. Imidazole was added to these supernatants to a final concentration of 40 mM prior to mixing with Ni-nitrilotriacetic acid-agarose (Qiagen) equilibrated in buffer A plus 40 mM imidazole, 0.1% Triton X-100, and 1 mM phenylmethyl-sulfonyl fluoride. After a 90-min incubation with rotation at 48C, Ni-nitrilotri-acetic acid-agarose resin was washed four times with the same buffer prior to elution of the His-E2 proteins with buffer A plus 150 mM imidazole and 0.1% Triton X-100. Then the purified His-E2 proteins were dialyzed against buffer B (20 mM HEPES-NaOH [pH 7.9], 20% glycerol, 50 mM KCl, 0.2mM EDTA, 2 mM dithiothreitol) prior to being flash frozen and stored at2708C.
Gel shift assays.For the experiment shown in Fig. 6, the annealed oligonu-cleotides discussed above were labeled by end-fill with the Klenow fragment of DNA polymerase I and [a-32
P]dCTP (Amersham). For the experiment shown in Fig. 7, primers were designed to amplify by PCR a 192-bp fragment containing four E2 binding sites from p2x2xE2BS-luc and this fragment was gel purified and labeled with T4 polynucleotide kinase and [g-32
P]ATP (Amersham). Both la-beled probes were purified away from unincorporated nucleotides with NucTrap columns (Stratagene). Binding reactions were performed in 20ml at 308C and included 20 mM HEPES-NaOH (pH 7.9), 50 mM KCl, 5 mM MgCl2, 0.1%
Nonidet P-40, 8 to 10% glycerol, 1 mM dithiothreitol, 1 mg of BSA per ml, the probe containing two or four E2 binding sites, and bacterially expressed His-6bE2 or His-16E2. Binding reactions were run out on 4% polyacrylamide–0.253 Tris-borate-EDTA gels, and dried gels were exposed to film and Storage Phos-phor screens (Molecular Dynamics).
RESULTS
Activation by E2 proteins in C33-A cells.In order to make
accurate comparisons of the functions of different E2 proteins, we cloned the E2 coding regions from HPV-6b, HPV-11, HPV-16, HPV-18, and BPV-1 into different vectors. We want-ed to measure intrinsic E2 activities and avoid including any flanking sequences outside the E2 coding regions which could lead to differential mRNA stabilities, splicing patterns, or translational initiation efficiencies. Therefore, we cloned PCR products containing papillomavirus DNA sequences between the initiation and termination codons of the E2 coding regions downstream of the CMV major immediate-early promoter. We measured the activities of the encoded E2 proteins in C33-A cells, which are derived from a cervical carcinoma but do not contain papillomavirus DNA (45), so that we could determine E2 activity in the absence of any other viral proteins. These pCMV-E2 expression constructs were transfected into C33-A
cells along with a plasmid containing an E2-dependent
lucif-erase reporter and a plasmid expressingb-galactosidase under
the control of a constitutive promoter in order to determine E2-dependent transcriptional activation relative to an internal control.
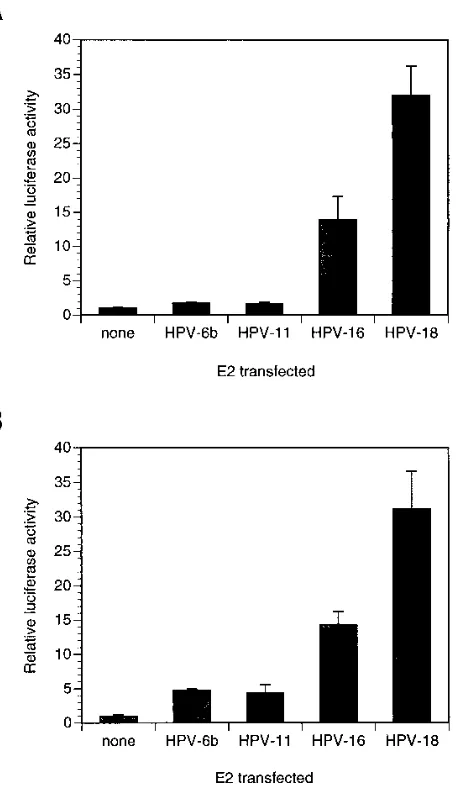
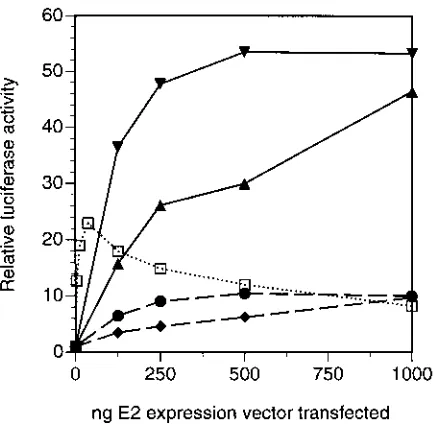
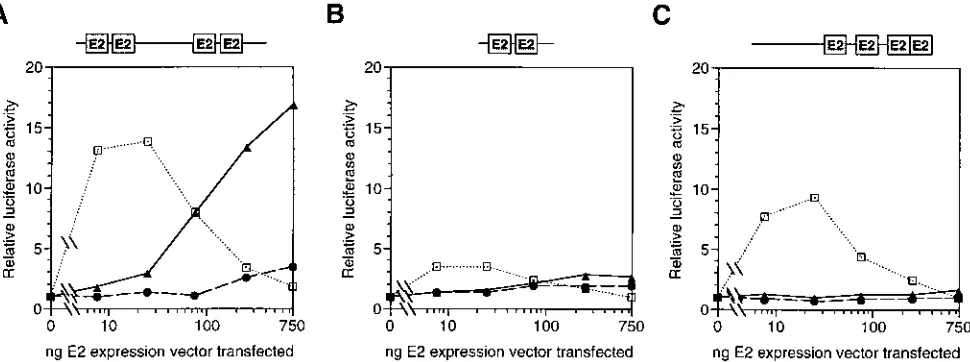
As shown in Fig. 1A, we titrated the pCMV-E2 constructs in order to perform a thorough characterization of the transcrip-tional regulatory properties of these E2 proteins. We found that expression of the HPV-16 and HPV-18 E2 proteins in C33-A cells resulted in 10- to 25-fold activation of E2-depen-dent luciferase activity at all of the input plasmid amounts tested (solid lines). Strikingly, however, expression of the HPV-6b and HPV-11 E2 proteins resulted in much lower (less than fourfold) activation of E2-dependent luciferase activity (dashed lines). We observed this large difference in activity even though the E2-responsive element in the reporter plasmid is derived from the E2-responsive elements in HPV-6b URR; if this reporter causes any bias in determining the activities of the E2 proteins, it should be in favor of the HPV-6b E2 protein and the closely related HPV-11 E2 protein. This categorization of E2 protein activities correlates directly with the classifica-tion of HPVs into high-risk and low-risk groups, with HPV-16 and HPV-18 being significant risk factors for cervical cancer and HPV-6b and HPV-11 not being associated with carcino-genesis (see reference 63 for a review).
