Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Hepatitis B Virus X Protein Acts as a Tumor Promoter in
Development of Diethylnitrosamine-Induced
Preneoplastic Lesions
CHARLES R. MADDEN,1MILTON J. FINEGOLD,2ANDBETTY L. SLAGLE1*
Department of Molecular Virology and Microbiology1and Department of Pathology,2Baylor College of Medicine,
Houston, Texas 77030
Received 16 November 2000/Accepted 19 January 2001
Chronic infection with hepatitis B virus (HBV) is one of the major etiological factors in the development of human hepatocellular carcinoma. Transgenic mice that express the HBV X protein (HBx) have previously been shown to be more sensitive to the effects of hepatocarcinogens. Although the mechanism for this cofactor role remains unknown, the ability of HBx to inhibit DNA repair and to influence cell cycle progression suggests two possible pathways. To investigate these possibilities in vivo, we treated double-transgenic mice that both express HBx (ATX mice) and possess a bacteriophage lambda transgene with the hepatocarcinogen diethylni-trosamine (DEN). Histological examination of liver tissue confirmed that DEN-treated ATX mice developed approximately twice as many focal lesions of basophilic hepatocytes as treated wild-type littermates. Treatment of mice with DEN resulted in a six- to eightfold increase in the mutation frequency (MF), as measured by a functional analysis of the lambda transgene. HBx expression was confirmed by immunoprecipitation and Western blotting and was associated with a modest 23% increase in the MF. Importantly, the extent of hepatocellular proliferation in 14-day-old mice, as measured by the detection of proliferating cell nuclear
antigen and by the incorporation of 5-bromo-2ⴕ-deoxyuridine, was determined to be approximately twofold
higher in ATX livers than in wild-type livers. These results are consistent with a model in which HBx expression contributes to the development of DEN-mediated carcinogenesis by promoting the proliferation of altered hepatocytes rather than by directly interfering with the repair of DNA lesions.
Liver cancer is the fourth-leading cause of cancer mortality worldwide and results in more than 400,000 deaths annually (41). One of the primary risk factors for the development of hepatocellular carcinoma (HCC) is chronic infection with hep-atitis B virus (HBV) (1, 53). For individuals chronically in-fected with HBV, concurrent exposure to dietary aflatoxin increases the probability of developing HCC by at least three-fold (57). The synergistic contributions of these two factors to the development of HCC has led to speculation that chronic HBV infection predisposes an individual to the detrimental effects of hepatocarcinogens.
Several studies using transgenic mouse lines provide exper-imental evidence to support the hypothesis that chronic HBV infection acts synergistically with environmental carcinogens. The increased sensitivity to aflatoxin B1 of mice that overex-press the HBV surface antigen (18) is believed to be due in part to elevated levels of cytochrome P450 isoenzymes (27) that metabolize aflatoxin B1 into a mutagenic epoxide. Trans-genic mice that express the HBV (54) or woodchuck hepatitis virus (13) X proteins (HBx or WHx, respectively) are also more sensitive to the effects of the hepatocarcinogen diethylni-trosamine (DEN). However, the levels of carcinogen-metabo-lizing enzymes do not appear to be elevated in mice expressing HBx (12). Therefore, the mechanism by which HBx influences
the development of liver cancer in mice following exposure to environmental carcinogens remains to be elucidated.
Several lines of evidence suggest that HBx could influence the development of liver cancer by promoting the survival and growth of transformed hepatocytes. HBx is a 17-kDa regula-tory protein necessary for the establishment of hepadnaviral infection in woodchucks and, presumably, in all mammals (11, 63). Detectable in both the cytoplasm and the nucleus of in-fected cells (14, 40), HBx interacts with numerous cellular proteins (reviewed in reference 16) and is capable of transac-tivating cellular and viral genes (reviewed in reference 9) and activating protein kinase signaling cascades (3, 14, 24, 28, 34, 39). Transactivation of cellular genes by HBx (8, 50, 55) and the induction of one or more signaling pathways (3, 14, 24, 28, 34, 39) may lead to changes in cell cycle progression and/or regulation. Indeed, HBx is reported to induce cell cycle pro-gression in Chang liver cells (4) and in quiescent skin fibro-blasts (30). By affecting cell cycle regulation, HBx could facil-itate the survival and proliferation of hepatocytes that were altered after exposure to a mutagenic agent.
