0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
A Preferred Region for Recombinational Patch Repair in the 5
9
Untranslated Region of Primer Binding Site-Impaired Murine
Leukemia Virus Vectors
JACOB GIEHM MIKKELSEN,1ANDERS H. LUND,1KAREN DYBKÆR KRISTENSEN,1
MOGENS DUCH,1MICHAEL SCHANDORF SØRENSEN,1POUL JØRGENSEN,1
ANDFINN SKOU PEDERSEN1,2*
Department of Molecular Biology1and Institute of Medical Microbiology and Immunology,2
University of Aarhus, DK-8000 Aarhus, Denmark
Received 25 September 1995/Accepted 15 November 1995
Transduction of primer binding site-impaired Akv murine leukemia virus-based retroviral vectors from the murine packaging cell linesC-2 andVE was studied. The efficiency of transduction of theneomarker of all mutated constructs was found to decrease by 5 to 6 orders of magnitude compared with that of the wild-type vector. Thirty-two of 60 transduced proviruses analyzed harbored a primer binding site sequence matching a glutamine tRNA primer. Sequence analysis of the regions flanking the glutamine tRNA primer binding site revealed a distinct pattern of nucleotide differences from the Akv-based vector, suggesting the involvement of a specific endogenous virus-like sequence in patch repair rescue of the primer binding site mutants. The putative recombination partner RNA was found in virions fromC-2 cells as detected by analysis of glutamine tRNA-initiated cDNA and by sequence analysis of regions at or around the glutamine tRNA primer binding site. We propose that the forced recombination of primer binding site mutants involves initial priming on endogenous viral sequences and requires template switching during minus-strand synthesis in the region between theneogene and the mutated primer binding site to allow correct second-strand transfer in reverse transcription. The system thereby selects for a reverse transcriptase-mediated recombination event in the 5* untranslated region. A panel of sequence differences between the recombination partners in this region has allowed mapping of the site of recombination for each transduction event. Interestingly, the majority of the recombination events were clustered within a narrow, 33-nucleotide region thought to be involved in genomic RNA dimerization.
Reverse transcription-mediated recombination is an impor-tant source of genetic variability in retroviruses (4). Retroviral particles carry a genomic RNA dimer consisting of two plus-strand RNAs noncovalently linked near the 59end of the viral genome, and copackaging of genetically distinct RNAs in a heterodimer is a prerequisite for observing retroviral recom-bination (15, 18, 42). Recomrecom-bination may result from template switching during minus- or plus-strand synthesis in reverse transcription (12, 16, 19, 30). However, the character of the first DNA strand transfer of the minus-strand strong-stop DNA also influences genetic variability, since both intra- and interstrand transfers have been reported (16, 17, 35). Studies of rare transductional patterns demonstrate that recombina-tion may involve viral RNA of both exogenous and endoge-nous origins (5, 6, 9, 26, 28, 39), as well as nonviral RNA, occasionally leading to the generation of highly oncogenic ret-roviruses (43, 44, 50). Endogenous viruses represent a rich source of functional retroviral sequences, and recombination with endogenous virus-like sequences may represent a major rescue mechanism for viral mutants. Thus, in murine systems, VL30 endogenous elements (28) and endogenous murine leu-kemia virus (MLV)-like sequences (5, 39) have been shown to be involved in recombinational repair of mutations affecting viral integration.
Initiation of reverse transcription is primed by the 39end of
a tRNA molecule annealed to the 18-nucleotide primer bind-ing site (PBS), which is located directly downstream from the U5 region of the retroviral genome. In MLVs a proline tRNA molecule matching PBS-Pro is utilized as a primer. However, proviral MLV-like sequences endogenous to the murine ge-nome harbor PBS sequences matching the 39terminus of glu-tamine tRNA (5, 6, 31, 33), as members of the VL30 retroele-ment family have PBS sequences matching tRNAGly(32). We
have previously shown that MLVs harboring PBSs perfectly matching tRNA3
Lysor tRNA 1
Glnreplicate with high efficiencies;
hence, different PBS-tRNA combinations may support viral replication (24). The interaction between the PBS and the tRNA 39terminus appears to be the primary determinant for primer selection in MLV reverse transcription, although a per-fect PBS-tRNA match is not required for replication (24). Among a panel of PBS mutants that were generated in our laboratory to investigate PBS-tRNA interactions by single-cy-cle vector replication, some were found to be strongly inhibited in viral replication. These mutants enabled us to selectively study rare transductional events in MLV replication. DNA sequence analysis of transduced proviruses revealed that rep-lication of PBS-mutated vectors involved aberrant reverse transcription or reverse transcription-mediated recombination. In this report we focus on the specific recombination pro-cesses that led to rescue of PBS-impaired vectors. Apparently, all recombinants have used the same endogenous virus-like sequence for patch repair. This MLV-like endogenous virus (MLEV) sequence harbors a functional PBS-Gln, as previously detected in a patch repair revertant of an integration-defective MLV mutant (5). We report that RNA of MLEV is found in * Corresponding author. Mailing address: Department of Molecular
Biology, University of Aarhus, C.F. Moellers Alle´, Bldg. 130, DK-8000 Aarhus, Denmark. Fax: 45 86196500. Electronic mail address: fsp@ mbio.aau.dk
1439
on November 9, 2019 by guest
http://jvi.asm.org/
particles released from the packaging cells, and we propose that the observed patch repair of PBS-mutated vectors involves initial priming on endogenous viral sequences and transfer to the 39end of the vector RNA, followed by template switching in the 59 untranslated region (59 UTR) during minus-strand synthesis. Our studies allowed us to map and quantify individ-ual recombination events and, most interestingly, point to a preferred region for recombination within target sequences of retroviral origin.
MATERIALS AND METHODS
Vector construction.PBS-modified retroviral vectors were generated by PCR-mediated site-specific mutagenesis as previously described (24). Mutagenesis was performed on the Akv MLV-based retroviral vector ptvAkv-neo (34), containing the Akv MLV long terminal repeats, the 59255 bp of the 59UTR, and the neomycin resistance gene (neo) flanked downstream by 480-bp Akv MLV se-quences. Briefly, mutations were introduced by a two-step PCR procedure: first, the 59part of the vector was amplified, generating a 618-bp fragment carrying the desired mutations in the PBS, and second, the mutated 59fragment was con-nected by PCR with the 39part of the vector. The amplified vectors carrying the mutated PBSs were digested with the appropriate restriction enzymes and cloned into the pUC19 cloning plasmid. PCR was performed as previously described (24). Taq polymerase and PCR buffer were obtained from Stratagene, and deoxynucleoside triphosphates were purchased from Pharmacia; all PCRs were performed in a water-based thermal cycler (DNA Racer; Microlab, Aarhus, Denmark).
