0022-538X/83/050513-10$02.00/0
Copyright C 1983,AmericanSociety forMicrobiology
Replication of Vesicular Stomatitis
Virus
Defective
Interfering
Particle RNA
In
Vitro: Transition
from
Synthesis
of
Defective
Interfering
Leader RNA
to
Synthesis
of
Full-Length
Defective
Interfering
RNA
GAIL W. WERTZ
DepartmentofBacteriology andImmunology, School ofMedicine, UniversityofNorth Carolina,Chapel
Hill, NorthCarolina 27514
Received 15November1982/Accepted25January1983
The replication of the RNA of vesicular stomatitis virus (VSV) defective interfering (DI) particles was established in a defined cell-free system. The transition from synthesis of only the DI-leader RNA to replication of the full-length DI RNA was effected in the systemby newly synthesized VSV proteins and occurred in the absence of VSV helper virus. Bothpositive- and
negative-polarityfull-lengthDI RNAweresynthesized. Furthermore,theproductsof RNA replication associated with newly synthesized viral proteins toform complexes thatwereindistinguishable from authentic DI particle nucleocapsidsonthebasis ofbuoyantdensity andresistancetoribonucleasedigestion. TheDI-leader RNA did not form ribonuclease-resistant structures. We conclude that this in vitro
systemsuccessfullyexecutesmanyof the reactions of VSV DI particle replication and assembly.
Oneof themajordifferences between thetwo
RNAsyntheticreactions, transcriptionand
rep-lication, that are carried out by the
negative-strand RNA virus, vesicular stomatitis virus (VSV), is thatreplication requiresviral protein synthesis, whereas transcription does not. The template for both RNAsyntheticreactions is the
negative-strandgenomicRNA(4 x 106 daltons)
in theform ofanucleocapsid structure, that is,
coated with the nucleocapsid protein, N, and
associatedwith thephosphoprotein, NS,and the large protein, L, which are components ofthe RNA polymerase. This structure is capable of
carryingouttranscriptionof thegenometoyield
leader RNAand the five VSVmRNAs(2, 10). Atpresent, we do not known precisely what
protein or proteins are required to effect and
maintain the transition from the synthesis of leader RNA and the discrete mRNAs (tran-scription) to the synthesis ofacomplete
read-through product to yield a full-genome-sized plus strand RNA, which issubsequentlyusedas
the template forsynthesis ofthe progeny
nega-tive-strandRNAgenomes(replication). Incells,
the full-length genomicRNAproducts of repli-cationarefoundonlyin the formof nucleocap-sids and are therefore resistant todigestion by
ribonucleases, whereas the mRNA products of
transcription are completely sensitive to
diges-tion by nucleases (30). Since the products of replicationareprotein-coatedRNAs, it has been
postulatedby numerousworkers thata
require-mentfor thenucleocapsid structural protein, N,
mayconstitute the need for continuous protein synthesis in negative-strand virusRNA replica-tion. It has been proposed that Nprotein may
play a specific role in catalyzing the transition from transcriptiontoreplication by bindingto a
site in leader RNAwhichmaybe thenucleation site for encapsidation (1, 14, 21). This event
would be dependent on the availability of N
protein and would determine the balance
be-tween replication and transcription. Ithasbeen proposed specifically that N functions to sup-press aterminationsignalatthe endof the leader
gene and that the binding of N within leader
simultaneouslystartsnucleocapsid assembly(5,
21).
Itis also possible that other newly synthesized
proteinsarerequiredtopromotereplication. For
example,theswitchtoreplicationmayrequirea newly synthesized L molecule, an L molecule
that has been modified by association with a
newly synthesized form ofthe phosphoprotein NS, which has been shown to exist in two
distinct phosphorylated forms (6, 15), orboth.
Additionally, itispossiblethat hostfactorsmay
playarole inreplication (24).
To investigate the requirement for protein synthesis in RNAreplication,adefined in vitro
system has been designed that supports both transcription and replication of the
negative-513
on November 10, 2019 by guest
http://jvi.asm.org/
strand genomic RNA of the rhabdovirus, VSV (8). The system consists of three components: (i) purified VSV nucleocapsids as templates for RNA synthesis; (ii)an mRNA-dependent rabbit reticulocyte lysate to support protein synthesis; and(iii) purified VSV mRNAs to direct protein synthesis, if required. By using this combination of components, the level of viralprotein synthe-sis can be controlled as desiredby omission or addition of various amounts of viral mRNA. In
thissystem,replicationof thegenomicRNAis a
function of the level ofviral protein synthesis,
thereby allowing us to investigate the protein
requirements for replication.
Since VSVnucleocapsid templates carry out both RNA transcription and replication in this system, wechosetoextend our studies of
repli-cationby using a defective interfering (DI)
parti-cle asthe template forRNA synthesisto focus
solely on the process of RNA replication. The RNAof theDIparticleweselected (calledDI-T, VSI-DI 0.25; genome molecular weight, 0.9 x
106) contains only the 5' 25% of the standard virus genome;it completely lacks genetic
infor-mation forthe N, NS, M, and G proteins and contains approximately half of the information
for the L protein (19, 31). This DI particle, therefore, doesnotdirectmRNAsynthesis. The major RNA product made in vitro by the DI
particles generated from the 5' end of the genome is a 46-base RNA encoded by the ex-treme3' endofthe DIparticle RNA andcalled theDI-leader RNA(9, 28, 29). In cells coinfect-ed with DI particles and standard helper virus,
both the DI-leader andgenome-lengthDI RNA ofpositiveandnegative polarityaresynthesized (20, 25, 26). An important feature of the DI
particle RNA, for our purposes, is that the 3' terminus of thepositive strand (the template for
negative-strand replication)is identicaltothe3' terminus of the standard VSV positive strand
(13, 18). Inaddition, the 5' and 3' termini ofthe
DI RNAsarecomplementary. Therefore,the
3'-terminalsequencesof bothpositiveandnegative
RNA strands, which are the initiation sites for
RNA synthesis, are identical. Thus, this DI
particle constitutes an excellent template for investigating replication, since it is small,
con-taining only a quarterofthe genome, and does
notdirect mRNA synthesis, and yet it has the
correct sites for initiation ofreplicationand can
replicate efficiently in cells coinfected with in-fectiousVSV. Itisassumed that therequirement for helper virus is as a source ofmRNAs to
direct viral protein synthesis.