We also determined how these HPV E2 activities compared with that of the BPV-1 E2 protein. As shown in Fig. 1A, the transfection of the construct expressing the BPV-1 E2 protein under the control of the CMV promoter resulted in a dramat-ically different pattern of activation (dotted line). The BPV-1 E2 protein was highly active as a transcriptional activator when very small amounts (60 ng or less) of E2 expression vector were transfected. With larger amounts of BPV-1 E2 expression vec-tor, transcriptional activation was dramatically reduced; in-deed, when 500 or 1,000 ng of the BPV-1 E2 expression vector was transfected, luciferase activity was no higher than that when the HPV-6b and HPV-11 E2 genes encoding the low activity E2 proteins were transfected (compare dashed and dotted lines in Fig. 1A). The most likely explanation for this dramatic dose-dependent decrease in transcriptional activation by the BPV-1 E2 protein is that high levels of E2 protein not bound to DNA in transfected cells titrate out a limiting factor required for E2-dependent transcriptional activation. This
squelching was not indiscriminate, as transcription ofb
-galac-tosidase from the internal control plasmid pCMV-b-gal was
actually slightly stimulated by expression of the BPV-1 E2 protein.
Because of this unusual pattern of activation by BPV-1 E2 and the lack of consensus in previous reports on activation by different E2 proteins (6, 29, 59–61), we also performed similar transfections of plasmids expressing the E2 proteins under the control of the Rous sarcoma virus (RSV) promoter. As shown in Fig. 1B, the BPV-1 E2 protein (dotted line) was an efficient dose-dependent activator protein in vivo when expressed from the RSV promoter. On the other hand, the activities of the HPV E2 proteins were all low level, although even in these assays the HPV-16 and HPV-18 E2 proteins (solid lines) were slightly more active than were the HPV-6b and HPV-11 E2 proteins (dashed lines). We have confirmed by immunoblot analysis that expression from the CMV promoter results in much higher steady-state protein levels than does expression from the RSV promoter (see Fig. 5). Thus, we conclude that the E2 proteins from high-risk HPVs are more efficient tran-scriptional activators in vivo than are the E2 proteins from low-risk HPVs and that the extent of transcriptional activation
on November 9, 2019 by guest
http://jvi.asm.org/
observed with the BPV-1 E2 protein is very sensitive to the amount and type of expression vector transfected.
Transcriptional activation by E2 proteins in other cells.The
transfection experiments described above were performed with C33-A cells, which are derived from a cervical carcinoma
(ref-erence 45 and ref(ref-erences therein). In order to determine whether the higher transcriptional activities of the HPV-16 and HPV-18 E2 proteins are specific to cervical keratinocytes, we performed transfections of A431 cells, which are derived from a vulvar carcinoma. As shown in Fig. 2A, we obtained results very similar to those with C33-A cells. The HPV-16 and HPV-18 E2 proteins were still much more active than were the HPV-6b and HPV-11 E2 proteins. Thus, the E2 proteins from high-risk HPVs are also more potent transcriptional activators when assayed in noncervical keratinocytes.
We also wanted to determine whether the activity differ-ences between the E2 proteins from high-risk and low-risk HPVs were related in some way to the transformed phenotypes of established cell lines or whether, in contrast, differences would also be observed in primary keratinocytes. When we transfected the same plasmids containing the HPV E2 genes under the control of the CMV promoter into primary human keratinocytes, we again found that the E2 proteins from
high-FIG. 1. Transcriptional activation by E2 proteins expressed from a CMV promoter or an RSV promoter. C33-A cells in 24-well plates were transfected with the amounts of pCMV-E2 (A) or pRSV-E2 (B) expression vector indicated along with the reporter p2x2xE2BS-luc (see Fig. 9 for details) and pCMV-b-gal. Cells were lysed 2 days after transfection, and the luciferase andb-galactosidase activities of lysates were determined. Data are luciferase activities normalized to
[image:4.612.68.324.74.551.2]b-galactosidase activities, with the results for lysates of control cells transfected with the reporter alone being set to a value of 1. These data and the transfection data shown in other figures are representative of experiments which were re-peated at least three times.h, pCMV-BE2 (A) and pRSV-BE2 (B);F, pCMV-6E2 (A) and pRSV-pCMV-6E2 (B); }, pCMV-11E2 (A) and pRSV-11E2 (B); å, pCMV-16E2 (A) and pRSV-16E2 (B);ç, pCMV-18E2 (A) and pRSV-18E2 (B).
FIG. 2. Transcriptional activation by E2 proteins in A431 cells and primary human keratinocytes. A431 cells (A) and primary human keratinocytes (B) were transfected in triplicate with a fixed amount of pCMV-E2 expression vector along with the p2x2xE2BS-luc reporter. Luciferase activities were determined, and the bar graphs show the averages of three determinations, with error bars indicating the standard deviations.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.328.556.281.674.2]risk HPVs were more active than were the E2 proteins from low-risk HPVs (Fig. 2B), indicating that these differences are found in primary keratinocytes as well as in two different epi-thelial cell lines.
Finally, given that E2 activities are conserved in divergent types of epithelial cells, we wanted to assay E2 function in a completely unrelated cell type. We chose to use the U373 MG cell line because their glial-cell origin clearly distinguishes them from keratinocytes and because they can be easily trans-fected by the same method used for the transfection of C33-A cells. We performed extensive titrations of the plasmids con-taining the E2 coding regions cloned downstream of the CMV promoter, and as shown in Fig. 3, the results obtained were quite similar to those presented in Fig. 1A for C33-A cells. Once again, the HPV-16 and HPV-18 E2 proteins (solid lines) were much more active than were the HPV-6b and HPV-11 E2 proteins (dashed lines), and expression of the BPV-1 E2 pro-tein (dotted line) resulted in activation with small amounts of input DNA followed by squelching with larger amounts, albeit with less extreme patterns than those observed with C33-A cells. Thus, the much higher activity levels of the E2 proteins from high-risk HPVs and the activation-squelching pattern observed with the BPV-1 E2 protein do not result from inter-actions with factors specific to epithelial cells but must instead be caused by differences in E2 functions that are conserved across highly divergent cell types.
