0022-538X/79/04-0132/09$02.00/0
Avian
Sarcoma Virus-Transformed Quail
Clones
Defective
in
the Production of
Focus-Forming Virus
WILLIAM S. MASON,1*TERESA W. HSU,' CAROL YEATER,' JAN L. SABRAN,2 GEORGE E. MARK,' AKIRA KAJI,2ANDJOHN M. TAYLOR1
The Institutefor CancerResearch, Fox Chase Center, Philadelphia, Pennsylvania 19111,' and Department
ofMicrobiology, University of Pennsylvania, Philadelphia,Pennsylvania 191742
Received for publication 20 December 1978
Quail embryo fibroblastswereinfectedatlowmultiplicity with aviansarcoma
virus, and transformedcells wereselectedby their abilitytoformcolonies inagar.
Five clones that failed to produce focus-forming virus were examined for (i)
intactness of the integrated proviral DNA, (ii) intracellular viral RNA production,
(iii)intracellular viral antigen production, (iv) production of virus particles, and
(v) rescue ofafunctional srcgene and ofparental hostrange determinants by
superinfectionwithRous-associatedvirus-60,anavian leukosisvirus of subgroup
E. Deletions in the integrated viral DNA were apparent in three of the five
nonproducer clones.Inoneclone producingfocus-forming virus, analysis of the
integrated viral DNA revealedaninsertion in the region of thegenomethat codes
forsrc.
In a previous publication we described the
state of the integrated proviral DNA in
trans-formed clonesofchicken, duck, andquailcells
isolated after low-multiplicity infection with
avian sarcoma virus (27). Within one group of
transformedquailclones derived fromthe cells
of a single embryo, we found that 7 out of 18
transforned clones failedtoproduce
focus-form-ing virus. The purpose of this manuscript is to
present a preliminary characterization of five of
these defective clones and of one interesting nondefective clone.
MATERIALS AND METHODS
Cells and viruse8. The sourcesofembryonated
eggs andtheproceduresfor thepreparationofchicken
andJapanesequail embryofibroblast cultureswere as
described (36).The chickenembryofibroblastswere
eitherC/EorC/BE,i.e.,resistanttoinfectionbyavian
tumorviruses ofsubgroupsEorBplusE,respectively. Thequail embryofibroblastswereQ/BD,resistantto
viruses of subgroups B and D, but susceptible to
infectionby viruses ofsubgroupsA,C,and E. For isolation of clones of transformedquailcellswe
used both the Prague strain of Rous sarcoma virus
(RSV)ofsubgroupA(wtPR-A)and theBratislava77
strain ofaviansarcomavirusofsubgroupC(wtB77).
Theproceduresandmedia for the isolation andgrowth
ofclones of transformed quail cellswere previously
described (27). Forrescueoftransformingvirusfrom defective clones we used Rous-associated virus-60
(RAV-60), anavian leukosis virus ofsubgroupE (8)
that isinfectious forJapanesequail embryofibroblasts butnotforC/EorC/BEchickenembryofibroblasts.
Cellculturewas at37-38°C. wtB77wasprovided by
E. Hunter (University of Alabama, Birmingham);
RAV-60wasobtained fromH. Hanafusa(Rockefeller University, New York, N.Y.). ABryan strain
RSV-infected quail cellline, Q-BH-RSV(-) cl. 3 (7), was provided by R.Friis,Justus Liebig Universitat, Gies-sen,WestGermany.
Virus a8says. Focus assays on chicken or quail embryo fibroblastswere done using standard
proce-dures (36),and concentrations of sarcoma viruses are
expressedasfocus-formingunits per milliliter. Trans-formation-defectivesarcomaviruses and avian leuko-sis virusesare notdetectedbythe focus assay.
To test for the presenceof transformation-defective
viruses or avianleukosis viruses,gs(-)chf(-) chicken embryofibroblastcultures(38) were infected with the
sampleinquestionandpassagedthree times. The cells were growninHam F10 medium supplemented with 10%(vol/vol)tryptosephosphate broth, 10% (vol/vol) calf serum, and 2% (vol/vol) heat-inactivated (1 h, 56°C) chickserum, exceptthat infection and transfer weredone in Ham F10supplemented with10% (vol/ vol)tryptosephosphate broth,5%(vol/vol)calf serum, and0.0002%(wt/vol) polybrene (GM+PB).After the third passage, the cellson a 100-mm-diameter petri dishwerewashed three times with10ml of ice-cold
phosphate-buffered saline (0.15 M NaCl, 0.0028 M
KH2PO4,0.0072 MNa2HPO4, pH7.1),scrapedinto0.5
ml ofphosphate-buffered saline,and storedat-80°C forsubsequentassay.Thesamplesweresonically dis-rupted priortoassayfor thegroup-specific(gs) antigen of aviantumorvirusesbythe COFAL assay(2, 28, 32). Forthis assay, antiserumwasobtained fromapigeon
bearinganRSV-inducedtumor.
Radioactivelabelingof viruses and cells.
La-belingwasdoneon100-mm-diameterpetridishes with monolayers that were ca. 70 to 90%confluent. The monolayerswerefirstwashed threetimeswith37°C Earle basal salt solution containing 0.056% (wt/vol)
132
on November 10, 2019 by guest
http://jvi.asm.org/
VIRUS-TRANSFORMED
NaHCO3 and 1% (vol/vol) dimethyl sulfoxide. For labeling of virus, the cellswereincubated for 16h in 5ml of HamF10 medium containing 2% the normal concentrationofmethionine, 5% (vol/vol) calfserum, 1%(vol/vol) dimethyl sulfoxide, and 100
f&Ci
of [3S]-methionine (200 to 300 Ci/mmol; New England Nu-clearCorp., Boston, Mass.).The virus was then har-vested from the tissue culture fluids as described be-low. To label thecells for immune precipitation, 1.5 ml of Earle saltsolution containing 0.056% (wt/vol) NaHCO3, 1% (vol/vol) dimethylsulfoxide and 100 uCi of[3S]methioninewasaddedfor20min at37°C.The label was removed, and the cells were washed twotimes with 5 ml ofprewarmed GM + 1% (vol/vol) dimethyl sulfoxide and then incubated for an
addi-tional 2.5 h. At the end of this incubation, the cells werewashed once with 10 ml ofice-cold phosphate-buffered saline containing1%(vol/vol) dimethyl sulf-oxide and stored at-80°C, priortolysisfor immune
precipitation.