In this report we describe the replication in vitro of full-length DI particle RNA of both
positiveandnegative polarity.Thereplicationof
the DI RNA occurs in the absence of helper virus and only requires viralprotein synthesis.
Additionally, the full-length DI RNAs produced in this system are encapsidated with protein to form nucleocapsids that are indistinguishable from authentic DI nucleocapsids on the basis of buoyant density and resistance to ribonuclease digestion.
MATERIALS ANDMETHODS
Cell cultures and virus. Virus was propagated in
monolayer cultures ofBHK-21/13 cells as described
previously (33). The Indiana serotype(San Juan strain)
of VSVwasusedasstandardVSV. Stocks of the DI
particle, DI-T(VSI-DI0.25; 5' 25% of VSV genome
[31]), weregenerously provided by R. Lazzarini
(Na-tionalInstitutes ofHealth, Bethesda, Md.).
Preparation of nucleocapsids.Intracellular DI
parti-clenucleocapsidswereprepared byinfection of BHK
cellswith standard VSV at amultiplicityof infection of
1 andVSV DI particles (VSI-DI 0.25 was used in all
cases) at multiplicities indicated. DI nucleocapsids
were extracted from mixedly infectedcells by Dounce
homogenization, and DI particle nucleocapsids were
separated from standardvirusnucleocapsidsby
veloc-ity sedimentation in 15 to30%sucrosegradients (2 hat
38,500 rpm; Beckman SW40 rotor). The band of DI
nucleocapsids was collected and sedimented through
25% sucrose (2 h at 44,000 rpm; Beckman SW50
rotor), andthepelleted nucleocapsids were suspended
inHGD buffer(10%glycerol, 10 mM
N-2-hydroxyeth-ylpiperazine-N'-2-ethanesulfonic acid [HEPES], pH
7.6, 2 mMdithioerythritol).
Analysis of template RNA.The amount of unlabeled
nucleocapsid template RNA added to each reaction
wasquantitated by parallel preparationof
[3H]uridine-labeled templates with eachpreparation ofunlabeled
templates. The specific activity of labeled template
RNA was quantitated, and this figure was used to
calculate the amountoftemplate RNA in unlabeled
preparations made under identical conditions.
Similar-ly, the ratio ofpositive to negative strand full-length
DI RNAintemplatepreparationswasdetermined by
densitometricscanningoffluorogramsorbyexcision
ofappropriatebands fromgelsasdescribed below. In allexperiments where DIparticlenucleocapsid
tem-plateswereused,approximately1,000 pgofDI
parti-cletemplateRNAwasadded per25-,ulreaction.
In vitrosynthesis ofviral RNA and proteins. Viral
RNA and protein synthesis were carried out in the
presenceof amicrococcal nuclease-treated rabbit
re-ticulocyte lysate as described previously (8) except
thattheincubation timeswerevariedasindicated in
thefigure legends.
Analysisof RNAproducts.3H-labeled productswere
analyzed by electophoresis on 1.75% agarose-6 M
urea gels as described previously (32) or on 20%o
polyacrylamide slabgelsaccordingto the method of
Laemmli and Faure (16). DI-leaderRNA
(character-ized andkindly provided by L. A. Ball, University of
Wisconsin, Madison, Wis.)and DI particletemplate
RNA wereelectrophoresedasinternalmarkers inall
gels, and theirpositions areindicated. After
electro-phoresis, gels were fixed with 10% acetic acid and
subjectedtofluorography accordingtothemethodof
Laskey (17). Theresulting fluorogramswerescanned
toquantitate theintensityof bandsby usinganLKB
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
515
2202Ul trascan densitometer interfaced with anApple
II computer. Preflashed film was used to ensure a
linear response to low levels of radioactivity. Addi-tionally, a calibration curve of the linear response
range forfilm darkening was constructed and used to
ensurethatintensity scans were alwayscarriedoutin
thelinear range. To measure the amount of
radioactiv-ity in 3H-labeled RNA species, appropriate bands
were cut outof adried gel, using the fluorogram as a
guide.Thedried gelfragmentsweredissolved inNCS solubilizer (Amersham Corp.) at 60°C for 16 h before analysis by liquid scintillation spectroscopy.
Materials. [5,6_3H]UTP (specific activity, 35 Ci/
mmol)wasfrom ICN Pharmaceuticals.