E2 protein levels in extracts of transfected cells.One
pos-sible mechanism for these differences in E2-dependent tran-scriptional regulation is that the E2 proteins from high-risk HPVs and BPV-1 accumulate to much higher concentrations in vivo than do the E2 proteins from low-risk HPVs. One approach commonly employed to compare protein concentra-tions involves adding an epitope tag and then using antibodies to this tag for immunoprecipitation or immunoblot analysis. We added an influenza virus hemagglutinin tag onto the N termini of the E2 coding regions cloned into the pCMV ex-pression vector, but we found that the addition of such a tag interfered with the ability of the E2 proteins to activate
tran-scription and significantly altered the steady-state levels of the E2 proteins (data not shown). We therefore decided to make direct measurements of the levels of untagged E2 proteins in extracts of transfected cells by performing immunoblot analy-ses with polyclonal antisera generated against the BPV-1, HPV-6b, and HPV-16 E2 proteins, with the last two serving as representatives of the proteins from low-risk and high-risk HPVs, respectively.
[image:5.612.69.286.69.281.2]As shown in the bar graphs of Fig. 4, transfection of pCMV-6E2 resulted in 5-fold transcriptional activation, while trans-fection of pCMV-16E2 resulted in 30-fold activation, similar to the results presented in Fig. 1A. The results of Western blot-ting performed with extracts from these cells are shown above each graph, with the panels at the right providing standards to calibrate antisera. We found that the HPV-6b and HPV-16 E2
FIG. 3. Transcriptional activation by E2 proteins in nonepithelial cells. U373 MG glial cells were transfected and analysis was performed as described in the legend to Fig. 1 for C33-A cells.h, pCMV-BE2;F, pCMV-6E2;}, pCMV-11E2;
å, pCMV-16E2;ç, pCMV-18E2.
FIG. 4. Immunoblot analysis of steady-state levels of HPV E2 proteins after transfection. C33-A cells were lysed for determinations of luciferase andb -ga-lactosidase activities as well as for Western analyses of E2 proteins after the transfection of cells in 100-mm-diameter dishes with 4.5mg of pCMV-6bE2, pCMV-16E2, or the control pCMV, along with p2x2xE2BS-luc and pCMV-b-gal. The graphs show luciferase activities normalized tob-galactosidase activities, and above the graphs are shown the results of Western blotting performed with antibodies against the HPV-6b (A) or HPV-16 (B) E2 protein. In order to normalize for the different titers of antibodies, Western blots also included separate lanes containing known amounts of bacterially expressed His-6bE2 protein (A) or His-16E2 protein (B); the relevant portions of these blots are shown on the far right.
on November 9, 2019 by guest
http://jvi.asm.org/
proteins accumulated to approximately the same concentration in vivo, with the amounts loaded onto the gels corresponding to 5.0 to 7.5 ng of protein when normalized to the bacterial controls. We performed several repetitions of this experiment and found that the variation in E2 protein levels was consis-tently less than twofold, differences that were always substan-tially smaller than the corresponding activity differences (six-fold or greater). In addition, both HPV-6b and HPV-16 E2 proteins in these extracts migrated at the expected molecular weight, making it unlikely that the lower activity of the HPV-6b E2 protein resulted from differential splicing of its mRNA to result in N-terminally-truncated transcriptional repressors. Thus, the higher-level activities of the E2 proteins from high-risk HPVs (i.e., HPV-16) compared with those of the E2 pro-teins from low-risk HPVs (i.e., HPV-6b) do not result from differences in steady-state protein levels.
We also wanted to determine how these protein amounts compared with those of the BPV-1 E2 proteins, but for the latter we performed more extensive titrations with both pRSV-BE2 and pCMV-pRSV-BE2 expression vectors because we had ob-served both activation and squelching of activation upon ex-pression of the BPV-1 E2 protein (Fig. 1). As shown in Fig. 5A, we obtained patterns of activation and squelching similar to those observed previously. We found that the highest levels of activation, resulting from transfections of large amounts of pRSV-BE2, corresponded to concentrations of E2 protein be-low the limit of detection (Fig. 5B, lanes 9 to 11). On the other hand, transfections of larger amounts of pCMV-BE2 resulted in readily detectable BPV-1 E2 protein in extracts of these cells (Fig. 5B, lanes 4 to 6), but this corresponds to an in vivo concentration of E2 protein which is well above the optimum for transcriptional activation (Fig. 5A). Note also that squelch-ing at these higher concentrations of pCMV-BE2 expression
plasmid did not result from the production of E2 repressor proteins lacking the N-terminal activation domain, as the pro-tein in extracts of transfected cells migrated at the expected molecular weight. From comparisons of the Western blots shown in Fig. 4 and 5 as well as those from other comparable experiments, we can conclude that the BPV-1 E2 protein ac-cumulates to significantly higher concentrations than do the HPV E2 proteins and that the BPV-1 E2 protein is also likely to be a more potent transcriptional activator than are the HPV E2 proteins even when normalized for differences in in vivo protein levels.
DNA-binding properties of the E2 proteins.Since these
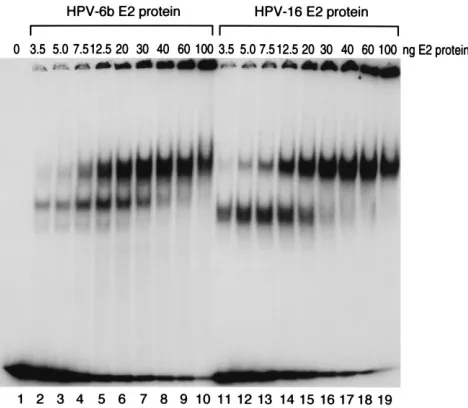
ex-periments indicated that differences in the steady-state levels of the HPV-16 and HPV-6b E2 proteins are not the cause of the divergence in their activation potentials, we decided to examine whether differences in DNA-binding properties could be the underlying mechanism. We expressed the HPV-6b and HPV-16 E2 proteins in bacteria with an N-terminal 6-His tag and purified the proteins by using a nickel-chelating resin. Shown in Fig. 6 are the results of gel shift assays performed with these E2 proteins and a probe containing two E2 binding sites in the configuration found in the HPV-6b URR. The addition of increasing amounts of either HPV-6b or HPV-16 E2 protein resulted in successive dose-dependent appearances of complexes containing the DNA probe with one or two binding sites occupied. The binding patterns observed for the HPV-6b and HPV-16 E2 proteins were very similar, indicating that there are no significant differences in DNA binding to a probe containing two E2 binding sites.