Oligonucleotides.The following oligonucleotides matching Akv MLV posi-tions 135 to 181 (47) were used for introduction of modified PBS sequences into ptvAkv-neo: 1, 59-CCTGGGCGGGGGTCTCCAACGCGTTAGATTCATCCC AAATGAAAGAC-39, introducing PBS-XXX; 2, 59-CCTGGGCGGGGGTCTC CAAGGGGTTCGAATCAGGTTTAATGAAAGAC-39, introducing PBS-UMU; 3, 59-CCTGGGCGGGGGTCTCCAAGCTGGGCCCATAACCAATGAAAGA C-39, introducing PBS-Met(i)intP; and 4, 59-CCTGGGCGGGGGTCTCCAAGCT GGACCCATAACCAATGAAAGAC-39, introducing PBS-Met(i)intM (mutations generating PBS modifications are underlined). Other primers employed in the mutagenesis procedure have been described previously (24). Oligonucleotides used in sequence analysis were as follows: 5, 59-TTCATAAGGCTTAGC CAGCTAACTGCAG-39, matching Akv MLV positions 7838 to 7865 (47); 6, 59-GGCGCCCCTGCGCTGACAGCCGGAACAC-39, matching positions 1656 to 1683 of the neo gene (3); 7, 59-CGCAGGCGCAAAAAGTAGATGC-39, matching Akv MLV positions 268 to 289 (47); and 8, 59-TCCGAATCGTG GTCTCGCTGATCCTTGG-39, matching Akv MLV positions 69 to 96 (47). The following oligonucleotides were used in semi-random-primed PCR (40): 9, 59-CTAGTTCAATTCGCGGCCGCTGGAGGTCCCACCGAGAT-39, matching PBS-Gln2; 10, 59-CGTCGGGAGGTAAGC-39, matching Akv MLV positions 196 to 210; and the degenerate primer 11, 59-CAGTTCAAGCTTGTCCAG GAATTCNNNNNGCGCT-39. Biotinylated primer 12, 59-GGGAGCTCTAG AGGT(C/T)CCATGGTGTAATGGTTAG-39, matching murine glutamine tRNA positions 1 to 22 (41), was used in reverse transcription-PCR. This primer recognizes the 59acceptor stem of tRNA1Glnand tRNA2Glnmolecules distinctive
in one nucleotide position within the 59acceptor stem, one position within the 39 acceptor stem, and one position within the anticodon loop (41). All oligonucle-otides were purchased from DNA Technology ApS, Aarhus, Denmark.
Cell culture, transfection, and virus infection.C-2 (25),VE (27), and NIH 3T3 cells were grown in Dulbecco modified Eagle medium with Glutamax-1 (Gibco BRL, Life Technologies) supplemented with 10% newborn calf serum, 100 U of penicillin per ml, and 100mg of streptomycin per ml. Cells were incubated at 378C in 90% relative humidity and 5.7% CO2. Transfections of virus
producer cell lines were performed by the calcium phosphate method (13), omitting a glycerol shock and leaving the Ca phosphate DNA precipitate on the cells for 12 to 16 h. At 48 h posttransfection, medium containing G418 (0.6 mg/ml) (active compound; Sigma) was added in order to select for stably inte-grated PBS-modified retroviral vectors. G418-resistant colonies appearing after 12 days of selection were pooled. Transductional efficiencies were determined as follows. Producer cells were seeded at maximum density (105cells per cm2) and
allowed to attach; the medium was renewed and left on the cells for 24 h. Virus-containing medium was centrifuged at 5003g to pellet cell debris, filtered
through a sterile 0.45-mm-pore-size filter, serially diluted, and eventually trans-ferred to NIH 3T3 target cells (seeded at 53103
cells per cm2
24 h preinfection) in the presence of Polybrene (6mg/ml; Aldrich Chemical Co., Inc). Medium containing G418 (0.6 mg/ml) was added at 48 h postinfection; resistant colonies were counted, individually isolated, and expanded after 10 days of selection.
Sequence analysis of proviral DNA.Genomic DNA from G418-resistant col-onies was prepared by freeze-thaw treatment and subsequent incubation in the presence of trypsin as previously described (24). The upstream U5 region, the PBS, and the 59UTR upstream from the neo gene of integrated proviruses were sequenced in order to characterize individual transduction events. By utilizing primers 5 and 6 (see above) for PCR amplification of vector sequences, sequence
analysis was performed on both the minus and plus strands of the transduced vector; biotinylated primers 5 and 6 were used in the PCR prior to sequencing of the plus strand and the minus strand, respectively, enabling Dynabead (Dynal Inc.)-mediated purification of PCR fragments. Primer 7, matching part of the 59 UTR, and primer 8, matching the upstream U5 region, were used as sequencing primers in sequence analysis of the plus and minus strands, respectively, which was performed on an automated DNA sequencer (373A DNA sequencer; Ap-plied Biosystems Inc.).
Virion RNA preparation and sequence analysis.Virus-containing medium from nontransfectedC-2 producer cells (seeded at maximum density the day prior to virus harvest) was cleared of cell debris by centrifugation and filtration through a 0.45-mm-pore-size filter; subsequently, virus particles were pelleted at 17,0003g in Eppendorf tubes for 1 h at 48C. Total viral RNA was prepared by adding 700ml of GTC buffer (4 M guanidinium thiocyanate, 25 mM sodium citrate, 0.1 M 2-mercaptoethanol) to pelleted virions; RNA was extracted the addition of 70ml of 2 M sodium acetate (pH 4), 700ml of water-saturated phenol, and 280ml of chloroform-isoamyl alcohol (49:1) and precipitated with isopro-panol. The RNA pellet was resuspended in 500ml of GTC buffer, reprecipitated, and washed prior to cDNA synthesis.