RESULTS
Preparation of DIparticlenucleocapsids. VSV
DI paticles lack complete information for the structuralgenes, soit isnecessary topropagate
them by mixed infection of cells with standard VSV helper virus. The following method was usedto prepare DIparticle nucleocapsid prepa-rations that did not contain standard virus
nu-cleocapsidsto use astemplates forRNA
replica-tionreactions. Mixedinfections to propagate DI
particles were carried out at multiplicities that
yielded the greatest amount ofDI particle
nu-cleocapsids while giving minimum yields of stan-dard virus without totally inhibiting the infec-tion.Optimummultiplicitiesweredetermined by assaying the types of RNA synthesized inBHK
cells infected with standard virus at a
multiplic-ity of infection of 1 while the input of DI
particlesvaried. AsshowninFig. 1, decreasing dilutions of the DIparticle preparationgavehigh yields ofDIparticleRNAs, whereas synthesis of standard 40S virion RNA was greatly dimin-ished. These RNAs were analyzed on 1.75%
agarose-urea gels, which can resolve the posi-tive and negative strands of the DI particle genomicRNAs(11). The dilution of DI particles which wesubsequently employed inour
experi-ments was1:3,800,adilution which reduced the 12-hyieldof standard virus by98.6%, using the
Cooperand Bellettassay (7).
DIparticlenucleocapsidswereextractedfrom
mixedly infected cells andseparated from
stan-dard virus nucleocapsids by velocity gradient sedimentation. Thepurityof the RNAs isolated
by this procedure is shown in Fig. 2. Only the positive-andnegative-strandRNAsof DI
parti-cle nucleocapsids were detectable even after
overexposure offluorograms ofgels on which theseRNAs were analyzed. These results indi-cated that our preparations ofDI particle
nu-cleocapsids were essentially free of standard
VSV nucleocapsids and mRNA. Further
evi-dence that thepreparations were devoid of
de-tectable standard VSVnucleocapsidscamefrom
ananalysis oftheRNA products synthesized by
these preparationsin vitro (see below).
MIXED INFECTION
1
2
3
4
40S
RNA-L
RN
---*
U
=
DI
RNA(|
-,_~~
"W
m--
NS-FIG. 1. Agarose-urea gel electrophoresis of RNA
from BHK cellsinfected with standard VSV and VSV
DI particles. BHK cellswere infected with standard
VSV at amultiplicity of infection of 1 and with VSV
DIparticlesatdilutions of: lane1,1:10,000; 2,1:7,500;
3, 1:5,000;and4,1:2,500. RNAlabeled with
[3H]uri-dinefrom thestartof infectionwasharvestedat12 h
postinfection fromcytoplasmic extractsandanalyzed byelectrophoresisin 1.75% agarosegels containing6 Murea.
Analysis of RNA products: effects of protein synthesis.RNAsynthesiswasdirected by the DI particle templates in the in vitro system, either in the absence orpresence ofconcomitant viral protein synthesis programmed by added VSV
mRNA. Theproducts of thereactionwere
ana-lyzed byelectrophoresison a1.75%agarosegel
containing 6 M urea (Fig. 3). This gel system
separatedthe DI particlegenome-size positive-andnegative-strandRNA aswellasretaining the 46-base DI-leader RNA and thereby allowed
simultaneous quantitation of both DI particle-specificproducts.
Themajor product synthesized by the VSVDI
particle nucleocapsids in the absence of viral protein synthesis was the 46-base DI-leader RNA (Fig. 3, lane 2). This finding confirmed previousreports that theDI-leader is themajor
product transcribed from the genomes of
puri-fied DI particles (9). In the presence of VSV
mRNA translation, two distinct changes in the
pattern ofRNAproductswereobserved:(i) full-length DI particle RNA of both positive and
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.490.277.423.75.327.2]Di
TEMPLATES
1
2
3
DI
RNA
Hparticle genomic RNA as well.Analysisof pro-teinsynthesisinthis system(datanotshown)(8) has demonstrated that all five of the
VSV-specific proteins are synthesized. Synthesis of
theVSVproteinsis linear withtimefor up to 3 h and approximately 5 to 10 pmol ofprotein is madeduring180minina25-,ul reaction.Totest
directlywhether this transition in the pattern of RNA products was due to viral protein synthesis
or was a direct effect of the addition of viral
mRNA, protein synthesis was inhibited by the addition of cycloheximide orcycloheximideand anisomycin. Under these conditions, where pro-tein synthesis was inhibited by more than 99%, noDI full-length RNA was synthesized and the amountofDI-leader RNA made increased(Fig.
3, lane 4). These results showed that viral pro-tein synthesis isresponsible for enabling the DI
[image:4.490.75.220.62.310.2]Di RNA
PRODUCTS
FIG. 2. Analysis of RNA fromDIparticle
nucleo-capsid preparations. DI particle nucleocapsids were
isolated from cytoplasmic extracts of BHK cells
in-fected with standard VSVat amultiplicityofinfection
of 1 and DI particles at adilution of1:3,800bytwo
cycles ofvelocity sedimentation.RNA wasextracted
from nucleocapsids and analyzed as described in the
legendtoFig. 1. Lane 1, Marker RNA from DIparticle
virions; 2, RNAfrom DI particle nucleocapsids
(fluor-ogramexposed 3days);3,same aslane 2 except that
thefluorogram was exposedfor7days.
negative polarity appeared (Fig. 3, lane 3), whichcomigrated with markertemplateDI
par-ticle RNA (Fig. 3, lane 5); and (ii) therewas a
marked reduction in the amount of DI leader that was produced. The appearanceof the full-length RNAswasdependentonthepresencein the reaction mixture of all four ribonucleoside
triphosphates. This findingindicated that these products were synthesized de novo and that
they were not generated by terminal incorpo-rationorexchange of labeled nucleotide.
At high concentrations of DI particle nucleo-capsids, the synthesis of full-length DI RNA
becameless efficientand, in someexperiments,
was notcompletelydependent on the addition of viral mRNA. The reasons for this areunknown,
but onepossibilityis that thehigh concentration ofviralproteinscontributedbythenucleocapsid preparation was able to support a limited
amountofRNAreplication.