However, other results (see Fig. 9) indicate that the HPV-16 E2 protein is not an efficient activator of a reporter containing only two E2 binding sites, so we wanted to determine whether cooperativity in DNA binding between sets of E2 proteins bound at multiple E2 binding sites could be responsible for the enhanced activity of the HPV-16 E2 protein. In order to ad-dress this possibility, we prepared a probe containing the four E2 binding sites in the configuration found in the reporter used in the experiments described above. As shown in Fig. 7, com-plexes corresponding to partial or full occupancy of the
[image:6.612.60.297.73.320.2]mul-FIG. 5. Immunoblot analysis of steady-state levels of the BPV-1 E2 protein after transfection and comparison to transcriptional activation. Transfections and subsequent analysis were performed as described in the legend to Fig. 4, except that the BPV-1 E2 expression vectors were titrated. The resultant lucif-erase activities normalized tob-galactosidase activities (A) and the immunoblot analysis (B) are shown. Molecular weights (in thousands) are given on the right.
FIG. 6. Binding of the HPV-6b and HPV-16 E2 proteins to a probe contain-ing two E2 bindcontain-ing sites. Gel shift assays were performed with a probe containcontain-ing two E2 binding sites and the amount of HPV-6b or HPV-16 E2 protein indicated above each lane.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.318.552.486.690.2]tiple E2 binding sites were formed at essentially the same protein concentrations when either the HPV-6b or HPV-16 E2 protein was added (compare lanes 2 to 8 with lanes 9 to 15). This demonstrates that the large difference between transcrip-tional activation by the HPV-16 and HPV-6b E2 proteins is not caused by intrinsic differences in DNA-binding affinities or cooperativity in DNA binding.
Transcriptional activation by chimeric E2 proteins.Since
the results from immunoblot and gel shift analyses indicated that enhanced transcriptional activation by the HPV-16 E2 protein relative to that by the HPV-6b E2 protein is not caused by differences in protein levels or DNA-binding activities, we were interested in determining more directly whether this en-hancement results from an intrinsic difference in the strengths of the respective transcriptional activation domains. We there-fore created chimeric E2 coding regions in the background of the same CMV promoter-containing expression vectors em-ployed for the previous studies. Transfection of pCMV-6/16E2 results in the expression of an E2 protein with the N-terminal transcriptional activation domain of the HPV-6b E2 protein and the C-terminal DNA-binding domain of the HPV-16 E2 protein; conversely, pCMV-16/6E2 encodes an E2 protein with the transcriptional activation domain of the HPV-16 E2 pro-tein hooked up to the DNA-binding domain of the HPV-6b E2 protein. As shown in Fig. 8, the E2 chimera with the HPV-16 activation domain was a significantly better transcriptional ac-tivator than was the chimera with the HPV-6b activation do-main, thus mapping the observed difference in transcriptional stimulation to this domain of the E2 proteins. Note, however, that the chimera with the HPV-16 activation domain was not as strong an activator as was the wild-type HPV-16 E2 protein and that the chimera with the HPV-6b activation domain was a slightly stronger activator than was the wild-type HPV-6b E2 protein. This indicates that the activation domain is the most important but not the sole determinant of the strength of activation by the HPV E2 proteins.
Dependence of transcriptional activation by E2 proteins on
the configuration of E2 binding sites.The E2-responsive
ele-ment in the reporter plasmid employed in the activation
stud-ies described above contains four E2 binding sites in a config-uration which is essentially a dimer of the region containing
two E2 binding sites at the 39 end of the HPV-6b URR (see
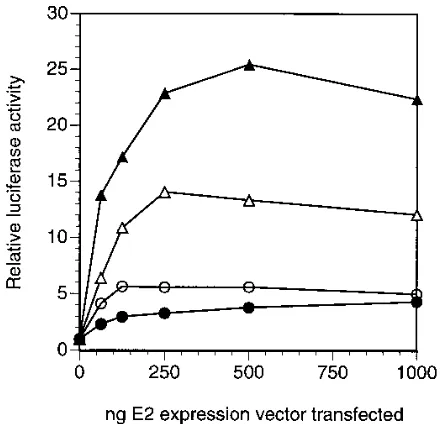
Materials and Methods for details). The sequences of these E2 binding sites and their positioning are highly conserved in genital HPVs; therefore, we would expect this reporter to be optimal for assaying the transcriptional activation function of E2 proteins. Nonetheless, in order to learn more about the mechanism of E2 function, we performed experiments to ex-plore the role of the number and geometry of E2 binding sites as determinants of transcriptional activation. As for the immu-noblot and gel shift analyses presented above, we focused on the BPV-1, HPV-16, and HPV-6b E2 proteins, with the last two serving as representatives of the proteins from high-risk and low-risk HPVs, respectively. As shown in Fig. 9A, more extensive titrations of E2 expression plasmids resulted in acti-vation patterns similar to those observed previously (Fig. 1A) when p2x2xE2BS-luc, the reporter with four E2 binding sites that was used in the experiments presented above, was
cotrans-fected. (Note the use of a log scale for the x-axis to allow a
better comparison of the activation properties of the BPV-1 and HPV E2 proteins.) Once again, both BPV-1 (dotted line) and HPV-16 (solid line) E2 proteins were found to be efficient transcriptional activators, but the amounts of input plasmid for optimal activation differed by at least 30-fold. Again, only weak
(;threefold) activation was observed upon expression of the
HPV-6b E2 protein (dashed line).
[image:7.612.68.290.67.286.2]When similar experiments were performed with p2xE2BS-luc, a reporter containing only a single copy of the region of the URR encompassing the two E2 binding sites, we found that activation by the BPV-1 and HPV-16 E2 proteins was signifi-cantly reduced (Fig. 9B). This is not surprising, since it has been noted that the E2 proteins are much more efficient acti-vators when the reporter construct contains an increased num-ber of the cognate DNA-binding sites (16, 24, 27, 38, 52). Interestingly, however, the extent of activation by the HPV-16
FIG. 7. Binding of the HPV-6b and HPV-16 E2 proteins to a probe contain-ing four E2 bindcontain-ing sites. Gel shift assays were performed as described in the legend to Fig. 6, except with a probe containing four E2 binding sites.