A second general mechanism by which HBx might increase the sensitivity of transgenic mice to the effects of hepatocar-cinogens involves its ability to inhibit DNA repair. Studies performed in transiently transfected cell cultures and in pri-mary transgenic mouse hepatocytes revealed that HBx expres-sion leads to a significant inhibition (25 to 60%) of global nucleotide excision repair (NER) following exposure to either UV light or aflatoxin B1 (2, 20, 23, 44). Although mapping studies suggest that HBx-mediated inhibition of NER corre-lates with its ability to interact with the NER component * Corresponding author. Mailing address: Department of Molecular
Virology and Microbiology, Mailstop BCM-385, One Baylor Plaza, Baylor College of Medicine, Houston, TX 77030-3411. Phone: (713) 798-3006. Fax: (713) 798-5075. E-mail: bslagle@bcm.tmc.edu.
3851
on November 9, 2019 by guest
http://jvi.asm.org/
DDB1 (32, 52), HBx has also been shown to interact with at least two other proteins or protein complexes that are also directly involved in DNA repair: p53 (17, 60) and TFIIH (46). These results suggest that HBx expression could cause an ac-cumulation of DNA mutations in vivo by compromising the repair ability of cells.
The purpose of the present study was to identify the cofactor role of HBx in transgenic mice exposed to DEN. Double-transgenic mice that express HBx (ATX mice) and possess an integrated lambda transgene, allowing measurement of muta-tion frequency (MF) (29), were used to measure the impact of HBx on repair of DEN-induced DNA damage. Although DEN-treated ATX mice developed 70% more expansile, ba-sophilic, focal lesions than DEN-treated wild-type mice, there was not a significant increase in the accumulation of DNA mutations. However, livers of ATX mice had significantly in-creased rates of hepatocellular proliferation as measured by immunohistochemical detection of proliferating cell nuclear antigen (PCNA) and by 5-bromo-2⬘-deoxyuridine (BrdU) in-corporation. These results support a hypothesis in which HBx contributes to the development of DEN-induced liver cancer by promoting the proliferation of altered cells rather than by increasing the frequency of DNA mutations.
MATERIALS AND METHODS
Transgenic mice.Transgenic mice harboring theXgene (nucleotides 1376 to 1840 of subtype adw2) under the control of the human␣-1-antitrypsin inhibitor
regulatory region (ATX mice) (33, 54) were maintained by the breeding of hemizygous ATX males (ICR⫻B6C3) with wild-type females (ICR). Hemizy-gous ATX females (ICR⫻B6C3) were then mated with homozygousmales (C57B1/6 Big Blue) (29) obtained from Stratagene Corporation, and the male F1
progeny were used for this study.
The genotypes of F1progeny were determined by Southern blot hybridization.
High-molecular-weight (HMW) DNA was purified from transgenic mouse tail samples using the Wizard genomic DNA purification kit (Promega). DNA was digested withBamHI, resolved on a 1% agarose gel, and transferred to a nylon membrane (Boehringer Mannheim).X-gene-specific probe DNA was prepared by PCR amplification of HBV plasmid DNA using the primer set 5⬘-ATGG
CTGCTAGGCTGTACTG-3⬘and 5⬘
-CTACAAGAGATGATTAGGCAGA-3⬘. Probe DNA for thecIIgene was amplified from 3g of homozygous Big Blue mouse DNA using thecII-specific primer set 5⬘-ACCACACCTATGGTGTAT GCA-3⬘ and 5⬘-GTCATAATGACTCCTGTTGATAG-3⬘. DNA was radiola-beled with [32P]dCTP (3,000 Ci/mmol) (ICN) using the Rediprime II random
priming labeling kit (Amersham/Pharmacia). Standard conditions for hybridiza-tions and X-ray film exposures were used (51).