Thedatapresentedabovesuggested that pro-tein synthesis directed by VSV mRNAs was
responsible forthe transition from synthesizing only leader RNA to synthesizing full-length DI
1
2 34
5
DI RNA
= -- (+)DI-Leader
additions:
mRNA
cyclo
0++
0./0+
FIG. 3. Agarose-urea gelelectrophoresis ofRNA
products synthesized byDI particlenucleocapsidsin
vitro.RNAproducts were labeled with[3H]UTP in the
cell-free systemprogrammed withthefollowing
com-ponents:lane 1,noDItemplates; 2, DItemplates, no
VSV mRNA; 3, DI templates, VSV mRNA; 4, DI
templates, VSV mRNA, and cycloheximide (50
,g/ml); 5, markerDItemplateRNA. Incubationwas
for 180 minat4°C. RNAwasextracted andanalyzed
asdescribed inthelegendtoFig.1.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.490.277.423.284.575.2]REPLICATION OF
particlenucleocapsidstocarryoutthesynthesis of full-length RNA copies.
Quantitation of RNA products. The reduction in DI-leader synthesis under conditions where thereplication offull-length RNAwasoccurring
promptedus toquantitate theamounts of these
two products. The relative amounts of full-length DI RNA and DI-leader RNAwere
quanti-tated by scanning laser densitometry of
fluoro-grams of gels of the separated products. The
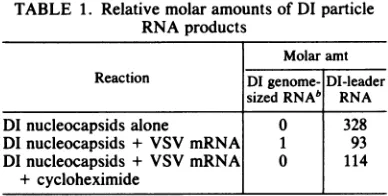
datarepresented in Table 1 show that for each 1 mol offull-length DI product (both positive and negative strand) produced in the presence of
protein synthesis, approximately 100 mol of DI-leaderwas synthesized. In the absence of
pro-tein synthesis, threefold more DI-leader was
synthesized than in the presence of protein synthesis. If cycloheximidewasaddedtoinhibit protein synthesis, more leader was made,
al-though less than was seen in the absence of added mRNA and inhibitor. These resultsarein agreementwiththeobservationthatthe amount
of negative-strand leader in infected cells is
increased in thepresence ofcycloheximide (5).
The ratio ofpositive to negative strand for full-sizedDIRNAproductwascomparedtothis
ratio forthe input template nucleocapsidRNAs.
Averaged data from five separate experiments showed that after 90minofsynthesis, the
prod-uct DI full-length RNA was composed of
ap-proximately 42% negative-strand RNA and 58% positive-strand RNA. Input template
prepara-tions contained anaverage of 57%negative and 43% positive strands. These data showed that synthesis of progeny full-sized RNA in this
systemclosely reflected the ratio ofinput
tem-plate RNAsatearly times in the reaction.Thisis theresultonewouldpredict if the full-length DI
productwascopied from the input template. At
later times in the reaction (3 h), the ratio of positive- to negative-strand products became
approximately equal. This finding may suggest
thatRNAssynthesizedatearly timescan serve as new templates for replication atlater times.
Additionally, the absoluteamountofinput
tem-plate RNAwascalculated andcomparedtothe
amountof DIfull-lengthRNAproduct by
exci-sion ofappropriate bands from gels. Approxi-mately1,000pgoftemplateRNAwasaddedper
25-IlI reaction andapproximately 400to700pg
ofDIgenome-lengthRNAwasproduced.
Kineticsof RNA synthesis. To investigate the
decreaseinDI-leadersynthesis with theonsetof RNA replication, we analyzed the kinetics of appearance ofbothDI-leader andfull-length DI RNA. For these experiments, the synthesis of DI-leaderRNAwasanalyzed byelectrophoresis ofsamples on20%oacrylamide gels followed by
[image:5.490.254.448.76.174.2]densitometric scanning of fluorograms of dried gels(Fig. 4). Initially,DI-leaderwasproducedat
TABLE 1. Relative molar amountsof DI particle
RNAproducts
Molar amt Reaction DIgenome- DI-leader
sizedRNAb RNA
DInucleocapsids alone 0 328
DInucleocapsids + VSV mRNA 1 93
DInucleocapsids + VSV mRNA 0 114
+ cycloheximide _I
a Relative molar amounts were calculated by
densi-tometric scanningof fluorograms of[3H]UTP-labeled
products separated by gel electrophoresis. Uridine
contentsof6.5% for DI-leader (28, 29) and31%for DI
particle genome RNA (27) were used in making calcu-lations.
bBoth positive- and negative-strand genome-sized
RNAs wereincludedin this calculation.
thesame ratewhether mRNAhadbeen addedto programviral protein synthesisor not.
Howev-er,after 20to40min,while DI-leader continued
toaccumulateat alinearrate in the absence of protein synthesis, therewas amarked decrease in its rate of accumulation in the presence of
ongoing protein synthesis. Indeed, there was
little additional DI-leader RNA accumulation after 40 min in reactions with ongoing protein synthesis.
Acorrespondingkineticanalysisoffull-length DI RNA synthesis showed that full-size RNA
was notdetectable until approximately60 to 90 min after the startof the reaction (Fig. 5). The accumulation of this productwasnotlinear with time, and the major accumulation of full-length DIRNAoccurredabruptly2 to3 h after thestart
of the reaction.