Reverse transcription-PCR-mediated amplification of the hybrid of the endo-genous U5 region as part of the minus-strand strong-stop DNA and the glu-tamine tRNA molecule annealing to the endogenous PBS sequence was per-formed by using primers 8 and 12. Prior to amplification, elongation from the annealed tRNAGlnprimer was allowed to occur by incubating viral RNA at 378C
for 1 h in the presence of Moloney MLV reverse transcriptase provided in a First-Strand cDNA Synthesis Kit (Pharmacia). Primer 8 was used as a sequenc-ing primer in sequence analysis of the amplified hybrid. Sanger dideoxy manual sequencing was carried out by using a Sequenase version 2.0 DNA sequencing kit (U.S. Biochemical Corp.) according to the manufacturer’s directions; sequence reaction mixtures were electrophoresed in a 6% denaturing polyacrylamide se-quencing gel. Random-primed cDNA synthesis was carried out with a First-Strand cDNA Synthesis Kit (Pharmacia) by following the directions of the manufacturer; first-strand synthesis was primed by a random hexadeoxynucle-otide primer provided in the kit. A semi-random-primed PCR approach was employed to selectively amplify PBS-Gln-containing endogenous sequences; the reactions were carried out as previously described (40) with the degenerate primer 11 and primer 10 as a sequencing primer. Structural analysis of the PBS and surrounding sequences of MLEV was performed by using primers 7 and 8 for PCR on viral cDNA and primer 7 as a sequencing primer.
RESULTS
Design of PBS-modified retroviral vectors. To investigate tRNA-PBS interaction during initiation of reverse transcrip-tion in MLV, nucleotide modificatranscrip-tions were introduced into the PBS of the Akv MLV-based transmission vector
ptvAkv-neo (34). During processing of cellular tRNA molecules in
eukaryotes, a CCA terminus (CCA tail) is added to the 39end of the molecule (reviewed in reference 8). In order to assess the implication of the conserved 39 CCA tail in initiation of reverse transcription, a PBS sequence was designed to retain the nucleotides complementing the 39CCA tail without having a complete match with the 39 end of any known tRNA. The design was based on an alignment of the 3918 nucleotides of all known murine tRNA molecules and tRNA genes (41); thus, generation of the modified PBS sequence (designated PBS-XXX) from PBS-Pro required modification of 13 of 18 nucle-otides. Also, a PBS sequence unlikely to match any tRNA molecule was generated; all positions in PBS-Pro were altered in order to establish this sequence, designated PBS-UMU. A cleaved form of cellular tRNAMet(i)has been reported to serve
as a primer for initiation of reverse transcription in Drosophila copia virus-like particles (20). To investigate whether this al-ternative priming mechanism involving no tRNA 39CCA tail mediates initiation of reverse transcription in MLV, we intro-duced a PBS sequence, PBS-Met(i)int, matching 15 bases of the anticodon arm corresponding to the 39-terminal nucleo-tides of the cleaved tRNAMet(i)molecule, in ptvAkv-neo. Two
PBS-Met(i)int constructs were designed, one with a perfect match to the internal tRNAMet(i) [PBS-Met(i)intP] and one
introducing a single nucleotide mismatch [PBS-Met(i)intM]; the latter was generated in order to be able to prove specific tRNA primer usage according to the most widely accepted model for reverse transcription (10).
1440 MIKKELSEN ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
PBS modifications were introduced into ptvAkv-neo by a two-step PCR procedure as previously described (24) and were analyzed with the wild-type PBS-Pro vector as a positive con-trol.
Restricted transduction of PBS-modified vectors.To study replication capacity and the consequences of introducing the mutated PBS sequences, we measured transductional efficiency fromC-2 orVE packaging cells stably transfected with retro-viral vectors harboring Pro, XXX, UMU, PBS-Met(i)intP, or PBS-Met(i)intM. TheC-2 andVE cell lines are NIH 3T3 cell-derived fibroblast cell lines harboring Moloney MLV-based packaging constructs. The transductional titer was determined by counting the number of G418-resistant colonies appearing per milliliter of virus-containing medium transferred to the target cells. As shown in Fig. 1, we observed a dramatic decrease in the titers of all PBS-modified vectors compared with that of the PBS-Pro control vector; thus, a high PBS-Pro titer (.106 CFU/ml) was obtained, while an approximately
105-fold decrease was observed for all mutated vector
con-structs (titers ranging from 0.13101to 6.83101CFU/ml).
Hence, neither retention of the three positions matching the 39 tRNA terminus nor inclusion of a PBS matching the internal primer fragment of tRNAMet(i)supports efficient replication in
an MLV-based vector system.
Sequence analysis of transduced PBS sequences.Because of the very low transduction efficiency observed for the various PBS-modified vectors, it was of interest to analyze the struc-tures of the transduced PBS sequences. Since our protocol for measurement of transfer capability did not involve passaging of G418-resistant target cells, we were able to study individual transduction events by separately analyzing resistant colonies. A 1.3-kb fragment was PCR amplified from genomic DNA prepared from individual colonies by using a primer set that
allowed amplification of only proviruses originating from rep-lication of the PBS-mutated vectors (24). The amplified frag-ments were subjected to automated DNA sequence analysis.
As expected, sequence analysis of a large number of resis-tant colonies indeed revealed rare transductional mechanisms. Thus, we found that the majority of the integrated proviruses (referred to as type 1 proviruses) harbored a PBS sequence matching the 39 terminus of tRNA1
Gln or tRNA 2
Gln (Fig. 1).
Sixteen of 22 PBS-Gln-harboring clones, shown in Fig. 2, car-ried PBSs matching PBS-Gln2 (i.e., they had a C residue in nucleotide position 152); four clones harbored PBSs with a perfect match to the other glutamine tRNA molecule, tRNA2
Gln(i.e., they had a T residue in nucleotide position 152);
and the remaining two clones contained mixed PBS-Gln1,2 sequences.
Among the remaining proviruses (referred to as type 2 pro-viruses), some harbored PBS sequences matching the original vector; for PBS-XXX, PBS-UMU, and PBS-Met(i)intP and -M constructs, 11 of 40, 2 of 6, and 3 of 14 resistant colonies, respectively, had retained the original PBS. Furthermore, we found that proviruses transduced from the PBS-XXX vector harbored mixtures of PBS-Pro and PBS-XXX (5 of 40) or PBSs matching PBS-Pro (7 of 40), both of which indicate specific tRNAProprimer usage.