Purification of viruses. Radioactive virus was
partiallypurified from tissue culturefluids,essentially
aspreviously described (5). Briefly, the media were
clarified,0.1mg ofpurifiedwtPR-A virus was added as carrier,the media werelayered over 3 mlof35% (wt/vol)sucrose in 0.1MNaCl-0.01M Tris-chloride-0.001 M EDTA (pH 7.4), and the viruses were sedi-mentedthroughthe sucroseby centrifugationfor 2h,
38,000 rpm,4°C,in aSpinco SW40rotor.Thepellets
were then suspended for sodium dodecyl sulfate
(SDS)-polyacrylamidegel electrophoresis (15).
Immunoprecipitation. Cells were lysed and
clar-ifiedpriortodirectimmunoprecipitation,asdescribed (5). To half of thecelllysate (900
id)
wereadded 5yg ofwtPR-Avirus and 0.1ml ofrabbitanti-gsserum.As acontrol,to anequalvolume ofcelllysatewasadded 5jg
of human serum albumin and 0.1 ml of rabbit anti-human serum albumin(Behring Diagnostics,So-merville, N.J.). The immunoprecipitates were col-lected after anovernightincubation at4°Cand
resus-pended for SDS-polyacrylamide gel electrophoresis.
Theanti-gagserum waspreparedby three subcu-taneous injections of a female New Zealand white rabbit at2-to3-week intervalswith 0.5 mg of
Nonidet-P40-disrupted avian myeloblastosis virus in Freund
adjuvant.Thefirst was incomplete adjuvant,the next two in incomplete adjuvant. Eight weeks after the thirdinjection,avianmyeloblastosisvirus wasinjected
intraperitoneally without adjuvant. The serum was
collected10dayslater. Avianmyeloblastosisvirus for these injections was gradient purified (23) from chicken plasma obtained from D. Bolognesi (Duke University,Durham, N.C.).
Gel electrophoresis. Discontinuous
SDS-poly-acrylamide gel electrophoresiswascarriedout as
de-scribedbyLaemmli (15), except that the spacer and
separationgelscontained twice the recommended
con-centrations of Tris-chloride and the ratio ofacrylamide
tobisacrylamide in the separationgel was adjusted
according to the formula of Blattler etal. (1). Gels
were prepared and fluorographed using preexposed
film,asdescribed(3, 16).
Determinationof theaverage number of mol-eculesof viral RNA per transformed cell. Total nucleic acidwasobtained from ca.4 x 107cellsusing
an SDS-Pronase treatment followed by two phenol
extractions (27). The total nucleic acid was annealed with 3P-labeled DNA probes to the viral RNA (34). A
representative probelabeledwith[nP]dATP (350Ci/ mmol; New England Nuclear) was prepared using
avian myeloblastosis virus reverse transcriptase, wtPR-C RSV 70S RNA, and calf thymus DNA
primers, allaspreviouslydescribed(35).Aprobefor those sequences located near the 3'terminus,
exclud-ingpolyadenylicacid[poly(A)],of theviralRNA was prepared using a similar reactionexcept that we used astemplatepoly(A)-containingviral RNA, selected by two passages through acolumn of
oligodeoxythymi-dylic acid-cellulose(21). As primer weused 5'-p(dT)8-rG-3' obtained from Collaborative Research (Wal-tham, Mass.), which was subsequently removed by treatmentwith alkali.
To determine the average number of viral RNA molecules per cell, the kinetics ofannealingdata were
entered into aDigital 11/70 computerprogrammed for thisanalysis(22).
Analysis ofintegrated viralDNA. The proce-duresforanalysisofintegrated viralsequences were described in a previous report (27). Briefly, DNA extracted from infected cells wassubjectedto restric-tion endonuclease digesrestric-tionfollowed by electropho-resis on slabgelsof 0.7% (wt/vol) agarose. The DNA was then transferred tonitrocellulose filters by the method ofSouthern (30), andviral sequences were detectedby hybridizationwith a3P-labeled
comple-mentary DNA (cDNA) probe (11). For detection of
ribosomalsequences, weprepared a cDNAprobeto a mixtureof purified 28S and 18S ribosomal RNA using calf thymus DNA primers (35). Unintegrated viral DNA (11), used as a marker, was a gift from R.
Guntaka,ColumbiaUniversity,NewYork,N.Y. It is
importantto notethatthese unintegratedviralDNA intermediates contain the large terminal repeat
(LTR), of which at least part is also maintained in
integratedviralDNA(11, 27).
RESULTS
'tNonproducer"
quail clones. As notedabove,7 outof18clones ofaviansarcoma
virus-transformed quail embryo fibroblasts failed to
produce focus-formingvirus.These isolatesare
designatedas"nonproducer" clones.Two ofthe
clones did not grow well, and were discarded. Theremaining cloneshave beensubjectedto a
detailed analysis of the integrated viral DNA and to a preliminary analysis of virus gene
expression, with the ain of establishing the
block(s)tovirusreplication. On theassumption
that thefailuretoproduceinfectious progeny is
dueto avirusmutation,wehavedesignatedthe
virusescarried by the clonesasreplication
de-fective(rd)(37).Accordingly,the viruses
infect-ing the five clonesaredesignatedrdPH9PR-A,
rdPH18PR-A, rdPH2B77-C, rdPH1OB77-C,
andrdPH11B77-C.The correspondingquail cell
clones are called Q-PrA-9, Q-PrA-18, Q-B77-2,
Q-B77-10, and Q-B77-11, respectively.
30,1979
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 1. Synthesis of viral RNA and avian tumor virus gsantigen
Relative Quail Viral RNAb(mol- g antigen particle clone' eculespercell) conCnf
produc-tiond
Q-PrA-9 1,500 5.7-11.4 0.4
Q-PrA-18 1,300 7.7-15.4 1.2
Q-B77-1 1,600(2,200) NDe ND
Q-B77-2 60(370) <0.5 0
Q-B77-10 500 7.4 0.7
Q-B77-11 1,000 8.6 1.6
Q-PrA-4 2,200 16.5 1
Q-B77-8 300 14.4 0.7
aThenonproducer quailclonesaredescribed in the text. Q-PrA-4, Q-B77-1, and Q-B77-8 are producer
clones isolatedfrom infectedcells of the same quail embryo asthenonproducerclones.
bThe amount ofviral RNA percellwasdetermined asdescribed in thetext,assuming25x10-12gof total RNApercellandusing arepresentativecDNAprobe. The numbers in parentheses weredetermined using a
probe specificfor sequences near the 3' terminus of theviralRNA(Fig. 1).