Association of RNA products with protein. StudiesofRNAreplicationin infected cells have shown that replication is dependenton protein
synthesis and that both positive- and
negative-strandgenome-sizedRNAsarefoundonly in the form of nucleocapsids, never as naked RNA. Having demonstrated that full-sized DI particle
RNAcould be synthesized in vitro and that its
productionwasdependentonprotein synthesis,
we nextexamined theability of the RNA
prod-ucts to associate with newly synthesized pro-teins. Theassociation was assayed in two ways.
First,thebehavior ofthe RNAproducts in CsCl density gradientswas analyzed. The
3H-labeled
DIfull-length product banded in CsClgradients
atthesamepositionasmarker DInucleocapsids (datanot shown), indicating thatthe RNA was notnaked but wasassociated withprotein and, also, that the RNA/protein ratio was
approxi-mately the same as thatof authentic
nucleocap-sids. Next, the susceptibilityof the RNA
prod-uct to digestion with ribonuclease was
VOL.46,1983
on November 10, 2019 by guest
http://jvi.asm.org/
DI-LEADER
a b c d e f
-.origin
-._
_-w
am-DI-Leader+ m0Ni
After 10 minat23°Cethylene
glycol-bis(Q-amin-oethylether)-N,N-tetraacetic acid was added to inhibit further nuclease activity. The reactions were terminated, and the RNA was extracted and analyzed inthe same manner as the undi-gested samples. Nuclease digestion of mRNA transcriptionproducts synthesized in the invitro
system by VSV virion nucleocapsids was per-formed under identical conditions to test
wheth-erthemicrococcal nuclease was active in these conditions. The results of this experiment are presented in Fig. 6.Only the full-length DI RNA products were resistant to nuclease digestion (Fig. 6, lane 6). The DI-leader RNA was digest-edby micrococcal nuclease even when made in the presence of ongoing protein synthesis. As
expected, the VSV mRNAs synthesized by
nucleocapsids from infectiousvirionswerealso
SYNTHESIS OF
DI
RNAs
A..
1
minutes 6 9 2 8
at 30 0 0 0 0 M
==
82+DI
[image:6.490.51.239.70.428.2]minutes at 30(
FIG. 4. Kinetics of accumulation of DI-leader
RNA. (A) DI-leader RNAsynthesiswascompared in
cell-free reactions programmedwith DI templates to
which VSV mRNAwas(b,d, andf)or wasnot(a,c,
and e)added. RNAproducts synthesizedin the
pres-ence of [3H]UTP added atthe start of the reaction
wereanalyzed at 20(a,b), 40(c,d),or80(e,f)minby
electrophoresis on 20% acrylamide gels. 3H-labeled product comigrated exactly with marker DI-leader
(position indicated). (B) Fluorogramsof the driedgels
were scanned with a laser densitometer, and the
relative amounts ofproductsynthesized duringeach
timeinterval wereplotted.
examined. RNA products were synthesized in
vitro in the presence orabsence of viralprotein synthesisasdescribedforFig.3.After90minof
incubation, the reaction mixtures weredivided
in half. Half of each reaction was immediately terminated by the addition of sodium dodecyl sulfate. The RNA was extracted and analyzed by gel electrophoresis. The other halfofeach
reactionmixture wasdigestedwithmicrococcal
nuclease in the presence of calcium chloride.
15-E
C3
10-0
E
'Z
5-a
6(0 20 120 lSC
minutes at 30
FIG. 5. Kinetics of appearance offull-lengtb DI
particle RNAs. (A)[3H]UTP-labeledproducts
synthe-sized in the in vitro system programmed with DI
templates and added VSVmRNAwereanalyzedat60,
90, 120, and180minafter thestartof thereaction by
electrophoresis as described in the legendtoFig.1. A
small amount of positive- and negative-strand
full-length product was detectableat60miinontheoriginal
fluorogram,but was not of great enoughquantitytobe
discernedonsubsequentphotographic exposure. Lane
Mshows marker DItemplate RNAs. (B) Fluorograms
of the dried gel were scanned as described in the
legendtoFig.4, and thekinetics of appearanceof
full-lengthproduct (bothpositive and negative strand) are shown.
A.
C c
0
c
C)
a)
B.
/~~~~~~~~~~~~~~~----J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.490.256.445.283.529.2]519
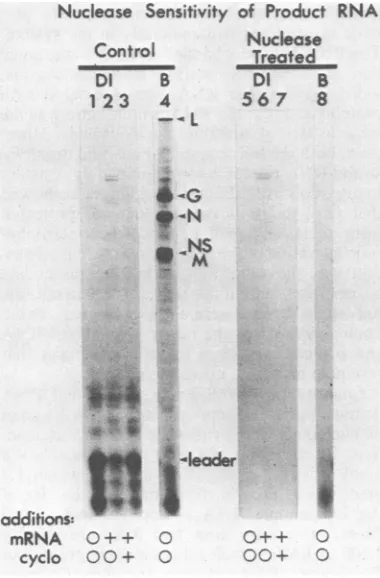
Nuclease Sensitivity of Product RNA
Control
DI B
123 4
- -L
Nuclease Treated
DI B
56 7 8
indicate that they contain N, NS, and L, the three viral proteins found inauthentic VSV and DI particle nucleocapsids (22).
DISCUSSION
.r
m
,i",p
:>
uaciitlonsu;
mRNA 0++
[image:7.490.49.239.74.363.2]cyclo 0 0+
FIG. 6. Nuclease
sizedby DI particle
RNAproducts synth
sids in thein vitros)
inthepresenceofno
5), added VSV mRI
mRNAplus cyclohe)
[3H]UTP-labeled pr(
VSVnucleocapsidsv
4and8)were analy2
after lanes 5 throug
nuclease,(10Fg/ml)
phoresisasdescribec
completely digeste 8).