FIG. 8. Transcriptional activation by chimeric E2 proteins. Transfections of C33-A cells were performed as described in the legend to Fig. 1 with plasmids expressing the HPV-6b E2 protein (pCMV-6E2 [F]), the HPV-16 E2 protein (pCMV-16E2 [å]), or chimeric E2 proteins consisting of the HPV-6b E2 N-terminal activation domain fused to the HPV-16 E2 C-N-terminal DNA-binding domain (pCMV-6/16E2 [E]) or the HPV-16 E2 N-terminal activation domain fused to the HPV-6b E2 C-terminal DNA-binding domain (pCMV-16/6E2 [Ç]).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.323.543.454.666.2]E2 protein was reduced to the level obtained when HPV-6b was expressed (compare solid and dashed lines in Fig. 9B), indicating that the enhanced activity of the E2 proteins from high-risk HPVs is dependent upon the presence of more than a minimal E2-responsive element containing two E2 binding sites. In addition, the similarity in the extent of transcriptional activation when either the HPV-16 or HPV-6b E2 protein was expressed in the presence of p2xE2BS-luc confirms our results from immunoblots (Fig. 4), indicating that the activation dif-ferences seen with p2x2xE2BS-luc are not simply a result of differences in E2 protein levels.
Most surprisingly, we found that the activation potentials of the HPV-16 and BPV-1 E2 proteins were quite sensitive to the configuration of E2 binding sites. In addition to the reporters described above, we constructed p4xE2BS-luc, which contains an E2-responsive element consisting of four copies of essen-tially the same consensus E2 binding site as that found in p2xE2BS-luc and p2x2xE2BS-luc but in a different configura-tion (see schematic drawings above the graphs in Fig. 9C). We obtained very different results when this reporter was cotrans-fected with vectors for the expression of E2 proteins. The BPV-1 E2 protein was almost as active with this reporter as it was with p2x2xE2BS-luc (compare dotted lines in Fig. 9A and C), whereas the HPV-16 and HPV-6b E2 proteins were com-pletely inactive (solid and dashed lines in Fig. 9C). In fact, note that activation by the HPV E2 proteins was even lower with p4xE2BS-luc (Fig. 9C) than it was with p2xE2BS-luc (Fig. 9B), despite the presence of twice as many E2 binding sites. We interpret these results to mean that the HPV E2 proteins have a more stringent requirement for a particular configuration of E2 binding sites related to that found in their cognate URRs, while the BPV-1 E2 protein is better able to activate promoters containing E2 binding sites regardless of their configuration.
E2-mediated transcriptional regulation of HPV URRs.Since
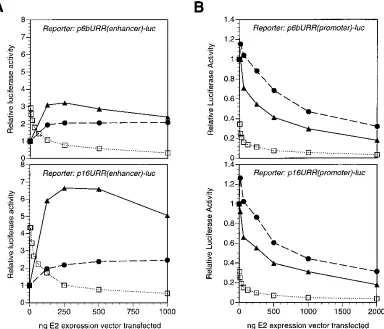
the experiments described above were performed with reporter plasmids containing minimal E2 binding sites cloned upstream of a heterologous SV40 promoter, we wanted to determine how the different E2 proteins would modulate transcription from reporters containing additional papillomavirus regulatory se-quences. We constructed p6bURR(enhancer)-luc and p16URR
[image:8.612.67.552.72.253.2](enhancer)-luc, which contain the URRs of 6b and HPV-16, respectively, cloned in an enhancer configuration upstream of the same SV40 promoter and luciferase coding regions present in p2x2xE2BS-luc and related plasmids. As shown in Fig. 10A, expression of the HPV-6b, HPV-16, or BPV-1 E2 pro-tein in the presence of either p6bURR(enhancer) or p16URR (enhancer) resulted in a pattern of transcriptional regulation very similar to what we observed with other reporters contain-ing only synthetic E2 bindcontain-ing sites, with the HPV-16 E2 protein being a stronger activator than the HPV-6b E2 protein and the BPV-1 E2 protein exhibiting an activation-squelching pattern. In fact, the only difference between E2-mediated regulation of the p6bURR and p16URR plasmids is a further enhancement of activation by the HPV-16 E2 protein on the homologous URR; otherwise, the results with these two reporters are es-sentially identical. This experiment demonstrates that the data obtained with the other reporters employed in our experiments are likely to be of physiological relevance for viral gene expres-sion.
While the E2 proteins have not been demonstrated to acti-vate transcription from natural HPV promoters, they have been shown to repress transcription from a promoter immedi-ately upstream of HPV E6 coding regions (5, 15, 44, 59, 60). In order to determine whether the differences we identified in the activation properties of the E2 proteins also extended to tran-scriptional repression, we constructed p6bURR(promoter)-luc and p16URR(promoter)-luc by cloning the URRs of HPV-6b and HPV-16 directly upstream of the luciferase coding region with no other promoter between them. As shown in Fig. 10B, these reporter constructs expressed luciferase in the absence of E2 protein expression and E2 expression resulted in dose-dependent inhibition of luciferase activity. The repression me-diated by the BPV-1 E2 protein was significantly greater than that by either the HPV E2 proteins tested, with the large extent of repression with small amounts of input plasmid mirroring the ability of the BPV-1 E2 protein to activate transcription when activatable reporters [i.e., p2x2xE2BS-luc and p16URR (enhancer)-luc] were used. On the other hand, we observed only a small difference between the extents of repression by the HPV-6b and HPV-16 E2 proteins. This is not unexpected,
FIG. 9. Dependence of transcriptional activation on the configuration of E2 binding sites in the reporter. Transfections of C33-A cells and analyses of luciferase andb-galactosidase activities were performed as described in the legend to Fig. 1, except that the E2-dependent reporters transfected were p2x2xE2BS-luc (A), p2xE2BS-luc (B), and p4xE2BS-luc (C). A schematic diagram of the configuration of E2 binding sites in the reporter is shown above each graph (see Materials and Methods for complete descriptions of the reporters). Note the use of a logarithmic scale for the amount of E2 expression vector transfected to provide a better comparison of the BPV-1 and HPV E2 activities.h, pCMV-BE2;F, pCMV-6E2;å, pCMV-16E2.
on November 9, 2019 by guest
http://jvi.asm.org/
since the differences in E2 transcriptional activation appear to map to the activation domain itself and not to the DNA-binding domain (Fig. 8) and since the DNA-DNA-binding properties of the HPV-6b and HPV-16 E2 proteins are similar (Fig. 6 and 7). Thus, if repression is largely a reflection of the E2 proteins binding to DNA in such a manner as to interfere with the binding of other transcription factors, the abilities of HPV-6b and HPV-16 E2 proteins to repress transcription should be roughly equivalent.