'TheCOFAL reactivity was expressed as the recip-rocal of thehighest sample dilution producinga
posi-tiveresponse,determined on twofold serial dilutions ofcelllysates. Theresults have been normnalizedby
dividingby the protein concentration(milligramsper
milliliter) in eachsample(19).
dTo quantitate the production of virus particles, virions were labeled with [3S]methionine, purified,
disrupted, and subjected to electrophoresis on 18%
(wt/vol) polyacrylamide-SDSslab gels,as described in the text.Afterfluorography,films were scanned at 620nm with adensitometer, and the areas under the
peaksweredetermined for themajorvirion proteins. The numberspresentedaretherelativeyieldsof virus
particlesdetermined fromthequantitationofthe
ma-jorvirionprotein,P27.Results obtainedby normaliz-ingto P19orP12/P15differedbyafactor of two or less from those presented. Q-B77-2 did not produce detectableyieldsof virusparticles.Theyieldof parti-clesproduced by Q-PrA-4wastaken as unity.
eND, Not done.
Tocharacterize the nature of the defect in the
five nonproducerclones, we first measured four
parameters ofvirus infection. These were
syn-thesis of viralRNA, synthesis of avian tumor
virusgsantigens, release of virus particles, and
rescue ofthe src gene by superinfection with
RAV-60 avian leukosis virus.
In the following section, we show that the
inabilitytoproduce infectious progeny virus can
beattributed neither to afailure to transcribe
the provirus into RNA nor to an inability to
translate that RNA into thetypical
group-spe-cific(gs) antigens of the avian tumor viruses.
Nonproducer clones synthesize viral
RNA and avian tumor virus gs antigens.
Theresults of the analysis for viral RNA and
gs-antigen synthesisarepresented in Table 1 and
in Fig. 1. Intracellular viral RNA was
quanti-tated by hybridization, in RNA excess, with a
cDNA probe representative of the entire viral
genome.Aviantumorvirusgsantigenwas
meas-ured in the COFALassayusingserumfroman
RSV tumor-bearing pigeon.
The results ofatypicalhybridization between
a cDNA probe representative of the viral
ge-nomeandnucleic acid isolated fromaproducer
clone, Q-B77-1, are shown in Fig. lb.
Qualita-tively similar resultswereobtained with four of
thenonproducer clones, PrA-9, PrA-18,
Q-B77-10, and Q-B77-11,aswellaswith the three
other producer clones, Q-PrA-4, Q-B77-1, and
Q-B77-8. The amount of viral RNA per cell
(Table 1) varied from 500 to 2,200 copies per
cell, with the exceptions of theproducer clone,
Q-B77-8 (300 copies percell), and the
nonpro-ducerclone, Q-B77-2 (ca.60copiespercell).
100
50
a w
-i
4
w
z z 4
100
z
:L
501.
W
-1 0 1 2
logio (Crt mole-sec/liter)
FIG. 1. Annealing kinetics of f2P]cDNA probes, representingtotal and3'-specificaviansarcomavirus
sequences,tonucleic acid extractedfrom transformed quailclones. [PP]cDNA probes (6,000cpm per
reac-tion), prepared by calf thymus-primed transcription (-4, total) or5'-p(dT)8-rG-3'-primed
transcrip-tion(x-x, 3') ofwtPR-CRNA, wereannealedto the total nucleicacid extractedfrom aproducer quail
clone(Q-B77-1) andanonproducer quailclone (Q-B77-2).At the indicated valuesofC,t, sampleswere
removed, and theformation of hybridswasassayed
byresistance tonucleaseSl. Eachannealingcurve
has been correctedto 100% annealingofthe
/P]-cDNAtoan excessofpurified70SRNAofwtPR-C. The actualextent of annealing ofeachprobe with virion RNAwasapproximately85%.
0. .
Q-B77-2 /I
/
3'C
/
/0.-31t/
X--- -
.O0
TOTALb.
-0-B77-1 14
/ -TOTAL
- _ S/I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.501.279.421.291.516.2]AVIAN SARCOMA VIRUS-TRANSFORMED QUAIL CLONES 135
In contrast to the hybridization of Q-B77-1
presentedinFig.lb, only40% ofarepresentative
cDNA probewasannealed, atalogio(Crt) of3,
to nucleic acid extracted from clone Q-B77-2
(Fig. la). A greater extent of annealing was
achieved withacDNA probe transcribed from
poly(A)-selected viral 35SRNA.This result
sug-geststhat it isprincipallythosesequences
spe-cifictothe 3'region of theviralgenomethatare
transcribedincloneQ-B77-2.
The results of molecular hybridization
indi-cated thateach nonproducer clone,except
pos-sibly Q-B77-2, synthesized RNA codingfor the
gs-antigenic determinants associated with the
gag gene products. Consistent with this, each
quail clone but Q-B77-2synthesized gsantigen
detectablein the COFALassay (Table 1). The
maximum variation between COFAL-positive
clones was ca. twofold, but the cells of the
COFAL-negative clone, Q-B77-2, accumulated
less than 1/10 theamountofgsantigen that the
cellsof theCOFAL-positiveclonesdid.
The positive results of the viral RNA and
COFAL assays (Table 1) allowed thatsomeof
thenonproducerclonesmightrelease
noninfec-tiousparticles. Totestthispossibility, the cells
were grown for 16 h in the presence of
[3S]-methionine, and any virus particles released
weresedimentedfrom the supernatantfluidsby
ultracentrifugationandanalyzed by
electropho-resisonSDS-polyacrylamide gels. By this
anal-ysis, each of the clones, except Q-B77-2,
pro-ducedapproximately equivalentamountsof
typ-ical virusparticles containing P27, P19, andP15
+ P12asdidthewild-type virus-infected clone,
Q-PrA-4 (Table 1). In addition, we have
dem-onstrated thatthese virusparticles, labeled by
growthinthepresenceof[3H]uridine, bandedat
adensityofca.1.15g/mlafterisopycnic
centrif-ugation in sucrose density gradients, and that
theseparticlescontained 70SRNA (G. Seal,C.
Yeater,and W.Mason,unpublished data). The
particles released by the clones were then
as-sayed for reverse transcriptase activity, using
calf thymus DNA as an exogenous template.