[image:3.612.66.551.75.324.2]Proviruses with PBS-Gln generated by recombinational patch repair.We examined the nucleotide sequences flanking the transduced PBSs to verify whether they were of vector origin or derived from endogenous sequences. For the type 2 proviruses the sequences flanking the PBS were of vector or-igin. Further structural analyses and assessment of possible mechanisms for transduction of the type 2 proviruses are a subject of ongoing investigations. Here we concentrate on transduction of the type 1 proviruses.
FIG. 1. Structures and functions of PBS-modified vectors. Mutations were introduced into the PBS of transmission vector tvAkv-neo by PCR-mediated site-directed mutagenesis. Modified PBS sequences are boldface type. Transduction titers were measured by counting the G418-resistant colonies appearing per milliliter of virus-containing medium transferred from stably transfectedC-2 orVE packaging cells to NIH 3T3 target cells. Ratios of transduced PBS-Gln1,2 sequences (Gln1,2/total) are given as the number of transduced vectors harboring PBS-Gln1,2 relative to the total number of vector sequences analyzed in G418-resistant colonies. Titers obtained for PBS-Pro, PBS-XXX, PBS-UMU, and PBS-Met(i)intP were all based on three experiments, while the titer for PBS-Met(i)intM was based on one experiment.
on November 9, 2019 by guest
http://jvi.asm.org/
Sequence analysis of the type 1 proviruses revealed a specific pattern of mutations in the upstream U5 region and the 59 UTR between PBS and neo, including deletions, insertions, and substitutions, compared with the sequence of Akv MLV (Fig. 2). Specifically, the PBS-flanking sequences did not cor-respond to the Moloney MLV sequences of the packaging constructs (data not shown). The mutational patterns flanking PBS-Gln1,2 were similar for all transduced vector sequences and also were related to an endogenous sequence (Fig. 2) recovered by Colicelli and Goff (5) in a recombinational re-vertant of a retroviral mutant in NIH 3T3 cells. This finding indicated that the introduced PBS-impaired vectors had re-combined with homologous, but distinct, endogenous se-quences harboring a functional PBS-Gln. Related endogenous sequences from genomic DNAs of NIH fibroblasts (6, 23) (clone 621 [Fig. 2]), BALB/c mice (33), and RFM/un mice (31) have been reported. However, RNA of this family of endoge-nous mouse viruses has not been detected previously (5).
Endogenous RNA with a functional PBS-Gln in virions.The most frequent mechanism of retroviral recombination involves copackaging of genetically distinct RNA species in viral parti-cles. To identify a possible recombination partner involved in transduction of the PBS-Gln-harboring proviruses, we looked at RNA in virus particles produced byC-2 packaging cells into which no vector DNA had been transferred.
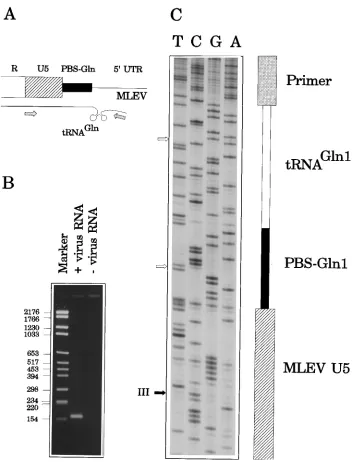
As a first step (Fig. 3), we searched for a relevant recombi-nation partner with a functional tRNA-Gln primer. RNA iso-lated from virus particles produced byC-2 cells was incubated with MLV reverse transcriptase to allow tRNA-primed synthe-sis of strong-stop cDNA. The tRNA-U5 DNA hybrid was am-plified by PCR with a tRNA-Gln1,2-specific primer and a U5 primer matching Akv positions 69 to 96 (Fig. 2) as well as the homologous regions in the PBS-Gln-harboring proviruses (Fig. 3A). Sequence analysis of the 163-bp PCR product (Fig. 3B) revealed the U5 sequence given in Fig. 4 as positions 1 to 48 of MLEV linked to the tRNAGln sequence (41) (Fig. 3C). We
FIG. 2. Nucleotide sequences of transduced proviruses harboring PBS-Gln1,2 and parent genomes. A PCR-amplified 1.3-kb fragment was partly sequenced in order to determine nucleotide sequences of the U5 region (positions 69 to 144), the PBS (positions 145 to 162), and the 59UTR (positions 163 to 402). Numbers refer to distances from the transcription start site. Comparison with homologous regions of the Akv MLV-based vector (Akv) and a PBS-Gln-harboring MLV-like endogenous sequence (MLEV), directly isolated from virus particles, indicates the site of recombination in each individual clone. The endogenous viral sequence (dl587rev) was obtained from reference 5. The sequence of clone 621, derived from NIH fibroblast genomic DNA, was obtained from reference 6. The MLEV sequence was obtained as illustrated in Fig. 4. Individual sequences originate from transduction of vectors harboring the following PBS sequences: 1 to 9 and 22, PBS-XXX; 10 to 12, PBS-UMU; 13 to 17, PBS-Met(i)intM; and 18 to 21, PBS-Met(i)intP. The mixed PBS-Gln1,2 sequences in XXX6 and XXX12 are indicated by insertion of a Y (C and T) in the position distinct in PBS-Gln1 and PBS-Gln2. Nucleotides homologous to positions in Akv are indicated by hyphens, deleted nucleotides in transduced vectors are indicated with a colon, and insertions are indicated by the introduction of a colon in the Akv sequence. N, nonidentified nucleotide positions. The nucleotide differences between Akv-neo and MLEV outside the PBS are marked I through XVIII. The relevant genetic markers for mapping of recombination sites are IV through XVIII. The Tn5 fragment encompassing the neo marker gene starts in the Akv vector at position 400.
1442 MIKKELSEN ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
conclude from these studies that reverse transcription was ini-tiated from a tRNAGlnmolecule annealing to an endogenous
MLV-like sequence packaged in MLV viral particles. The se-quence, shown in Fig. 3C, indicates that tRNA1Glnwas utilized
as a primer for initiation of reverse transcription.
Second, PBS-Gln and downstream sequences were analyzed in random-primed cDNA derived from C-2 virion RNA. A primer matching PBS-Gln2 was utilized together with a partly degenerate primer in a semirandom PCR amplification strategy. Direct automated DNA sequencing of the PCR product gave the sequence given as MLEV positions 116 to 317 in Fig. 4.