DISCUSSION
Our results demonstrate that the E2 proteins from a variety of genital HPVs and from the fibropapillomavirus BPV-1 are capable of activating transcription in vivo when assayed with various reporter constructs containing sequences derived from HPV URRs. Importantly, the resultant patterns of activation separate the E2 proteins into the following three classes: (i) strong activators which can result in the squelching of activa-tion at high expression levels (BPV-1), (ii) strong activators which do not result in squelching (HPV-16 and HPV-18), and (iii) weak activators (HPV-6b and HPV-11). The differences between the activities of the E2 proteins from high-risk HPVs (class II) and those from low-risk HPVs (class III) are not cell type specific and do not appear to result from differences in steady-state protein levels or DNA-binding properties. In-stead, the differences map primarily to the transcriptional
ac-tivation domain. Thus, the functional distinctions between these classes of E2 proteins are likely to result from down-stream steps in the activation process, such as cooperativity in the interactions between E2 proteins and cellular coactivators or basal transcription factors. We believe that these differences are likely to be of physiological importance in the respective viral life cycles.
Three categories of E2 transcriptional activators.
Papillo-mavirus E2 proteins have been reported to be activators and repressors of transcription (5, 9, 13, 15, 17, 25, 28–30, 40, 41, 54, 59, 60). Short BPV-1 E2 proteins not containing the N-terminal transcriptional activation domain can inhibit activa-tion by dimerizing with the full-length protein (10, 13, 17, 34, 62), and similar results have been obtained with HPV E2 proteins (6, 9, 13). In contrast, the roles of full-length E2 proteins in transcriptional regulation are less clear. A number of studies have suggested differences between the regulatory properties of full-length E2 proteins from different papilloma-viruses (6, 9, 29, 59–61). However, only the most recent of these studies (6, 61) have employed E2 expression constructs made equivalently with respect to different E2 coding regions. In any case, previous reports have focused on differences be-tween the BPV-1 E2 protein and the HPV-16 and/or HPV-18 E2 protein and have not systematically addressed the question of whether different HPV E2 proteins have distinguishable activities. We have now compared the in vivo activation
func-FIG. 10. Transcriptional regulation of HPV URRs by E2 proteins. Transfections and subsequent procedures and analyses were performed as described in the legend to Fig. 1, except that the reporter plasmids contained the URRs of HPV-6b (top) or HPV-16 (bottom) cloned either in an enhancer configuration upstream of the SV40 early promoter and the luciferase coding region (A) or in a promoter configuration directly upstream of the luciferase coding region (B).h, pCMV-BE2;F, pCMV-6E2;
å, pCMV-16E2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.121.505.71.400.2]tions of the BPV-1, HPV-6b, HPV-11, HPV-16, and HPV-18 E2 proteins by cloning the relevant ORFs into mammalian expression vectors in an identical fashion, and the results of these experiments have allowed us to divide these E2 proteins into three classes.
Class I E2 proteins, exemplified by the BPV-1 E2 protein, are effective transcriptional activators of E2-dependent report-ers containing E2 binding sites arranged in different configu-rations. We found that the BPV-1 E2 protein activated tran-scription almost equally well from p2x2xE2BS-luc, which contains four E2 binding sites in a configuration closely related to that in the URRs of genital HPVs, and from p4xE2BS-luc, which contains four E2 binding sites in an arbitrary configura-tion (Fig. 9). In comparison with the HPV E2 proteins, the BPV-1 E2 protein also accumulated to much higher steady-state levels after transfection into C33-A cells. Since the ex-pression plasmids used are identical outside of the E2 coding regions, this difference indicates that the HPV E2 mRNAs and/or proteins are likely to be much less stable in human cells than is their BPV-1 counterpart. It could well be of importance for the viral life cycle that E2 proteins do not accumulate to high levels in infected cells, since this might cause inappropri-ate transcriptional regulation of the viral genome. One mech-anism for ensuring that E2 proteins do not accumulate to high levels in vivo would be for them to have sequences which ensure high rates of degradation, and the BPV-1 E2 protein might not have a sequence which would be recognized as such in human cells. Whatever the mechanism of its accumulation, BPV-1 E2 protein expression resulted in the squelching of activation when moderate to large amounts of pCMV-BE2 were transfected (Fig. 1, 5, 9, and 10). The pattern of activation with small amounts of input plasmid in transfections followed by the squelching of activation with larger amounts was ob-served even with reporter constructs containing complete URRs as the E2-dependent reporter element (Fig. 10), dem-onstrating that this aspect of BPV-1 E2 function is not ob-served solely with reporters containing synthetic E2 binding sites. Squelching has been observed in a number of other systems and has been postulated to result from the titration of limiting coactivators or basal transcription factors by transcrip-tional activators which are not bound to DNA (see reference 42 for a review). We expect that this is the cause of squelching by high-level expression of the BPV-1 E2 protein, but since transcription from control plasmids not containing
E2-depen-dent promoters (i.e., pCMV-b-gal) was not squelched, the
factor being titrated is not one required for activity from all promoters.
Class II E2 proteins, which include those from the high-risk viruses HPV-16 and HPV-18, are also effective transcriptional activators; however, they have a more stringent requirement for a particular configuration of E2 binding sites. As shown in Fig. 9, the HPV-16 E2 protein efficiently activated the HPV URR-derived construct p2x2xE2BS-luc but was completely in-active on p4xE2BS-luc, which contains a different arrangement of four E2 binding sites. The HPV-16 E2 protein was also only weakly active on p2xE2BS-luc, which contains two E2 binding sites in the configuration found in the HPV URRs. These results demonstrate that efficient activation by class II E2 pro-teins requires more than the minimal two E2 binding sites. Since we do not have any evidence for greater cooperativity in the binding of the HPV-16 E2 protein than of the HPV-6b E2 protein to probes containing four E2 binding sites in an opti-mal configuration (Fig. 7), we interpret these results to mean that a particular arrangement of multiple dimers of class II E2 proteins forms a complex at the promoter which is especially effective at activating the cellular transcription machinery. In
any case, the HPV-16 E2 protein was also more active than was the HPV-6b E2 protein when assayed with reporters contain-ing complete HPV URRs (Fig. 10). In regard to the qualitative and quantitative differences between activation by class I and class II E2 proteins, we demonstrated that the BPV-1 E2 protein accumulated to higher concentrations than did the HPV E2 proteins after transfection (Fig. 4 and 5 and data not shown). However, this is not a complete explanation for the differences between class I and class II E2 proteins. In fact, Bedrosian and Bastia (4) previously showed that the BPV-1 E2 protein binds to DNA with a less stringent sequence specificity than does the HPV-16 E2 protein. Such differences are likely to be important as determinants of E2 function.