DNA polymerase activity wasdetected in the
particles released by Q-PrA-18, Q-B77-10, and
Q-B77-11, butwasnotdetected in the particles
released by Q-PrA-9. Unfortunately, we were unabletodetermine whether
'S-labeled
Q-PrA-9virus contained theaand/i subunits ofreversetranscriptase, since a high background of
cell-specific proteins in the purified virus
prepara-tions reducedtheresolutioninthe region of the polyacrylamide gel containing these subunits.
Thepresence oftypical C-type particles
sug-gested that some of the clones might produce
transformation-defective virus.Despitethe
pro-duction of virus particles by four out of five nonproducer clones, none of the nonproducer
clones produced infectious but
transformation-defective virus. The supematant fluids from the nonproducer clones did not transfer
transfor-mation-defective, replication-competent avian
tumorvirusestocultures ofsusceptiblechicken
embryo fibroblasts.
Rescueof transforming viruses by
super-infection withRAV-60.Tofurther
character-ize virus expression in the transforned quail
clones, we haveattempted to rescue viral
trans-forming
andhost-range
determinantsby
super-infectionwith RAV-60 avianleukosisvirus. The
rescueof viral RNA sequences
specifying
trans-formation (src) wasdetected in the focus assay.
Simultaneous rescue ofsubgroup A or C host
rangedeterminants was recognized by theability
ofthe rescuedtransforming viruses to forn foci
onC/BE cells resistanttothesubgroupEhost
rangeof RAV-60. Wewereabletorescue
focus-forming virus fromallof thenonproducer clones
(Table2). The absolute efficiency of rescue of
focus-forming virus fromQ-B77-2was approxi-mately 100-fold lowerthan from any otherclone, suggesting a defect either in the packaging of
the transforming gene into virions or in the
utilization of thesesequencesduringinitiation of infection in the focus assay. Moreover,
trans-formingvirusrescued fromQ-B77-2did notform
foci on C/BE, indicatingthatsubgroup Chost
range determinants were absent or defective in
this clone. Therelativelylowefficiencyof rescue
ofsubgroup C virus fromQ-B77-10suggeststhat
thisclone, also, mightcarryadefectiveenvgene. Fortheremainingnonproducerclones, the
effi-ciency of rescue of transforming virus of the
parentalhost rangemoreclosely
paralleled
thatseen with the producer clones. We have not
determined whetherrescueoftheparental host
rangeoccurred bycomplementationor as a
con-sequence of intragenic recombination in env
(39).
Theseresults suggest that viral RNA
synthe-sizedby allthenonproducerclones,except
pos-sibly Q-B77-2,wasefficiently packagedinto
vir-ions released aftersuperinfectionwith RAV-60.
Thus, these mutantsdiffer from thepackaging
mutant, rdSE21PR-E-(M. Linial, E. Medeiros,
and W.S.Hayward, Cell,inpress).Inaddition,
all the mutantsexceptpossibly Q-B77-2 coded
for the determinants of the host range of the
parental viruses.
Analysis ofintegrated viral DNA.
Proba-blythemostinformativeproperty ofthe
defec-tive quailclones that wehave examined is the
statusoftheintegrated viralDNA.Forthiswe
haveuseddigestion ofcellDNAby one of
sev-VOL. 30,1979
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 2. Rescueoftransformingvirusesby superinfection withRAV-60a
Virus production
Quailclone" (FFU/ml)
assayed
(Q/BD)/(C/Quailcln'on: BE)C
C/BE Q/BD
Q-PrA-4 3.4 x105 9.3 x104 0.27
+RAV-60 1.3x 105 6x 105 4.6
Q-PrA.9 <10d <10
+RAV-60 7.1x 104 2.6x 104 0.37
Q-PrA-18 <10 <10
+RAV-60 2x lo, 2.8 x105 1.4 Q-B77-1 1x 104 9.4x 102 0.094
+RAV-60 2.1x 104 2.8x 105 13.3
Q-B77-2 <10 <10
+RAV-60 <10 6.8x102 >68 Q-B77-8 6x103 ca. 3x 102 ca. 0.05
+RAV-60 5x 103 9.8x104 19.6
Q-B77-10 <10 <10
+RAV-60 1.1x 103 5.8x 104 52.7
Q-B77-11 <10 <10
+RAV-60 2.8x 104 1.9X 105 6.8 Q-BH-RSV(-)cl. 3 <10 <10
+RAV-60 <10 1.8x 105 >1.8x 104 Controls
wtPR-A 1.7x 106 6.4x 105 0.38
wtB77 8x105 1.lX105 0.14
'Atotal of106cells per 60-mm-diameterpetri dish were infectedwith 0.2ml of RAV-60 andpassagedtwice at a 1:2 dilutionat 6- to11-day intervals.Supernatant fluids harvested 3 to5days after the secondpassage wereassayed for focus-forming units(FFU)permilliliteronC/BE chicken embryo fibroblastsor onquail embryofibroblasts.
h Data arepresented for cellsthatreceivedRAV-60aswell asforcells thatwere notsuperinfected. Q-PrA-4, Q-B77-1, andQ-B77-8areproducerclones.Q-BH-RSV(-)clone 3 has beendescribed(7).
CRelativeplating efficiencies ofsubgroups A, C, and E avian viruses onC/BEandquailembryo fibroblasts.wtPR-A andwtB77weregrown onchickenembryo fibroblasts.
d<10, Focus-formingunits were not detected in 0.3 ml of supernatantfluids.
eral restriction endonucleases, followedby
aga-rosegelelectrophoresis of theDNAfragments.
As in apreviousstudy (27), unintegrated viral
DNA wasnotobserved in thetransformedquail
clones (datanotshown).
In a previous study of the integrated viral
DNA ofwtPR-A or wtB77 productively
trans-formed clones ofchicken, quail,orduckembryo
fibroblasts,weobserved alargeterminalrepeat
(LTR) ofatleast300basepairsateach endof
the integrated viral DNA (27). The restriction
endonuclease PvuI makesone cut in the LTR
and releases fromintegratedviral DNA a
frag-ment of unit length (5.85 x 106 daltons [5.85
Mdal]); that is, a fragment the size of a transcript
ofthe viral RNA, minus the poly(A) and one
copy of the short terminal repeat. Digestion of the integrated DNA of the nonproducer clones also yielded a single fragment, equal to, or in
some caseslarger or smaller than, unit size (data
notshown). This result suggested that the
inte-grated viral DNA of the nonproducers also
con-tained theflankingcopies of the LTR but that
insome clonesinterveningsequences had been
lost. Moreover, as with the producer quail
clones, analysisoffragmentsproducedby
diges-tionwith the restriction endonuclease KpnI
sug-gested that each clone containedasingle
inte-grated provirus.