Carcinogen treatment.At 12 days old, male mice were given a single injection (intraperitoneal) of DEN (Sigma) at 2g/g of body weight as previously de-scribed (54). At appropriate time points (14, 30, 90, and 240 days old), mice were sacrificed, and portions of three liver lobes were fixed in 10% neutral buffered formalin for 16 h and then stored in 70% ethanol. Tissues were paraffin embed-ded, and coded hematoxylin-and-eosin-stained sections were submitted for his-tological analysis by M.J.F. Remaining tissue was frozen in liquid nitrogen and stored for later experiments.
Quantification of mouse liver tissue foci.The size and number of basophilic, nodular foci were determined by microscopic examination of hematoxylin-and-eosin-stained liver tissue sections. These values were used in conjunction with the overall area of the tissue section examined to calculate the number of foci per cubic volume of liver tissue according to the method of Pugh et al. (45).
Immunoprecipitation and Western blot verification of HBx expression.Liver extracts were prepared by homogenizing tissue in extraction buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1% NP-40). The protein concentration of each liver extract was determined using the DC Protein assay system (Bio-Rad Lab-oratories) according to the manufacturer’s directions. Subsequent immunopre-cipitation and Western blot analysis, to verify expression of HBx, were performed using 2 mg of total liver protein and have been described previously (54). Briefly, following electrophoresis on sodium dodecyl sulfate–15% polyacrylamide gels, separated proteins were transferred to nitrocellulose filters. The presence or
absence of HBx was verified using rabbit anti-HBx polyclonal serum, an avidin-biotin detection kit (Vector Laboratories), and enhanced chemiluminescence (Amersham/Pharmacia).
Packaging of lambda phage DNA and plaque assays.Liver HMW DNA for use in the Big Blue mutagenesis assay was isolated using the RecoverEase system (Stratagene/Biocrest) under conditions recommended by the supplier. Impor-tantly, the RecoverEase system uses no phenol or chemical extractions that may further damage the DNA. Liver HMW DNA was subsequently incubated with Transpack (Stratagene/Biocrest) to excise and package the lambda phage ge-nome. Packaged DNA was then incubated with a suspension (optical density at 600 nm⫽0.5) ofEscherichia colistrain G1250. Determination of the relative MF was accomplished by assaying for inactivating mutations (cII⫺) in the
bacterio-phage lambdacIIgene (22). A portion of diluted culture (1/100) was plated and incubated at 37°C for 24 h to determine the total (cII⫺mutant pluscII⫹) number
of PFU. The remaining culture was plated and incubated at 24°C for 48 h, conditions allowing growth only ofcIImutant plaques. The MF was calculated as the ratio of mutant PFU to total PFU. All mutant plaques were subsequently isolated and replated under the same conditions to verify the mutant phenotype.
DNA sequencing.DNA obtained from cored mutant plaques (heated to 95°C for 5 min) was amplified by a standard PCR procedure usingcII-specific primers (listed above). AmplifiedcIIDNA was then sequenced using a Thermoseque-nase cycle sequencing kit (Amersham/Pharmacia), and products were resolved by electrophoresis on 6% polyacrylamide-urea gels.
PCNA detection.Immunohistochemical detection of PCNA was performed using formalin-fixed, deparaffinized mouse liver tissue sections and a mouse anti-PCNA monoclonal antibody (PC-10; Santa Cruz Biotechnology). Sections were heated in 10 mM sodium citrate (36) and incubated in hybridization buffer (100 mM Tris [pH⫽7.5], 500 mM NaCl, 0.05% Tween 20, 5% reconstituted milk, anti-PCNA [4g of PC-10/ml]) at 37°C for 1 h. Antibody-protein com-plexes were visualized using an avidin-biotin detection kit (Vector Laboratories) and 3,3⬘-diaminobenzidine according to the manufacturer’s directions. PCNA-positive hepatocytes located within altered hepatic foci were excluded from quantitation. For Western blot analysis, liver tissue was homogenized in RIPA buffer (50 mM Tris [pH⫽7.5], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA), and extracts were clarified by incubation with formalin-fixed, heat-inactivatedStaphylococcus aureus. Following electrophoresis on sodium dodecyl sulfate–12% polyacrylamide gels, separated proteins were transferred to Immobilon-P (Amersham/Pharmacia) filters. Immobilized PCNA was subsequently visualized using a primary anti-PCNA antibody (PC-10) fol-lowed by an avidin-biotin detection kit (Vector Laboratories) and enhanced chemiluminescence (Amersham/Pharmacia) according to the manufacturer’s di-rections.