These results de length DI positive
are synthesizedin
thesis-dependentri
teins made are a
newlysynthesized
are stable in 7 M
density gradientsN
cleocapsids, andti
digestion with nu4
characteristic ofni
such structures ar tem. Furthermore, found in these n
The results presented above demonstrate that it ispossible to carry out the replication of full-length VSV DI particle RNAs ofboth positive
*-G and
negative
polarity
in vitro. The ratios of@
ZN
positivetonegative
strandprogenyRNAclosely
reflected the ratio of input nucleocapsid
tem-NS
plate RNA at earlytimes in thereaction. At later M times (3 h), approximately equal amounts ofpositive and negative strand RNAs were pro-duced. The DI particle nucleocapsid templates
were able to make the transition from synthesiz-ingonly the DI-leader RNA in vitro to synthesiz-ing full-length, DI genome-sized RNA by viral protein synthesis. This transition was blocked byinhibitorsof protein synthesis. These results
show that DI RNA replication is regulated by
viral protein
synthesis
in this system, as itis inAeader. ~~~~infected cells.
The events involved in replication of DI RNA can beconsidered in more detail. In the absence
o 0+ + 0 of
protein
synthesis,
the DI-leader is theonly
0 00+ 0 product synthesized by the DI nucleocapsid template. A strong termination signal is present
sensitivity of products synthe- at the endoftheleadergene. Theoretically,any
nucleocapsids. [3H]UTP-labeled read through or elongation ofproduct beyond
iesizedby DIparticlenucleocap- theDI-leaderboundarycanbe considered
repli-ystemduring a 90-minincubation cation. The successful elongation of the product
)addedVSVmRNA (lanes1 and to yield a full genome-sized RNA, however, may NA (lanes 2 and 6), added VSV be aprocess involving different protein require-ximide (50,ug/ml)(lanes 3 and 7). ments than the initial read-through event. For
oducts synthesized by standard example, the transition from transcription to
with noadded VSV mRNA (lanes
eplcathe
mayrequire ticorichiometo
zed before(lanes 1 through 4) or
replication
mayrequire catalytic or stoichiomet-,h 8) digestion with micrococcal ric (or both) amounts of a certain protein(s) or a for 10 min at23°C by gel electro- short-lived intermediate (see above), whereas dinthelegend to Fig. 1. the efficient elongation ofproduct to yield full-length progeny may require only a constantsupply of the nucleocapsid protein N. We have notassayed directlyfor theformer eventinthis
dby thenuclease(Fig. 6, lane system at this time. Rather, we chose to assay only for the complete read-through product, a
mmonstrate not only that full- full-lengthDI molecule. Therefore, based on the >- and negative-strand RNAs studies presented here, we can say that for
thissystem in a protein syn- efficient synthesis of full-length product to oc-eaction, but also that the pro- cur, a certain concentration of viral protein must
tble to associate with these be available. The findings reported here are RNAs to formstructuresthat consistent with the requirement for continuous I CsCl, that coband in CsCl proteinsynthesistomaintaingenome RNA syn-withauthentic DIparticle nu- thesis in VSV-infectedcells (23,33).
hatarecompletely resistantto A striking aspect of the
quantitation
ofRNAclease. These properties are productswas the finding that as protein synthe-ucleocapsidsand suggestthat siswasinitiatedin the system and the synthesis eformed in this in vitro sys- offull-length DI RNA occurred, the amount of analyses of the viralproteins DI-leaderRNA decreased threefold. When the ewly synthesized structures kinetics of appearance of the DI-leader RNA
VOL.46, 1983
_>of:s:..
on November 10, 2019 by guest
http://jvi.asm.org/
were compared in the presence and absence of viral protein synthesis, it was observed that the rateofaccumulation was similar for the first 20 minof the reaction. After that time, however, while DI-leader continued to accumulate at a linear rate in the absence ofprotein synthesis, there was a marked decrease in itsaccumulation in the presence ofprotein synthesis.The appear-ance of full-length DI RNAcould not be detect-ed untilapproximately 60to90min inreactions in whichprotein synthesiswas initiatedattime 0. These datashowed that the sharpdecrease in therate of accumulation of DI-leader RNA was
followed by the appearance of full-sized DI RNA. These two events,however, did not occur untilapproximately 40 min after the start of the reaction.
Inconsidering these results, it is important to note that viralprotein synthesis was being pro-grammed from the start of the reaction and,
therefore, that nopreexisting poolof free virus proteins existed. One explanation for the results of these kinetic studies may beasfollows. After protein synthesis is initiated in the system and proteinsbegin to accumulate, anecessary
con-centration ofprotein is achieved such that the transition can be made fromtermination atthe endofthe DI-leadertoproductionoffull-length product. As this occurs, one might predict a
decrease inthe appearanceofdiscreteDI-leader
RNA as this molecule is elongated to produce
the full genome-sized product. However, this
explanationcanaccountforonlyasmall part of the decrease in accumulation of discrete DI-leader (Table 1). We do not know what other factors may be involved. It ispossiblethat the rateof initiation in the system slows downasthe result ofproduction of some new viralprotein,
orperhapstheinvolvementofpolymerase mole-cules in elongation of the DI product restricts the amount ofpolymerase available for initia-tion. Anotherpossibilityis that there isapause
atthejunctionbetween DI-leader and therestof the DIgenome,which may result inaslowingof
polymerase movement and new initiation
events. Iverson and Rose (12)haveshown that thereisadistinctpause ofapproximately 5min in the rate of VSV RNA transcription ateach intercistronicjunction. Taken together,the sim-plestexplanation of these results and onethatis consistentwith our knowledgeofreplication in theinfected cell is thatthetransitionto replica-tion offull-length RNAin this system requires
notsimplyongoing protein synthesisbut rather theavailability ofacertain concentration of viral protein before it ispossibletodetect the synthe-sis of the full-sized RNA.