KpnI cleaves viral DNA once,approximately
in the middle of the genome (33; P. Shank, S.
Hughes, H. J.Kung,R. V. Guntaka, H.E.
Var-mus, and J. M. Bishop, personal
communica-tion). Thus, if viral DNA within a cloned
popu-lation is integrated at a unique site, two
cell-virusjunctionfragments should bereleased by
KpnI, the size of thefragmentsdepending upon
the location ofadjacent cellularKpnI sites
rel-ative to the site ofintegration. In Table 3 we
show that for all the clones but Q-B77-2 two
cell-virusjunctionfragmentswereobtained. The
fragment sizes are different between clones,
sug-gesting that the integration site on the host
DNAis different in each.Q-B77-2 yielded only
asingle fragmentof9.5Mdal. Results presented
below suggest that Q-B77-2 has lost the viral
KpnIsite,implying thatthe 9.5-Mdalfragment
contained the entireprovirus. Theresultswith
KpnIindicate that eachclonecontains one
pro-virusintegrated atasingle site, butdoesnot rule
outthe possibilitythatthe clones also contain
some viral sequences integrated at random
within each clonedpopulation.
The restrictionendonuclease EcoRI cuts the
integrated viral DNAat a site in the LTR, as wellas at two intemalsitesonthe viralDNA, yieldingthreerestrictionfragmentsof 2.35 Mdal
(A), 1.96 Mdal (B), and 1.53 Mdal (C),
respec-TABLE 3. Massof cell-virus DNA junction
fragmentsreleasedbythe restrictionendonuclease Kpnja
Quaflclone KpnIjunctionband (Mdal)
Q-PrA-9 ... 12.2,6.4
Q-PrA-18 ... 8.1,7.1
Q-B77-1 ... 13.7,11.5
Q-B77-2 ... 9.5
Q-B77-10 ... 6.9,3.3
Q-B77-11 ... 13.0,6.6
Q-PrA-4 ... .... 9.3,3.1
Q-B77-8 4.4,3.8
aSeetextfor details.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.501.263.454.530.656.2]AVIAN QUAIL CLONES 137
tively, representing the entire viralgenome(11)
(see Fig. 3). Relative to the 3' terminus of the
viral RNA genome, these fragments appear in
the order B, A,C (33).
The resultsofan EcoRIanalysis of the
inte-grated viral DNAs of the defective quail clones
are shown in Fig. 2. Lane 1 shows an EcoRI
digestion of unintegrated linear viral DNA in-termediates which yielded fragments A, B, and
C. Lane 2 shows the EcoRI digest of the
pro-ducerclone, Q-B77-1, with the surprising result
thatfragment B isreplaced byanewfragment
(B') which is 0.17 Mdal larger than B. The
EcoRI analysis of the nonproducer clone,
Q-B77-10 (lane 3),suggeststhatadeletion of0.09
Mdal has occurred infragment B, yielding
frag-mentB". InQ-PrA-9cells(lane4),fragmentsA
andCaremissing, whilea newfragment,A +C,
appears. A + C is 0.6 Mdal smaller than the
summed molecular massesoffragmentsAand
C, suggestingthat adeletion in either A,C, or
both has occurred.CloneQ-B77-2haslost
frag-mentsAandC, indicatingdeletion of substantial
portion(s) of both thesefragments (lane 5). In
lane 6 we show that certain common bands
observed in all the clones alsohybridizedwitha
ribosomalcDNAprobe, indicating thatourviral
cDNA probe had some contamination with
ri-bosomalsequences. As discussed in aprevious
report(27),even1% contamination of theprobe
withrRNA-specificsequencesduetopackaging
ofasmallamount ofrRNA into virions would
leadtodetection of ribosomal DNA because of
the reiteration of these sequences within the
cellulargenome.The EcoRIfragmentsof clones
Q-PrA-18andQ-B77-11 wereofnormalsize, at
thelevelofresolutionaffordedbythisanalysis.
Tomorepreciselylocate the deletions and the
insertion/duplication detected by our EcoRI
analyses of the defectivequail clones, we
exam-ined theproductsofdigestionof each clone with
the restriction endonucleases PvuI, XhoI, and
BglII, as appropriate. The cleavage sites for
these enzymes on integrated viral DNA are
shown in Fig. 3. The results ofour analysis of
each defective clone are also shown in Fig. 3,
which presents a schematic representation of
the size and location of each deletion and inser-tion orduplication. As shown, Q-B77-1 has an
insertion of0.17 Mdal, in src, Q-B77-10 has a
deletion of 0.09 Mdal in env, Q-PrA-9 has a
deletionof 0.41 Mdalinpoland/orenv, and
Q-B77-2hasadeletion of3.97Mdal encompassing
gag,pol, andenv. Although the Q-B77-10
dele-tion maps near the env-src junction, we have
placed this deletion in env because this clone
does notproduce infectious virus; however,some
overlapintosrccannotyet be ruled out.
Q-PrA-1 2 3 4 5
A
B'
B
B"
6
7
1-14.7
5.85
4.09
2.63
1.40
1.22
FIG. 2. Agarose gel electrophoresis of EcoRI re-strictionfragmentsof integrated proviral sequences in transformed quail clones. From 4 to 8pgof cell DNA was digested with 4 units ofEcoRIper Ag,
followed byslabgel electrophoresis, transferto nitro-cellulosefilters, andhybridizationwith[3P]cDNA.
Lanes 1-5,hybridizationwith a cDNAprobetoviral 70SRNA;lane6,hybridizationwith a cDNA probe topurifiedrRNA. Lane7, HindIIIdigest ofphage A DNAlabeled with[32P]dCTP,in vitro, as described (33). Lane 1,unintegrated intermediates (11); lane 2,
Q-B77-1; lane 3,B77-Q-10; lane 4, Q-PrA-9;lane 5,
Q-B77-2;lane 6, Q-B77-2. The numbers on theright arethe masses inmegadaltonsoftheHindIII frag-ments of phageA DNA, used as molecular weight markers. The notations on theleftareexplained in the text. The twofragmentsatthe topof the gels, not detected with a ribosomalprobe, are also seen in uninfected quail cells, as are the ribosomal
frag-ments.