BrdU injection and detection.DEN-treated mice were injected intraperitone-ally (i.p.) at 14 days old with BrdU (Sigma) in sterile phosphate-buffered saline at a dose of 100 mg/kg of body weight (58). Upon sacrifice 2 h later, liver tissues were fixed in neutral buffered formalin (16 h) prior to being embedded in paraffin. Deparaffinized sections were heated in 10 mM sodium citrate to en-hance the detection of BrdU epitopes according to the procedure of McGinley et al. (36). Incorporated BrdU was visualized using the BrdU Detection Kit II (Boehringer-Mannheim) according to the manufacturer’s directions.
Statistical analysis.Probability calculations for the PCNA staining, foci count-ing, and MF assays were performed using a two-tailed Student’sttest (Microsoft Excel software package). Deviation and mean values were calculated using the same software.
RESULTS
Double-transgenic mice. Double-transgenic mice used in
this study possess both anXtransgene and multiple copies of an integrated bacteriophage () reporter gene. For the pur-poses of clarity, single-transgenic mice harboring only are referred to as wild-type mice, while double-transgenic mice harboring both and X transgenes are referred to as ATX mice. Mice were generated as described in Materials and Methods and genotyped by Southern blot hybridization (data not shown). Recent studies have established that under normal conditions (i.e., no exogenous DNA damage), the expression of HBx in these double-transgenic mice has no apparent del-eterious effects and does not influence the accumulation of spontaneous mutations (35). The expression of HBx was
on November 9, 2019 by guest
http://jvi.asm.org/
firmed for all ATX mice used in this study by immunoprecipi-tation (IP) and Western blot hybridization (Fig. 1). For this system, increasing the amount of antibody used for IP does not result in a noticeable increase in HBx, indicating the IP is likely being performed in an excess of antibody. It should be noted that the carefully optimized IP step is required prior to West-ern blot detection of HBx in ATX livers. Indeed, the 2 mg of ATX liver extract required for detection of HBx in the present study is very similar to the amount of total protein needed to obtain a similar HBx signal from 5⫻ 106pSVX-transfected
HepG2 cells (2) or from 4⫻106chronically woodchuck
hep-atitis virus-infected woodchuck hepatocytes (13). Together, these results demonstrate that HBx is expressed throughout the course of this study and at levels comparable to those observed in chronically infected liver tissue.
HBx-associated increase in DEN-induced liver lesions.
Pre-vious studies with DEN-treated transgenic mice revealed that expression of HBx (and WHx) leads to a statistically significant increase in the development of expansive nodules of basophilic hepatocytes and that these focal lesions reliably predict the development of hepatocellular carcinoma (13, 54). To confirm the cofactor effect of HBx in the double-transgenic mice used in this study, male ATX and wild-type mice were treated at 12 days old with a single dose of DEN. All mice appeared normal at the time of sacrifice. However, histological examination of liver tissue sections revealed the development of expansile nodules of basophilic hepatocytes in both ATX and wild-type mice by 240 days old (Fig. 2A). The incidence of foci per cubic volume of liver tissue was increased by approximately 70% in ATX mice relative to that in wild-type mice (Fig. 2B) (P⬍
0.012). In addition to the clusters of small, basophilic hepato-cytes observed in all foci, some altered hepatohepato-cytes also pos-sessed globular eosinophilic inclusions. These inclusions were found in both ATX (four out of seven) and wild-type (two out of nine) animals. All other histological abnormalities cited (e.g., mild anisocytosis, isolated foci of inflammation, and de-creased glycogen content) appeared in a random distribution and were consistent with observations reported for untreated mice of the same lineage and age (data not shown) (35). These observations establish that the double-transgenic mice (ATX/)
used for this study are more sensitive than wild-type littermates () to the carcinogenic effects of DEN.