[image:5.612.130.487.68.530.2]Finally, we performed sequence analysis of a PCR product spanning the PBS-Gln. The PCR product was generated from random-primed cDNA derived fromC-2 virion RNA by using two PCR primers corresponding to sequences upstream and downstream of PBS-Gln as determined above. Sequence anal-ysis of the PCR product gave the sequence given as MLEV positions 1 to 165 (Fig. 4C), thus providing a perfect overlap with the U5 sequences determined from tRNA-Gln-primed cDNA as well as with the 59UTR sequences derived from the product of the semirandom PCR. Moreover, this last PCR sequence analysis indicates that the detected MLEV harbors a PBS-Gln2 sequence.
FIG. 3. Analysis of glutamine tRNA-primed cDNA inC-2 cell virion RNA. (A) Reverse transcription-PCR approach utilizing primers 8 and 12, matching the 59 terminus of the U5 region and the 59terminus of murine glutamine tRNA, respectively. The tRNA molecule was initially used as a primer in first-strand cDNA synthesis prior to PCR amplification. (B) Aliquots of PCR products were electrophoresed in a 2% agarose gel and stained with ethidium bromide. Left lane, DNA marker; middle lane, DNA amplified when viral RNA with MLEV strong-stop DNA was used as a template for PCR; right lane, negative control with no template included in the PCR. Numbers on the left indicate base pairs. (C) Sequence analysis of the obtained 163-bp PCR product. The fragment contains sequences of both MLEV and tRNAGln
origin, as illustrated to the right of the autoradiogram. The black arrow indicates the position of MLEV genetic marker III; white arrows point to positions distinct in Gln1 and Gln2 tRNAs.
on November 9, 2019 by guest
http://jvi.asm.org/
The overlapping sequences derived by direct sequence anal-ysis of the products of the three PCR approaches define one major RNA species with a functional PBS-Gln in virions from
C-2 cells. In addition, the use of primers on both sides of the PBS matching Akv as well as MLEV sequences did not reveal a predominant RNA species with PBS-Pro or with other patches of distinct Akv-like sequences.
Recombination between MLEV and Akv-neo can explain
PBS-Gln transductions. The determined MLEV sequence
(Fig. 4) has the characteristic pattern of non-Akv sequences found in the PBS-Gln-harboring proviruses and, furthermore, is homologous to the sequence of clone 621 (6, 23) and iden-tical to the sequence of dl587rev (5), except for the position distinct in PBS-Gln1 and PBS-Gln2 (1152) and two nucleotide differences at positions1178 and1377 (Fig. 2).
Nucleotide differences or clusters of differences between Akv MLV and MLEV outside the PBS were designated I to XVIII (Fig. 2). Five base pair substitutions were found in the U5 region (markers I to III in Fig. 2), while up to eight mu-tations or groups of mumu-tations (markers IV to XI) were found in the 59UTRs of the proviruses analyzed. With the possible exception of one provirus (XXX5 [Fig. 2]), all of the patch repair sequences embedded in Akv-neo sequences corre-sponded exactly to the MLEV sequence, and the sequences of all recombinants could be explained by precise homologous
recombination events. In case of XXX5, the crossover event has taken place within or around marker VIII in a region characterized by a stretch of TG repeats, which makes an unambiguous interpretation impossible.
The structures of all transduced PBS-Gln proviruses can thus be explained by recombination between Akv-neo and a specific MLEV sequence that is found as a predominant PBS-Gln-containing RNA species in the virions. The likely mecha-nism (Fig. 5A) is that MLEV minus-strand strong-stop DNA during the first template shift is transferred to the R region of Akv-neo. A selection for structures allowing efficient base pair-ing between the plus-strand copy of part of the tRNA and the minus-strand PBS copy during second-strand transfer will re-quire a template switch back to a PBS-Gln-containing RNA during minus-strand elongation. Since the colonies of trans-duced cells were isolated on the basis of selection for neo expression, this forced recombination system selects for a tem-plate switch in the 59UTR between PBS and neo.
A preferred region for recombination.To map the specific site of recombination in each individual transduction event, the 59 UTR sequences of the proviruses from 22 G418-resistant colonies harboring a PBS-Gln were analyzed (Fig. 2). The mutations or groups of mutations (IV to XVIII) served as genetic markers dispersed throughout the ca. 240-nucleotide 59 UTR flanking the PBS-Gln and the neo gene, enabling deter-FIG. 4. Sequence analysis of MLEV. Sequencing of MLEV was performed by three different approaches (see Materials and Methods). (A) Reverse transcription-PCR with primer 8 as a sequencing primer (for details, see Fig. 3); (B) semi-random-primed transcription-PCR with degenerate primer 11 together with PBS-Gln primer 9 and with primer 10 as a sequencing primer; (C) PCR amplification with primers 7 and 8 and with primer 7 as a sequencing primer. The Y at position 80 indicates the finding of a mixture of C and T in this position.
1444 MIKKELSEN ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.130.483.72.419.2]mination of the site of recombination. Interestingly, recombi-nation had occurred between markers XI (1308) and XII (1333) in 14 of 22 transduced proviruses, while 6 of 22 ana-lyzed sequences demonstrated a template shift between mark-ers X (1299) and XI (1308). In the remaining two colonies, the site of recombination was localized to the region between markers V (1198) and VI (1220) and within marker VIII (1259), respectively (Fig. 5B). These results reveal that the template shift during minus-strand synthesis tends to occur in a 33-nucleotide region located approximately 140 nucleotides downstream from the PBS between markers X and XII. We conclude that PBS-impaired MLV retroviral vectors may rep-licate by recombining with an MLV-like endogenous sequence and that a majority of the recombination events leading to successful transduction are clustered in a narrow region of the 59UTR.
DISCUSSION
We have described a single-cycle transfer protocol to study the process of reverse transcription-mediated homologous re-combination between introduced PBS-modified, replication-defective retroviral vectors and endogenous sequences harbor-ing a functional PBS. Extensive modifications of the PBS in MLV-based retroviral vectors caused a decrease of 5 to 6 orders of magnitude in replication efficiency, showing that PBS-tRNA base pairing based solely on the conserved tRNA 39CCA terminus or on an internal tRNA fragment may not be sufficient to support MLV replication. These results are in general agreement with reports on the effect of PBS mutations on the replication of human immunodeficiency virus 1 (7, 22, 29, 38, 48).