Class III E2 proteins, which include those from the low-risk viruses HPV-6b and HPV-11, were found to be only weak transcriptional activators in vivo. The HPV-6b E2 protein was expressed at levels comparable to those of the HPV-16 E2 protein (Fig. 4), and it was not impaired in its ability to bind DNA in any obvious way (Fig. 6 and 7). These results implicate the transcriptional activation domain as being the important determinant of E2 activity in vivo, and data from expression of chimeras of the HPV-6b and HPV-16 E2 proteins support this model (Fig. 8). Since we also found that the HPV-6b E2 pro-tein was less active than was the HPV-16 E2 propro-tein when assayed with reporter constructs containing complete URRs from either HPV-6b or HPV-16 cloned in an enhancer config-uration (Fig. 10), we believe that the much lower activation potentials of category III E2 proteins are of physiological sig-nificance.
Mechanisms of E2 function.The E2 proteins are unusual
transcriptional activators in at least one respect: activation apparently requires a minimum of not only two E2 binding sites (16, 24, 27, 52) but also a nearby binding site for a cellular transcription factor, such as Sp1 or AP-1 (22, 35, 61). This requirement for a cellular factor is not very specific, as a num-ber of different factors can fulfill this role (22, 61). The BPV-1 E2 protein has also been demonstrated to interact with the TATA-binding protein (15, 24, 43, 53) as well as the basal transcription factor TFIIB (43). It will be of importance for a mechanistic understanding of E2 function to determine which of these (or other) protein-protein interactions involving E2 differ for the classes of E2 proteins that we have identified. Similarly, it will be of interest to determine whether these differences in E2 function with respect to transcriptional acti-vation have any parallel in the role of the E2 proteins in viral genome replication. Studies performed to date have indicated that the function of E1 and E2 in promoting DNA replication is conserved across different papillomavirus types (8, 14, 57), but potentially significant differences in the efficiency of repli-cation with origins of replirepli-cation and/or E1 and E2 proteins from different papillomaviruses were also observed in these studies. In addition, replication of the evolutionarily more dis-tant HPV-1a was reported not to require the E2 protein (19), indicating that there are likely to be important differences in the requirements for viral proteins in the replication of papil-lomaviruses. Given the tight links between transcription and replication in many systems, particularly viral ones, we expect that the differences in E2 transcriptional activity that we have identified might have relevance for the control of replication as well.
Physiological relevance of E2 activity differences.Because of
the difficulty involved in recapitulating the HPV life cycle in vitro, the roles of the HPV E2 proteins in their respective viral life cycles are unclear. Transcription start sites in HPVs have
been mapped to a few sites in the region extending from the 39
end of the URR to the 39end of the E7 ORF (11, 32, 46, 50,
on November 9, 2019 by guest
http://jvi.asm.org/
51, 55, 58). While previous studies have shown that E2 proteins can be effective repressors of transcription from papillomavi-rus promoters (5, 15, 44, 59, 60), transcription from promoters in their natural configurations in HPV genomes has not as yet been demonstrated to be activated by E2 proteins in any sys-tem. We consider it unlikely that the HPV E2 proteins do not activate transcription from their own viral genomes during the normal course of infection, since the transcriptional activation domain is highly conserved between papillomaviruses and vi-ruses with such small genomes are unlikely to retain unneces-sary coding sequences. Instead, we favor the model that E2 proteins become transcriptional activators at the later stages of viral infection which have been experimentally difficult to study. Hummel et al. (32) have mapped a transcription start site in HPV-31b which is induced after the differentiation of keratinocytes in an organotypic raft system; it is possible that E2 activates transcription from this promoter or other promot-ers in conjunction with keratinocyte-specific differentiation factors, and our results indicate that there could be important differences between the interactions of E2 proteins from high-risk and low-high-risk HPVs with these cellular factors. Our results further suggest that while the E2 proteins from high-risk HPVs may function somewhat more effectively as transcriptional re-pressors than do the E2 proteins from low-risk HPVs, the more prominent difference is in transcriptional activation and that this has potentially significant implications for the role of the E2 proteins in HPV-infected cells.
In summary, we have found that there are substantial qual-itative and quantqual-itative differences between E2 proteins. These differences correlate with the biological activities of the respec-tive papillomaviruses and are likely to be important in the different roles that the proteins perform in the viral life cycles. It will be of interest to map the sequence and structural de-terminants of these differences, to determine which protein-protein interactions are affected, and to investigate the physi-ological relevance of these differences in papillomavirus-infected cells.
ACKNOWLEDGMENTS
We thank Elliot Androphy for the gift of antibodies against the BPV-1 E2 protein, David Breiding for the pCGE2B construct, and Peter Ghazal for U373 MG cells. We also thank Felix Wettstein for a critical reading of the manuscript, Paul Todd for helping with the preparation of plasmids used for transfections, and Bernd Stein for his assistance in the preparation of figures.
This work was supported in part by grant CA55703 from the Na-tional Cancer Institute to M.S.B.
REFERENCES
1.Androphy, E. J., D. R. Lowy, and J. T. Schiller.1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature (London)325:70–73.
2.Barbosa, M. S., and R. Schlegel.1989. The E6 and E7 genes of HPV-18 are sufficient for inducing two-stage in vitro transformation of human keratino-cytes. Oncogene4:1529–1532.
3.Barbosa, M. S., W. C. Vass, D. L. Lowy, and J. T. Schiller.1991. In vitro biological activities of the E6 and E7 genes vary among human papilloma-viruses of different oncogenic potential. J. Virol.65:292–298.
4.Bedrosian, C. L., and D. Bastia.1990. The DNA-binding domain of HPV-16 E2 protein interaction with the viral enhancer: protein-induced DNA bend-ing and role of the nonconserved core sequence in bindbend-ing site affinity. Virology174:557–575.