9mayalsocontain amutationnearthe junction
ofgag andpol, since the EcoRI cleavage site location in this region is missing in Q-PrA-9.
Although Fig.3 shows thedeletions and
inser-tion(s) in the mutants as single changes, it is
importanttonotethat thechangesinsize of the
DNAbetweenadjacentrestrictionendonuclease
sites mayactuallyresultfromanumberof
non-contiguousdeletionsor insertions.
VOL. 30,1979
A+C
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.501.259.449.72.378.2]138 MASON ET AL.
PH1877 rdPH10877.
rdPH9PR-A -0.41 t 0.04
rdPH2B77 -3.97t0.11
oPvuI
10- KO env
o Xho I
Eco RI 6 4
V§jE MW (Md)
Kpn I
10 5
Kb
FIG. 3. Thesizesandlocationsof
deletions on theintegratedproviral Q-B77-1, Q-B77-10, Q-PrA-9,and
Q
mutationsinPHIB77-C, rdPHIOB77andrdPH2B77-C.Cleavagesitesof
nucleases PvuI, XhoI, EcoRI, BglII personal communication), andKpni
ontheproviral DNAgenome. The ci
correspondstothe 5'terminusofthe
complementary totheviralRNA. (
additionalsequenceswerelocated b
siteat1.09Mdal and the XhoI site ai
ofthese sites are located on src ( personalcommunication).The size o
duplication,0.17+0.04Mdal,wasd
parison of EcoRIand BglII fragm with those of unintegrated DNA
(rdPH1OB77-C)The deletionwasloc XhoI site at 1.45Mdal and the Ec
Mdal. The sizeofthisdeletion, 0.a was determinedfrom analysis ofI
digests. (rdPH9PR-A) The deletion theleftoftheKpnI siteat2.71Mda experimentindicated that thedeleti
therightofanHpaIsiteat4.22M
TheBglII siteat 3.26Mdal is mis
that the mutation encompasses thi
Thesizeofthedeletion, atleast0.
was derivedfrom thefragmentspr and adoubledigestionwithPvuI an
theEcoRIdataindicateda
slightl)
0.6Mdal. Although themajordele asmappingtotherightoftheHpaI
analterationhas alsooccurredtot)
since the EcoRI site at4.4Mdalt
(Fig.2).(rdPH2B 77-C) Digestion wi
fragment of
size 1.76Mdal, wherecEcoRIyielded a singlefragmentc presenceofsrcwas indicated by ti
gene by superinfection with RAV-6
believe, therefore,that the1.9-Mdal is released bycleavagesatthe Eco Mdal andeither2.06or 5.95Mdalt nus.Inthefigure,theopen brackets tionsofthedeletions andinsertion/
the hatchedorfilled brackets ind should be noted that the mutations
tplesites within the indicatedlocat
+0.17t0.04 In thefollowingsection, we present a
prelim--0.090±0.025
mary
analysis
of theintracellular
viralproteins
synthesized by one of the nonproducer clones, Q-PrA-9.
Clone Q-PrA-9 synthesizes a defective form of Pr180. The resultsof our analysisof
0 integrated viral DNA indicated that Q-PrA-9
has adeletionmapping near the junction of env
src andpol. If the DNAsequences detected by our
| analysis were transcribed into viral mRNA
spe-2 0 cific forpol,the reversetranscriptase precursor,
Pr180 (9, 20, 26), would not be synthesized.Cells
o were labeled with[35S]methionine, and
cell-as-sociated viralproteinswereimmunoprecipitated
'theinsertions or with anantiserumto whole virus. As
expected,
IDNA inclones
Prl80
was notsynthesized in Q-PrA-9cells(Fig.?-B77-2,
defining
4).Instead,
wedetected a novel protein(desig-r-C, rdPH9PR-A, natedX) withamolecularweight of ca. 130,000
restrictionendo- to
140,000.
Pr76 appears to be ofnormal
size;Iare alsoshown however, processing of Pr76 to thegag structural
ircleontheright
!DNA transcript a b c d e f g h
PHIB77-C). The
PetweentheBglII Aft..
tl45MdalBoth 1 -u '. X
P.Shanketal., PriSO8 )ftheinsertionor X Serivedfrom
com-wents of Q-B77-1
iintermediates. Pr
76-atedbetween the coRI site at 2.06
VhoIand EcoRI;qgi V
twas located to
il.Apreliminary ..
on waslocated to ...
rdal(not shown).
ssing, suggesting
s site, as shown.
.41 ± 0.04Mdal, roduced by XhoI dKpnI, although
ylargerdeletion,
rtionis indicated site at 4.22 Mdal ieleftofthissite,
was not detected th PvuIyielded a isdigestion with
)f1.9Mdal. The he rescueofthis 50 (Table2). We EcoRIfragment
)RIsites at0.095 fromthe 5' termi-sdefinethe loca-'duplication, and 'icate the size. It could be at mul-tions.
P19~~~~~~
P12/15-inbdb
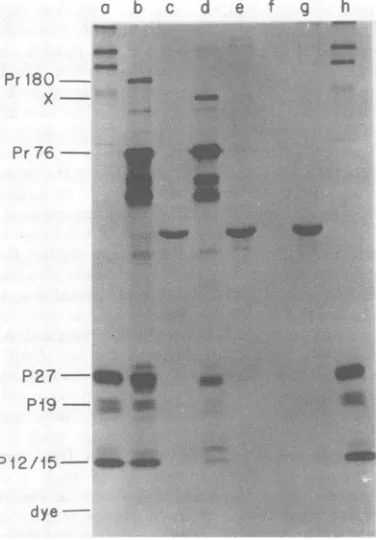
dye-FIG. 4. Immune precipitation of viral proteins
fromlysatesof Q-PrA-4, Q-PrA-9, and Q-B77-2. The procedure is described in the text. Electrophoresis was ona12.5%(wt/vol)polyacrylamidegel.Lanesa,
h:WtPR-A,grown in chickenembryofibroblastsand
purifiedasdescribedin thetext. Lane b: Q-PrA-4, anti-gs. Lane c: Q-PrA-4, anti-human serum albu-min. Lane d:Q-PrA-9, anti-gs.Lanee:Q-PrA-9, anti-human serum albumin. Lane f Q-B77-2, anti-gs. Laneg: Q-B77-2, anti-human serum albumin.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.501.61.251.55.214.2] [image:7.501.267.455.290.560.2]proteins P27, P19, P12,and P15 may be partially
defective. Byradioactivelabeling, Q-PrA-9 cells contained lessprocessedgagproteins and
pro-ducedslightly less virus than Q-PrA-4cells
(Ta-ble 1). Since the apparent reduction in the size
of Prl80 (ca. 40,000 to 50,000daltons) is greater
than predicted from the size of the deletion,
which issufficienttoaccountfor ca. 21,000
dal-tons, atleast part of the deletion probably maps
at aninternallocation in pol, rather than at the
3' endofpol or across the pol-envjunction.