Sequence analysis of transduced vectors showed that repli-cation of severely PBS-impaired vectors was mediated pre-dominantly through recombination with endogenous se-quences. Thirty-two of 60 colonies analyzed harbored a PBS-Gln flanked by sequences originating from an MLEV. Thus, the 59UTR downstream from PBS-Gln1,2 and the U5 region of the 59long terminal repeat was found to contain a specific pattern of nucleotide differences closely related to sequences previously found to be involved in retroviral recombination (5). In this work we have identified the putative recombination partner as an endogenous virion-associated RNA that uses tRNA-Gln as a primer for reverse transcription. The nucle-otide sequence of this endogenous RNA corresponds exactly to the patch repair sequences found in the PBS-Gln-harboring proviruses. MLEV was found to harbor a PBS-Gln2 sequence; however, tRNA1Gln was the only tRNA species found to be
associated with the PBS of the endogenous virus. These ob-servations explain the occurrence of PBS-Gln1 and PBS-Gln2 sequences and mixtures thereof in transduced proviruses, sup-porting the notion that a perfect PBS-tRNA match is not required for tRNA priming (2, 24).
[image:7.612.61.299.76.640.2]Our observations support previous results suggesting that defective retroviruses may restore function by recombination
FIG. 5. Recombinational patch repair of PBS-modified vectors. (A) Model for reverse transcription-mediated minus-strand recombination. 1, forced initia-tion of minus-strand synthesis on endogenous virus harboring a funcinitia-tional PBS. 2, selection for a second template shift within the 59UTR during minus-strand synthesis. One of several possibilities for a first template switch (during the minus-strand jump) is shown. 3, synthesis of plus-strand strong-stop DNA, ob-taining complementarity between the PBS sequence generated in minus-strand synthesis and the PBS sequence arising as a copy of the tRNA molecule. 4, final double-stranded provirus containing sequences of both exogenous (Akv) and endogenous (MLEV) origins. Thin lines indicate RNA; thicker lines indicate
DNA. (B) Mapping of recombination sites in the 59UTR between PBS and neo characterizes a preferred region for recombination. The numbers IV through XVIII refer to the relevant genetic markers shown in Fig. 2. In 20 of 22 analyzed colonies containing PBS-Gln, the recombination sites were mapped within mark-ers X and XII, in a region possibly involved in genomic RNA dimerization (11, 36, 46). This proposed dimerization region harbors a potential stem-loop struc-ture located immediately upstream from the putative double stem-loop strucstruc-ture (harboring markers XII through XVIII) required for RNA encapsidation (49). As indicated, the marker differences between Akv and MLEV, X through XVIII (F), are predicted not to affect the proposed secondary structure (1, 11, 21, 46, 49).
on November 9, 2019 by guest
http://jvi.asm.org/
with retroviral sequences endogenous to the host genome (5, 9, 26, 28, 39). The PBS mutants are severely impaired at the initiation of reverse transcription and therefore are forced to initiate the minus-strand synthesis on a copackaged endoge-nous viral sequence harboring a functional PBS. The suggested mechanism for PBS patch repair favors the copy choice model for recombination (4, 16), in which reverse transcriptase switches template during minus-strand synthesis (Fig. 5A). The cDNA initiated on the endogenous virus is transferred to the
neo vector RNA template, most likely during the first jump,
and minus-strand synthesis continues through the neo gene. However, a template shift within the 59 UTR is required to ensure sufficient base pairing for the second-strand transfer step of reverse transcription, although a few mismatches in this interaction may be tolerated in viral replication (37). Never-theless, to obtain complementarity between the PBS arising as a copy of the tRNA molecule and the PBS generated in minus-strand synthesis, we specifically select for a shift of template downstream from PBS. The combination of a low replication efficiency and a high frequency of recombination-mediated transduction of PBS-impaired vectors further stresses that ini-tiation of minus-strand synthesis from the vector PBS is strongly inhibited.
The presence of several genetic markers dispersed through-out the ca. 240-nucleotide 59 UTR retained in the vector al-lowed us to specifically map the site for each individual recom-bination event. In 20 of 22 clones the site of recomrecom-bination was mapped to a 33-nucleotide region located approximately 140 bases downstream from the glutamine PBS (Fig. 5B). This clustering of recombination events was not caused by uneven lengths of the regions of homology between individual mark-ers, since the sites of nucleotide differences are fairly evenly distributed throughout the 59UTR. For the following reasons we can also rule out the formal possibility that the clustering of recombination points reflects a bias caused by selection for vector transcription and translation. Various combinations of exogenous and endogenous viral sequences have been found to efficiently support expression of viral genes (5, 9, 26, 28, 39). The replication-competent revertant observed by Colicelli and Goff (5) had obtained a leader sequence nearly identical to the leader sequence of MLEV, suggesting that minor alterations in the 59 UTR do not affect expression of downstream genes. Also, this particular 59 leader region has been shown not to interfere with neo expression in embryonic stem cells (14). Thus, it seems very unlikely that inclusion of genetic markers XII to XVIII (Fig. 2) of MLEV origin in the integrated pro-virus would affect transcription of the neo gene. Furthermore, since the most-downstream genetic markers of the endogenous sequence do not introduce start codons or exclude stop codons, we suggest that no open reading frame may influence the level of translation from the viral mRNA.
We conclude that the narrow distribution of crossover sites between markers X and XII within the 59UTR reflects prop-erties solely of reverse transcriptase-mediated recombination. Moreover, our results indicate that transfer of the minus-strand growing point to homologous sequences of the copack-aged RNA template is exact. Thus, with the possible exception of one ambiguous case, no deletions nor insertions were found at the recombination site in any of the proviruses analyzed, despite the lack of selection for a functional 59UTR to medi-ate proper RNA dimerization and packaging.
Dimerization and packaging of MLV genomic RNA are controlled by highly conserved cis elements (21) located within the packaging signal (C) downstream from the PBS in the 59 UTR (36, 45, 46). The two RNA subunits are linked together in the dimer linkage structure, presumably because of specific
secondary and tertiary structures in this region of the genome (1, 11, 36, 46). Hence, nucleotide positions 329 to 394 (Fig. 2) form a stable double stem-loop structure (1, 46, 49) required for encapsidation (49), while the positions directly upstream from this stable secondary structure (positions 291 to 324) around genetic markers X and XI seemingly form a single stem-loop structure, which is thought to be of major impor-tance in dimerization of the viral genome (1, 11, 36, 46).