Theworkdescribed abovealsodemonstrated that the full-length DI positive- and
negative-strand RNAs, made in a protein
synthesis-de-pendentreaction, could associate with the pro-teins synthesized concomitantly in the system. The RNA productsbanded in CsCl at the same buoyant density as marker DI nucleocapsids, indicating that the RNA was associated with proteinsand that the RNA/protein ratio was the sameas that ofauthentic nucleocapsids. More-over,both the full-length positive- and negative-strand RNAproducts were completely resistant todigestion with ribonuclease. Our data showed that all of thereplicated product was protected from nuclease digestion. Since the electropho-reticmigration of the RNAs after nuclease diges-tion was the same as that before, these data suggestthat, within the limits of this assay, the full-length RNAs were fully protected. These findings show that the newly synthesized RNA and proteins associate to form structures that resembleauthentic nucleocapsids.
Quantitation offull-length DI product RNA demonstrated that approximately 1.4 to 2.1 pmol ofnucleotide was synthesized in a 25-,lI reac-tion. In this same reaction, approximately 4.8
pmol of viral protein was made, ofwhich 1.7 pmol was N protein. Assuming that the size of the DI genome RNA is approximately 3,000 bases, these data show that there are roughly 2,500 to3,600 N moleculesavailableper mol of DI product RNA. Bishop and Roy (3) have calculated that thereareapproximately2,000 N molecules per standard VSV genome. The DI genome is approximately one-quarter this size and, based on the above figure, should
require
approximately500Nmolecules to coat each full-size DI genome. Thus, the replication system reported here produces sufficient N protein to coat the progeny DI RNAs. This finding is consistent with thefact that all the DIfull-length
RNAis nucleaseresistant,and itshows that this system can produce viral proteins and RNA in quantities appropriate to successfully carry out RNAreplication andnucleocapsidassembly.
The DI-leader RNA, in contrast to the full-lengthDI RNA, however, was not resistant to
digestionwith ribonuclease. Amodel has been put forward (5, 21) which proposes that the
availability of N protein controls the balance between replication and transcription by the
binding ofN protein to a site in leader RNA, whichthereby attenuates the termination at the
end of leader and allows read through while
simultaneously creating a nucleation site for
encapsidation;failuretobindNresultsin termi-nation and release ofleader is adiscrete
mole-cule. Weobservedthatthe accumulationof free DI-leaderRNA wasdecreased inthepresence of protein synthesis and full-length RNA
replica-tion. Moreover, we have shown that all full-length DI RNA is encapsidated with protein, whereas free leader is not. These findings are
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
compatible with, but donot prove,theidea that Nprotein binding to aleader RNA canresultin elongation to yieldgenome-length RNA, where-as failure of N to interact with leader results in termination and release offree leader. In
con-trast to our finding that free leader was not
encapsidated in our system,Blumberg and Kola-kofsky (4) reported that approximately three-quarters of the total intracellular leader RNA
was encapsidated. In relation to the model
pro-posed, theirfindingsuggests thatassociation of
N with leader may not always result in read throughor thatencapsidation offreeleadermay
occur after release from the template in the infected cell, or both. We suggest thatin our in
vitro system, RNAsynthesis occurs under con-ditions where proteinconcentration may not be
as great as in the infected cell. Under these circumstances, it may be that free leader is revealed as a less efficient competitor for N
protein than a nascent leader molecule still at-tached to its template.
In conclusion, an in vitro system has been
establishedwhich supportsthe synthesis of full-length VSV DI particle RNA and carries out its encapsidation to form nucleocapsids. In this system, the transition fromsynthesis of only the DI-leader RNA, a transcription event, to the replication offull-length DI RNA is made as a
function of viralprotein synthesis, whichcanbe controlled by omission or addition of viral mRNA. Thus, individual VSV mRNAs can be added alone or in combination to program pro-tein synthesis, and the effects of theseproteins on the transition to RNA replication can be evaluated. This approach providesamethod for assaying the involvement of individual proteins in the process of RNAreplication.
ACKNOWLEDGMENTS
I thank Nancy Davis for helpful advice anddiscussions, John Glass for artwork, and Steve Loechel for excellent technical assistance.
This research was supported by Public Health Service grants AI12464 and AI15134 from the National Institute of Allergy andInfectious Diseases and grant CA19014 from the National CancerInstitute.
LITERATURECITED
1. Ball, L. A.1980.Geneorder,p.61-73. In D. H. L.Bishop
(ed), Rhabdoviruses.CRC Press, Inc.,Cleveland,Ohio. 2. Banerjee, A., G.Abraham,and R.Colonno.1977.
Vesicu-lar stomatitis virus: mode oftranscription. J. Gen. Virol. 34:1-8.
3.Bishop, D. H. L., and P. Roy. 1972. Dissociation of vesicular stomatitis virus and relation of the virion pro-teins to the viral transcriptase. J. Virol. 10:234-243. 4. Blumberg, B. M., and D. Kolakofsky.1981. Intracellular
vesicular stomatitis virus leader RNAs are found in nu-cleocapsid structures. J. Virol. 40:568-576.
5. Blumberg, B., M. Leppert, and D. Kolakofsky. 1981. Interaction of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell 23:837-845. 6. Clinton, G. M., B. W. Burge, and A. S. Huang. 1978.