As predicted from the results of the DNA
mappingand COFALexperiments, Q-B77-2did
notsynthesize any gsproteinsrecognized by an
antiserum raised against total virion proteins
(Fig. 4).
DISCUSSION
We have describedapreliminary
characteri-zationoffivetransformedquailclones with
non-conditionaldefects in virusreplication.Inthree
instances, restriction enzyme analysis has
re-vealed that the defect is a consequence of a
deletionin theviral genome. The mechanism of
formation of thesedeletionsis unknown. A
com-mon feature of the defective clones we have
studied is the maintenance ofan LTR. This
maintenance is consistent with the proposed
importance of the LTR in the initiation and
termination oftranscription ofintegratedviral
DNAinto RNA, as discussed inaprevious
re-port(27).
Although 7nonconditionalmutants were
ob-tainedoutof18clonesexamined,inasubsequent
experiment withcells ofadifferentquail embryo
andusinga newfocus clone ofwtPR-A, only1
transformedclone in35failedtoproduce
prog-enyvirus. A similar low level of defectiveswas
reported previously in RSV-transformed quail cells, and mutantswithnonconditional defects
inpol (18) and in thepackaging of viral RNA
(Linial et al., Cell, in press) were described.
Modified viral genomes in RSV-transforned
mammnlian
cellshave been foundbyP.Shankand co-workers (P. Shank, S. Hughes, H. J.
Kung,J. E.Majors,N.Quintrell,R. V.Guntaka,
J. M.Bishop,and H. E.Vannus,personal
com-munication)andbyK.Steimerand D.Boettiger (personal communication; 31). In addition, S.
Martin and co-workers (personal
communica-tion) have obtained a number ofdeletion
mu-tants similar tordPH2B77-C, using UV-inacti-vated RSV.
Irrespectiveof themechanismsbywhich
dele-tionsandinsertionswereformed, several
poten-tiallyusefulmutantsaredescribedinthisreport. Q-B77-1 is transformed and produces
phenotyp-ically wild-type virus (PHlB77-C), yet has an
insertion in src sufficientto code for an
addi-tional80 aminoacids. It is difficult to envision theretentionofafunctionalsrcproduct
follow-ingsuch alarge alteration, and it will be
inter-esting to see whether or not these cells
synthe-size alarger form of the src gene product, P60
(4, 12). Altematively, the additional sequences
may be removed by RNA splicing or protein
processing.
Theprovirus of rdPH9PR-A has a deletion in
polandaphenotype reminiscentof the
Schmidt-RuppinRSVmutant,rdNY8a (13).Unlike this
mutant,however, rdPH9PR-A has afunctional
envgene, and may be similar to a48T, isolated
as a recombinant between the Bryan strain of
RSV and RAV-49 avianleukosis virus (25). In
contrast to a48T, rdPH9PR-A has a
nondefec-tive parent and should be useful for precise
mappingofpolonthewild-typegenome.
Finally, rdPHlOB77-Chas adeletion near the
3' end of env and may, therefore, be a gp37
mutant(14, 29). This mutantcouldproveuseful
inanalyses of the processing of the glycoprotein
precursor Pr95(6, 9, 14, 24, 29)togp85 and gp37,
and in the assembly of the gp85-gp37 dimer,
VGP (17).
ACKNOWLEDGMENTS
We aregrateful toH.Hanafusa (Rockefeller University, New York, N.Y.), E. Hunter (UniversityofAlabama, Bir-mingham,Ala.),andR.Friis(JustusLiebig Universitat, Gies-sen, WestGermany)forprovidingvirusesand to R. Guntaka (Columbia University, New York, N.Y.) for providing the unintegrated viral DNA intermediates.
Thiswork wassupported bygrantsVC-155C and NP-241 from the AmericanCancerSociety, byPublicHealth Service grantsRR-05539, CA-06927, CA-16641, and CA-19497from the NationalInstitutes ofHealth, bygrantPCM76-81525from theNational Science Foundation, andbyanappropriation from theCommonwealthofPennsylvania.
LITERATURE CITED
1.Blattler,D.P., F.Garner,K.VanSlyke,andA. Brad-ley. 1972. Quantitative electrophoresis in polyacryl-amidegelsof 2-40%.J.Chromatogr.64:147-155. 2. Bonar,R. A., R. Ishizaki, and J. W. Beard. 1972.
Immunoelectrophoretic analysis of avianribonucleic acidtumorvirusgroup-specific antigens.J.Virol. 9:90-95.
3. Bonner,W.M.,and R.A.Laskey.1974.Afilm detection methodfor tritium-labeledproteinsand nucleic acids in polyacrylamide gels. Eur.J. Biochem.46:83-88. 4.Brugge,J.S.,and R. L.Erickson.1977.Identification
ofatransformation-specific antigeninducedbyan avian sarcomavirus. Nature269:346-348.
5. Eisenman, R., R. Shaikh, and W. S. Mason. 1978. Identification ofanavianoncovirus polyproteinin un-infected chick cells.Cell14:89-104.
6. England,J.M.,D. P.Bolognesi,B.Dietzschold,and M.S.Halpern.1977.Evidence thataprecursor glyco-proteinis cleaved toyieldthemajor glycoprotein of aviantumorvirus. J. Virol. 21:810-814.
7. Friis, R. R., W. S. Mason,Y. C. Chen, and M. S. Halpern.1975. Areplicationdefectivemutantof Rous sarcomavirus whichfailstomake afunctionalreverse
on November 10, 2019 by guest
http://jvi.asm.org/
140 MASON ET AL.
transcriptase. Virology 64:49-62.
8. Hanafusa,T.,H.Hanafusa,and T.Miyamoto. 1970. Recoveryofanewvirus fromapparentlynormal chick cells by infectionwithavian tumor viruses. Proc.Natl. Acad. Sci. U.S.A.67:1797-1803.