Interestingly, our results demonstrate a preferred region for recombination coinciding precisely with the proposed dimer-ization domain located upstream from the double stem-loop structure required for encapsidation (Fig. 5B) (11, 36, 46). Close RNA-RNA interactions in this particular region of the genome around markers X to XII may therefore favor the recombinational events observed in this study. Additionally, structural features may influence recombination probabilities through effects on donor template fragility or acceptor tem-plate accessibility.
Alternatively, an imperfect dimerization structure in a het-erodimer may favor a template switch of reverse transcriptase. Of interest in regard to this possibility is that markers VIII and X (Fig. 2) both show deletions of 6 nucleotides in the endog-enous RNA partner relative to the Akv sequences. These dif-ferences may affect the secondary and tertiary structures in the dimer linkage region and might affect a template switch. Al-though the exact mechanisms that may favor such a switch are purely speculative, independent support for this possibility comes from the work of Stuhlmann et al. (43), who found that deletion of the packaging signal decreased RNA encapsidation but increased recombination efficiencies.
In summary, we have selected for recombination events within a window of homology of about 240 nucleotides and found 20 of 22 recombination events to be located in a 33-nucleotide region. This uneven distribution of recombination sites may be a characteristic feature of the 59UTR or it may also apply to other sequences of retroviral origin. Previous in vivo studies of retroviral recombination (15, 16, 42) have been based primarily on the use of nearly identical vectors differing in only a few nucleotide positions and have not allowed a detailed recombination mapping as reported here. Finally, it should be emphasized that the observed pattern of recombi-nation events may be affected by the fact that the vectors used harbor only part of the sequences involved in RNA packaging and dimerization.
ACKNOWLEDGMENTS
This work was supported by the Danish Biotechnology Programme, the Danish Cancer Society, the Novo Foundation, the Danish Natural Sciences Research Council, and the Karen Elise Jensen Foundation.
REFERENCES
1. Alford, R. L., S. Honda, C. B. Lawrence, and J. W. Belmont. 1991. RNA secondary structure analysis of the packaging signal for Moloney murine leukemia virus. Virology 183:611–619.
2. Barklis, E., R. C. Mulligan, and R. Jaenisch. 1986. Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinomal cells. Cell 47:391–399.
3. Beck, E., E. A. Ludwig, R. Auerswald, B. Reiss, and H. Schaller. 1982. Nucleotide sequence and exact location of the neomycin phosphotransferase gene from transposon Tn5. Gene 19:327–336.
4. Coffin, J. M. 1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 42:1–26.
5. Colicelli, J., and S. P. Goff. 1987. Isolation of a recombinant murine leuke-mia virus utilizing a new primer tRNA. J. Virol. 57:37–45.
6. Colicelli, J., and S. P. Goff. 1987. Identification of endogenous retroviral sequences as potential donors for recombinational repair of mutant retro-viruses: positions of cross-over points. Virology 160:518–522.
7. Das, A. T., B. Klaver, and B. Berkhout. 1995. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers
1446 MIKKELSEN ET AL. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
other than the natural tRNA3Lys. J. Virol. 69:3090–3097.
8. Deutscher, M. P. 1990. Ribonucleases, tRNA nucleotidyltransferase, and the 39processing of tRNA. Prog. Nucleic Acids Res. Mol. Biol. 39:209–240. 9. DiFronzo, N. L., and C. A. Holland. 1993. A direct demonstration of
recom-bination between an injected virus and endogenous viral sequences, resulting in the generation of mink cell focus-inducing viruses in AKR mice. J. Virol. 67:3763–3770.
10. Gilboa, E., S. W. Mitra, S. Goff, and D. Baltimore. 1979. A detailed model for reverse transcription and tests of crucial aspects. Cell 18:93–100. 11. Girard, P.-M., B. Bonnet-Mathonie`re, D. Muriaux, and J. Paoletti. 1995. A
short autocomplementary sequence in the 59leader region is responsible for dimerization of MoMuLV genomic RNA. Biochemistry 34:9785–9794. 12. Goodrich, D. W., and P. H. Duesberg. 1990. Retroviral recombination during
reverse transcription. Proc. Natl. Acad. Sci. USA 87:2052–2056.
13. Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467.
14. Grez, M., E. Akgu¨n, F. Hilberg, and W. Ostertag.1990. Embryonic stem cell virus, a recombinant murine retrovirus with expression in embryonic stem cells. Proc. Natl. Acad. Sci. USA 87:9202–9206.
15. Hu, W.-S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 87:1556–1560. 16. Hu, W.-S., and H. M. Temin. 1990. Retroviral recombination and reverse
transcription. Science 250:1227–1233.
17. Hu, W.-S., and H. M. Temin. 1992. Effect of gamma radiation on retroviral recombination. J. Virol. 66:4457–4463.
18. Jones, J. S., R. W. Allan, B. Seufzer, and H. M. Temin. 1994. Copackaging of different-sized retroviral genomic RNAs: little effect on retroviral repli-cation or recombination. J. Virol. 68:4097–4103.
19. Junghans, R. P., L. R. Boone, and A. M. Skalka. 1982. Retroviral DNA H structures: displacement/assimilation model of recombination. Cell 30:53– 62.
20. Kikuchi, Y., Y. Ando, and T. Shiba. 1986. Unusual priming mechanism of RNA-directed DNA synthesis in copia retrovirus-like particles of Drosophila. Nature (London) 323:824–826.
21. Konings, D. A. M., M. A. Nash, J. V. Maizel, and R. B. Arlinghaus. 1992. Novel GACG-hairpin motif in the 59untranslated region of type C retrovi-ruses related to murine leukemia virus. J. Virol. 66:632–640.
22. Li, X., J. Mak, E. J. Arts, Z. Gu, L. Kleiman, M. A. Wainberg, and M. A. Parniak.1994. Effects of alterations of primer-binding site sequences on human immunodeficiency virus type 1 replication. J. Virol. 68:6198–6206. 23. Lowy, D. R., E. Rands, S. K. Chattopadhyay, C. F. Garon, and G. L. Hager.