Effects ofphosphorylationandpHontheassociation of
NS protein with vesicular stomatitis cores. J. Virol.
27:340-346.
7. Cooper, P.,andA.Belett.1959. A transmissable
interfer-ingcomponent of vesicularstomatitisviruspreparations. J.Gen.Microbiol. 21:485-497.
8. Davis, N. L., and G. W. Wertz. 1982. Synthesis of
vesicularstomatitis virusnegative-strandRNAinvitro: dependenceonviralproteinsynthesis. J. Virol. 41:821-832.
9. Emerson,S.U.,P. M.Dierks,andJ.T.Parsons.1977. In vitrosynthesisofauniqueRNAspecies byaTparticleof vesicular stomatitis virus. J. Virol. 23:708-716. 10. Emerson, S. U. and Y.-H. Yu. 1975. Both NS and L
proteins are required for in vitro RNA synthesis by vesicularstomatitisvirus. J. Virol. 15:1348-1356. 11. Huang, A., D. Rao, and G. Lanman. 1980. Defective
interfering particlesofvesicularstomatitis virus:structure
function relationships. Ann. N.Y.Acad. Sci.
354:238-250.
12. Iverson,L.,andJ.Rose.1981. Localized attenuation and
discontinuoussynthesisduringvesicular stomatitis virus
transcription.Cell 23:477-484.
13. Keene, J.,M.Schubert,R.Lazzarini,andM.Rosenberg. 1978. Nucleotide sequencehomologyatthe 3'terminiof
RNA from vesicular stomatitis virus and its defective
interfering particles. Proc. Natl. Acad. Sci. U.S.A. 75:3225-3229.
14. Kingsbury,D.1974.The molecularbiologyof paramyxo-viruses. Med.Microbiol.Immunol. 16:73-83.
15. Kingsford, L.,andS. U.Emerson.1980.Transcriptional
activities of differentphosphorylated speciesof NS
pro-tein purifiedfromvesicular stomatitis virions and
cyto-plasm ofinfected cells. J. Virol. 33:1097-1105. 16. Laemmli, U.,and M. Faure.1973.Maturation of the head
ofbacteriophageT4. I. DNApackagingevents.J. Mol.
Biol. 80:575-599.
17. Laskey, R. 1980. The use of intensifying screens or
organicscintillators forvisualizingradioactive molecules
resolved by gel electrophoresis. Methods Enzymol.
65:363-371.
18. Lazzarini, R., J. Keene, and M. Schubert. 1981. The
originsofdefectiveinterferingparticlesof the
negative-strand RNAviruses. Cell26:145-154.
19. Leamnson, R.,andM. E.Reichmann.1974. The RNA of
defectivevesicular stomatitis virusparticlesin relationto viral cistrons. J. Mol. Biol. 85:551-568.
20. Leppert,M.,andD.Kolakofsky.1980.Effect of defective
interfering particles on plus- and minus-strand leader
RNAs invesicular stomatitis virus-infected cells. J. Virol. 35:704-709.
21. Leppert,M.,L.Rittenhouse,J.Perrault,D.Summers,and
D.Kolakofsky.1979.Plus and minus strand leader RNAs
innegativestrand virus-infected cells. Cell 18:735-747. 22. Patton, J. T., N. L.Davis,andG.W.Wertz. 1983.
Cell-freesynthesisandassemblyof vesicular stomatitis virus
nucleocapsids.J.Virol.45:155-164.
23. Perlman,S.M.,and A.S.Huang.1973. RNAsynthesisof
VSVV.Interaction betweentranscriptionandreplication.
J.Virol. 12:1395-1400.
24. Prlngle, C. 1978. The tdCE and hrCEphenotypes: host
range mutants of vesicular stomatitis virus in which
polymerasefunctionisaffected. Cell15:597-606.
25. Rao, D.,andA.Huang.1979.Synthesisofasmall RNA in cells coinfected by standard and defective interfering
particlesof VSV. Proc. Natl. Acad. Sci. U.S.A.
76:3742-3745.
26. Rao, D., and A. S. Huang. 1980. RNA synthesis by vesicular stomatitis virusX.Transcriptionandreplication
bydefectiveinterfering particles.J. Virol. 36:756-765. 27. Repik, P.,and D. H. L.Bishop.1973.Determination of the
molecularweightof animal RNA viral genomesby
nucle-asedigestions.J.Virol. 12:969-983.
28. Schubert, M., J. Keene, R. Lazzarini,-and S. Emerson. 1978. The complete sequence ofaunique RNA species
on November 10, 2019 by guest
http://jvi.asm.org/
synthesized byaDIparticle of VSV. Cell 15:103-112. 29. Senler, B., J. Perrault, J. Abelson,andJ. Holland. 1978.
Sequence of a RNA templated by the 3' OH RNA terminus of defective interfering particles of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 75:3923-3931.
30. Soria, M., S. Little, and A. Huang. 1974. Characterization ofvesicular stomatitis virus nucleocapsids. I.
Comple-mentary40S RNA molecules in nucleocapsids. Virology
61:270-280.
31. Stammnger,G., and R.Lazzarinl. 1979. Analysis of the
RNA of defective VSV particles. Celi 3:85-93. 32. Wertz, G. W., and N. L. Davis. 1979. RNase IIIcleaves
vesicular stomatitis genome-length RNAs, but fails to cleaveviral mRNA's. J. Virol. 30:108-115.
33. Wertz,G. W., and M. Levine. 1973. RNA synthesis by vesicular stomatitis virus and a small plaque mutant: effectsof cycloheximide. J. Virol. 12:253-264.
on November 10, 2019 by guest
http://jvi.asm.org/