9. Hayman, M. 1978. Synthesis and processing of avian sarcoma virusglycoproteins. Virology85:475-486. 10.Hayman,M.J. 1978.Viralpolyproteinsinchickembryo
fibroblasts infected with avian sarcoma leukosis viruses. Virology85:241-252.
11.Hsu, T.W.,J. L.Sabran, G.E.Mark,R. V.Guntaka, and J.M.Taylor.1978.Analysisofunintegrated avian RNAtumorvirus double-stranded DNAintermediates. J.Virol.28:810-818.
12.Jay,G., R.P.C.Shiu, F. T. Jay,A. S.Levine, andI. Pastan. 1978. Identification ofa transformation-spe-cificprotein inducedbyaRous sarcoma virus.Cell13: 527-534.
13. Kawal, S., and H. Hanafusa.1973.Isolation ofdefective mutantofaviansrcomavirus. Proc.Natl.Acad. Sci. U.S.A.70:3493-3497.
14.Klemenz, R., and H. Diggelmann 1978.Thegeneration of the twoenvelope glycoproteinsof Rous sarcomavirus froma common precursorpolypeptide. Virology 85:63-74.
15.Laemmli, U.K. 1970. Cleavage of structural proteins during theassembly of thehead ofbacteriophageT4. Nature(London)227:680-685.
16.Laskey,R.A., andA. D.Mills.1975.Quantitativefilm detectionof3H and"4Cinpolyacrylamide gels by fluo-rography.Eur. J. Biochem.56:335-341.
17.Leamnson, R.N., and M. S.Halpern. 1976.Subunit structure of theglycoproteincomplexof avian tumor virus. J.Virol.18:956-968.
18.Linial, M., S.Brown, andP.Neiman.1978.A noncon-ditionalmutantof Roussarcomaviruscontaining de-fectivepolymerase.Virology87:130-141.
19. Lowry, 0.H.,N. J.Rosebrough,A.LFarr, andR. J. Randall. 1951. Protein measurement withthe Folin phenolreagent. J. Biol.Chem.193:265-275.
20.McGinnis, J.,A.Hizi,R.E. Smith, and J. P. Leis. 1978.Invitrotranslationof a180,000-dalton Rous sar-comavirusprecursorpolypeptide containing both the DNApolymerase and the group-specific antigens. Vi-rology84:518--522.
21. McLaughlin, C. S.,J. R.Warner,M.Edmonds,H. Nakazato, andM.H.Vaughan. 1973.Polyadenylic acidsequences in yeast messenger RNA.J. Biol. Chem. 248:1466-1471.
22. Mark,G.E.,J. M.Taylor,B.Broni,andR. M.Krug. 1979. Nuclear accumulation of influenza viral RNA
transcriptsand the effects ofcycloheximide, actinomy-cinD, and a-amanitin. J. Virol.29:744-752.
23. Mason, W.S., and C. Yeater.1977.A mutantof Rous sarcomaviruswithaconditionaldefect in the determi-nant(s)ofviral hostrange.Virology77:443-456. 24. Moelling, K.,and M.Hayami.1977.Analysisof
precur-sors to theenvelopeglycoproteins of avian RNA tumor
virusesinchicken and quailcells.J. Virol.22:598-607. 25.Murphy,H. M. 1977. A newreplication-defectivevariant oftheBryanhigh-titer strain of Rous sarcoma virus. Virology77:705-721.
26. Oppermann, H.,J.ML Bishop,HE E.Varmus, andL. Levintow. 1977. A jointproductof the genes gagand pol ofavian sarcoma virus: a possible precursor of reversetrnscriptase.Cell12:993-1005.
27. Sabran,J. L., T.W.Hsu, C. Yeater, A. Kaji,W.S. Mason,andJ. M.Taylor. 1979. Analysis of integrated avian RNA tumorvirus DNAintransformed chicken, duck, and quail fibroblasts. J. Virol.29:170-178. 28.Sarma,P.S., T. Log,R. J.Huebner, andH.C. Turner.
1969. Studies of avianleukosis group-specific comple-mentfixingserumantibodies inpigeons.Virology37: 480-483.
29. Shealy,D.J., andR. R.Rueckert. 1978.Proteinsof Rous-associatedvirus 61, anavian retrovirus: common precursor for glycoproteins gp85 and gp35 and use of pactamycin tomaptranslational order of proteins in the gag,pol,andenvgenes. J.Virol.26:380-388. 30. Southern, E. M. 1975.Detection ofspecificsequences
among DNAfragmentsseparated by gel electrophore-sis.J. Mol.Biol. 98:503-517.
31. Steimer,K.S., andD.Boettiger. 1977. Complementa-tion rescueof Roussarcoma virus fromtransformed
mammalin cells bypolyethyleneglycol-mediated fu-sion. J. Virol. 23:133-141.
32. Suzawa,H.,T. Sugimuri,Y. Moira, and T.Shimiza. 1966.Specific complementfixation test of Roussarcoma with pigeon serum. Natl. Inst. Anim. Health Q. 6:208-215.
33. Taylor,J. M., T. W. Hsu, andM. M.C. Lai. 1978. Restriction enzyme sites on the avian RNA tumor virus genome. J.Virol.26:479-484.
34. Taylor,J.M.,R.Illmensee,S.Litwin, LHerring,B. Broni,andR.M.Krug. 1977. Useofspecificprobes to studytranscriptionandreplication oftheinfluenzavirus genome. J.Virol.21:530-540.
35.Taylor,J. M.,R.[Umensee,and J. Summers. 1976. Efficient transcrption of RNA into DNA by avian sarcomaviruspolymerase.Biochim. Biophys. Acta442: 324-330.
36.Vogt, P.K. 1969. Focus assay of Rous sarcomavirus,p. 198-211.In K.Habel and N. P.Salzman(ed.), Funda-mental techniques invirology. Academic Press, New York.
37. Vogt, P. K., R.A. Weiss, and H. Hanafusa. 1974. Proposal for numberingmutantsof avianleukosis and sarcoma viruses. J. Virol.13:551-554.
38. Weiss, R.A., W. S. Mason, and P. K. Vogt. 1973. Genetic recombinantsandheterozygotesderivedfrom endogenous andexogenousavianRNA tumorviruses. Virology 52:535-552.
39. Wyke, J. A., J. G.Bell, andJ. A.Beamand. 1974. Genetic recombination among temperature sensitive mutants of Rous sarcomavirus. Cold SpringHarbor Symp.Quant. Biol.39:897-905.
on November 10, 2019 by guest
http://jvi.asm.org/