1980. Molecular cloning of infectious integrated murine leukemia virus DNA from infected mouse cells. Proc. Natl. Acad. Sci. USA 77:614–618. 24. Lund, A. H., M. Duch, J. Lovmand, P. Jørgensen, and F. S. Pedersen. 1993.
Mutated primer binding sites interacting with different tRNAs allow efficient murine leukemia virus replication. J. Virol. 67:7125–7130.
25. Mann, R., R. C. Mulligan, and D. Baltimore. 1983. Construction of a retro-virus packaging mutant and its use to produce helper-free defective retrovi-rus. Cell 33:153–159.
26. Martinelli, S. C., and S. P. Goff. 1990. Rapid reversion of a deletion mutation in Moloney murine leukemia virus by recombination with a closely related endogenous provirus. Virology 174:135–144.
27. Morgenstern, J. P., and H. Land. 1990. Advanced mammalian gene transfer: high titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587– 3596.
28. Murphy, J. E., and S. P. Goff. 1994. Forced integration of Moloney murine leukemia virus DNA with a mutant integration site occurs through recom-bination with VL30 DNA. Virology 204:458–461.
29. Nagashunmugan, T., A. Velpandi, C. S. Goldsmith, S. R. Zaki, and V. S. Kalyanaraman.1992. Mutation in the primer binding site of the type 1 human immunodeficiency virus genome affects virus production and infec-tivity. Proc. Natl. Acad. Sci. USA 89:4114–4118.
30. Negroni, M., M. Ricchetti, P. Nouvel, and H. Buc. 1995. Homologous re-combination promoted by reverse transcriptase during copying of two dis-tinct RNA templates. Proc. Natl. Acad. Sci. USA 92:6971–6975. 31. Nikbakht, K. N., C.-Y. Ou, L. R. Boone, P. L. Glover, and W. K. Yang. 1985.
Nucleotide sequence analysis of endogenous murine leukemia virus-related proviral clones reveals primer-binding sites for glutamine tRNA. J. Virol. 54:889–893.
32. Norton, J. D., J. Connor, and R. J. Avery. 1984. Unusual long terminal repeat sequence of a retrovirus transmissible mouse (VL30) genetic element: iden-tification of functional domains. Nucleic Acids Res. 12:3445–3460. 33. Ou, C.-Y., L. R. Boone, and W. K. Yang. 1983. A novel sequence segment and
other nucleotide structural features in the long terminal repeat of a BALB/c mouse genomic leukemia virus-related DNA clone. Nucleic Acids Res. 11: 5603–5620.
34. Paludan, K., H. Y. Dai, M. Duch, P. Jørgensen, N. O. Kjeldgaard, and F. S. Pedersen.1989. Different relative expression from two murine leukemia virus long terminal repeats in unintegrated transfected DNA and in inte-grated retroviral vector proviruses. J. Virol. 63:5201–5207.
35. Panganiban, A. T., and D. Fiore. 1988. Ordered interstrand and intrastrand DNA transfer during reverse transcription. Science 241:1064–1069. 36. Prats, A.-C., C. Roy, P. Wang, M. Erard, V. Housset, C. Gabus, C. Paoletti,
and J.-L. Darlix.1990. cis elements and trans-acting factors involved in dimer formation of murine leukemia virus RNA. J. Virol. 64:774–783.
37. Pulsinelli, G. A., and H. M. Temin. 1994. High rate of mismatch extension during reverse transcription in a single round of retrovirus replication. Proc. Natl. Acad. Sci. USA 91:9490–9494.
38. Rhim, H., J. Park, and C. D. Morrow. 1991. Deletions in the tRNALys
primer-binding site of human immunodeficiency virus type 1 identify essen-tial regions for reverse transcription. J. Virol. 65:4555–4564.
39. Schwartberg, P., J. Colicelli, and S. P. Goff. 1985. Recombination between a defective retrovirus and homologous sequences in host DNA: reversion by patch repair. J. Virol. 53:719–726.
40. Sørensen, A. B., M. Duch, P. Jørgensen, and F. S. Pedersen. 1993. Ampli-fication and sequence analysis of DNA flanking integrated proviruses by a simple two-step polymerase chain reaction method. J. Virol. 67:7118–7124. 41. Sprinzl, M., T. Hartmann, J. Weber, J. Blank, and R. Zeidler. 1989. Com-pilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 17(Suppl.):r1–r172.
42. Stuhlmann, H., and P. Berg. 1992. Homologous recombination of copack-aged retrovirus RNAs during reverse transcription. J. Virol. 66:2378–2388. 43. Stuhlmann, H., M. Dieckmann, and P. Berg. 1990. Transduction of cellular
neo mRNA by retrovirus-mediated recombination. J. Virol. 64:5783–5796.
44. Swain, A., and J. M. Coffin. 1992. Mechanism of transduction by retroviruses. Science 255:841–845.
45. Torrent, C., C. Gabus, and J.-L. Darlix. 1994. A small and efficient dimer-ization/packaging signal of rat VL30 RNA and its use in murine leukemia virus-VL30-derived vectors for gene transfer. J. Virol. 68:661–667. 46. Tounekti, N., M. Mougel, C. Roy, R. Marquet, J.-L. Darlix, J. Paoletti, B.
Ehresmann, and C. Ehresmann.1992. Effect of dimerization on the confor-mation of the encapsidation Psi domain of Moloney murine leukemia virus RNA. J. Mol. Biol. 223:205–220.
47. Van Beveren, C., J. Coffin, and S. Hughes. 1985. Nucleotide sequences complemented with functional and structural analysis, p. 790–805. In R. Weiss, N. Teich, H. Varmus, and J. Coffin (ed.), RNA tumor viruses, vol. 2. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
48. Wakefield, J. K., H. Rhim, and C. D. Morrow. 1994. Minimal sequence requirements of a functional human immunodeficiency virus type 1 primer binding site. J. Virol. 68:1605–1614.
49. Yang, S., and H. M. Temin. 1994. A double hairpin structure is necessary for the efficient encapsidation of spleen necrosis virus retroviral RNA. EMBO J. 13:713–726.
50. Zhang, J., and H. M. Temin. 1993. 39junctions of oncogene-virus sequences and the mechanisms for formation of highly oncogenic retroviruses. J. Virol. 67:1747–1751.