5.Bernard, B. A., C. Bailly, M. C. Lenoir, M. Darmon, F. Thierry, and M. Yaniv.1989. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J. Virol.63:4317–4324.
6.Bouvard, V., A. Storey, D. Pim, and L. Banks.1994. Characterization of the human papillomavirus E2 protein: evidence of activation and trans-repression in cervical keratinocytes. EMBO J.13:5451–5459.
7.Brewer, C. B., and M. G. Roth.1991. A single amino acid change in the
cytoplasmic domain alters the polarized delivery of influenza virus hemag-glutinin. J. Cell Biol.114:413–421.
8.Chiang, C. M., M. Ustav, A. Stenlund, T. F. Ho, T. R. Broker, and L. T. Chow.1992. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA89: 5799–5803.
9.Chin, M. T., R. Hirochika, H. Hirochika, T. R. Broker, and L. T. Chow.1988. Regulation of human papillomavirus type 11 enhancer and E6 promoter by activating and repressing proteins from the E2 open reading frame: func-tional and biochemical studies. J. Virol.62:2994–3002.
10. Choe, J., P. Vaillancourt, A. Stenlund, and M. Botchan.1989. Bovine pap-illomavirus type 1 encodes two forms of a transcriptional repressor: struc-tural and functional analysis of new viral cDNAs. J. Virol.63:1743–1755. 11. Chow, L. T., M. Nasseri, S. M. Wolinski, and T. R. Broker.1987. Human
papillomavirus types 6 and 11 mRNAs from genital condylomata acuminata. J. Virol.61:2581–2588.
12. Cole, S. T., and O. Danos.1987. Nucleotide sequence and comparative analysis of the human papillomavirus type 18 genome: phylogeny of papil-lomaviruses and repeated structure of the E6 and E7 gene products. J. Mol. Biol.193:599–608.
13. Cripe, T. P., T. H. Haugen, J. P. Turk, F. Tabatabai, P. G. Schmid, M. Durst, L. Gissmann, A. Roman, and L. P. Turek.1987. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratinocyte-dependent enhancer and by viral E2 trans-activator and repressor gene products: im-plications for cervical carcinogenesis. EMBO J.6:3745–3753.
14. Del Vecchio, A. M., H. Romanczuk, P. M. Howley, and C. C. Baker.1992. Transient replication of human papillomavirus DNAs. J. Virol.66:5949– 5958.
15. Dostatni, N., P. F. Lambert, R. Sousa, J. Ham, P. M. Howley, and M. Yaniv. 1991. The functional BPV-1 E2 trans-activating protein can act as a repres-sor by preventing formation of the initiation complex. Genes Dev.5:1657– 1671.
16. Gauthier, J. M., N. Dostatni, M. Lusky, and M. Yaniv.1991. Two DNA-bound E2 dimers are required for strong transcriptional activation and for cooperation with cellular factors in most cells. New Biol.3:498–509. 17. Giri, I., and M. Yaniv.1988. Structural and mutational analysis of E2
trans-activating proteins of papillomaviruses reveals three distinct functional do-mains. EMBO J.7:2823–2829.
18. Giri, I., and M. Yaniv.1988. Study of the E2 gene product of the cottontail rabbit papillomavirus reveals a common mechanism of transactivation among papillomaviruses. J. Virol.62:1573–1581.
19. Gopalakrishnan, V., and S. A. Khan.1994. E1 protein of human papilloma-virus type 1a is sufficient for initiation of viral DNA replication. Proc. Natl. Acad. Sci. USA91:9597–9601.
20. Grossel, M. J., F. Sverdrup, D. E. Breiding, and E. J. Androphy.1996. Transcriptional activation function is not required for stimulation of DNA replication by bovine papillomavirus type 1 E2. J. Virol.70:7264–7269. 21. Guan, K. L., and J. E. Dixon.1991. Eukaryotic proteins expressed in
Esch-erichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathioneS-transferase. Anal. Biochem.192:262–267. 22. Ham, J., N. Dostatni, F. Arnos, and M. Yaniv.1991. Several different up-stream promoter elements can potentiate transactivation by the BPV-1 E2 protein. EMBO J.10:2931–2940.
23. Ham, J., N. Dostatni, J. M. Gauthier, and M. Yaniv.1991. The papilloma-virus E2 protein: a factor with many talents. Trends Biochem. Sci.16:440– 444.
24. Ham, J., G. Steger, and M. Yaniv.1994. Cooperativity in vivo between the E2 transactivator and the TATA box binding protein depends on core promoter structure. EMBO J.13:147–157.
25. Haugen, T. H., T. P. Cripe, G. D. Ginder, M. Karin, and L. P. Turek.1987. Trans-activation of an upstream early gene promoter of bovine papilloma virus-1 by a product of the viral E2 gene. EMBO J.6:145–152.
26. Haugen, T. H., L. P. Turek, F. M. Mercurio, T. P. Cripe, B. J. Olson, R. D. Anderson, D. Seidl, M. Karin, and J. Schiller.1988. Sequence-specific and general transcriptional activation by the bovine papillomavirus-1 E2 trans-activator require an N-terminal amphipathic helix-containing E2 domain. EMBO J.7:4245–4253.
27. Hawley-Nelson, P., E. J. Androphy, D. R. Lowy, and J. T. Schiller.1988. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J.7:525–531.
28. Hermonat, P. L., B. A. Spalholz, and P. M. Howley.1988. The bovine papillomavirus P2443 promoter is E2 trans-responsive: evidence for E2 au-toregulation. EMBO J.7:2815–2822.
29. Hirochika, H., T. R. Broker, and L. T. Chow.1987. Enhancers andtrans -acting E2 transcriptional factors of papillomaviruses. J. Virol.61:2599–2606. 30. Hirochika, H., R. Hirochika, T. R. Broker, and L. T. Chow.1988. Functional mapping of the human papillomavirus type 11 transcriptional enhancer and its interaction with the trans-acting E2 proteins. Genes Dev.2:54–67. 31. Hubbert, N. L., J. T. Schiller, D. R. Lowy, and E. J. Androphy.1988. Bovine
papilloma virus-transformed cells contain multiple E2 proteins. Proc. Natl. Acad. Sci. USA85:5864–5868.
32. Hummel, M., J. B. Hudson, and L. A. Laimins.1992. Differentiation-induced