Effect of HBx on in vivo MF.Previous studies have
[image:3.612.73.276.72.140.2]estab-lished that HBx can inhibit the ability of cells to repair dam-aged DNA (2, 20, 23, 44). A similar inhibitory effect of HBx in vivo should lead to an increase in the accumulation of DNA mutations in DEN-treated mice. To test this hypothesis, we measured the relative MF in liver tissue samples obtained from DEN-treated ATX and wild-type mice (Table 1). Compared to the MF reported for untreated mice (35), exposure to DEN FIG. 1. Detection of HBx in transgenic mouse liver tissue. Shown is
a representative result using immunoprecipitation and Western blot hybridization to detect HBx in 14-day-old (lanes 1 and 2), 30-day-old (lanes 3 and 4), 90-day-old (lanes 5 and 6), and 240-day-old (lanes 7 to 10) mice. HBx expression was clearly demonstrated in all ATX mice (lanes 2, 4, 6, and 8) and absent in all wild-type mice (lanes 1, 3, 5, and 7). No protein bands were detected for 240-day-old ATX and wild-type mice when nonspecific rabbit serum was substituted for rabbit anti-HBx polyclonal serum during the Western blot hybridization proce-dure (lanes 9 and 10). All mice used in this study were similarly screened for HBx expression by this method (data not shown). IgG, immunoglobulin G.
FIG. 2. Incidence of DEN-induced foci in transgenic mouse liver tissue. (A) Appearance of basophilic liver nodule (foci) in a 240-day-old ATX mouse liver tissue section at a magnification of⫻200. (B) Mean number of foci per cubic centimeter in 240-day-old ATX (n⫽7) and wild-type (WT) (n⫽9) animals. Values were obtained by mea-suring foci area in three liver tissue sections (chosen at random) per animal, as described in Materials and Methods. The standard deviation is indicated by a vertical error bar.
on November 9, 2019 by guest
http://jvi.asm.org/
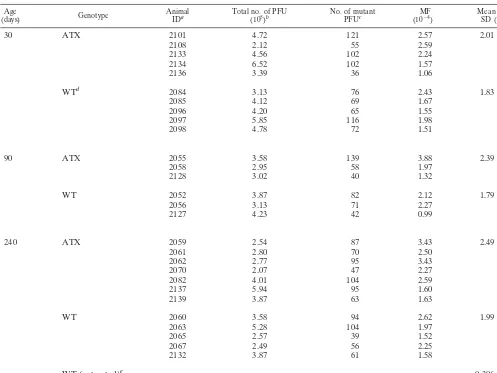
[image:3.612.315.544.81.517.2]resulted in a six- to eight-fold increase in the MF, confirming that this assay reliably detects changes in mutation accumula-tion. A slight elevation in the mean MF for ATX mice was found at each time point (30, 90, and 240 days old); however, this increase was not statistically significant (allPvalues were
⬎0.21). A comparison of the mean MF among all ATX (n⫽
15) and wild-type (n⫽13) mice revealed a 23% increase in the MF that was also not significant (P⬎0.1). These results dem-onstrate that the expression of HBx in vivo does not result in a significant increase in the accumulation of DEN-induced mutations.
Determination of DNA mutation spectrum.While HBx
ex-pression did not result in a measurable increase in the MF, it was conceivable that it could cause a change in the spectrum of mutations induced by DEN by inhibiting the repair of only a certain subset of DNA lesions. To examine this possibility, mutant phage derived from DEN-treated ATX and wild-type mice were picked at random, and thecIIgenes were sequenced (n⫽63) to establish the mutation spectrum (Table 2).
Com-pared to the mutation spectrum reported for untreated mice (49), DEN treatment resulted in a relatively large increase in transition and transversion events at A and T base pairs, a finding consistent with the long half-life and mutagenicity of O4-ethyldeoxythymine (O4-EtT) moieties formed by DEN
(t1/2⫽11 days) (48). In addition, there was an approximately
twofold increase in the incidence of transitions from G or C to A or T base pairs in ATX relative to wild-type animals. These results establish that HBx expression does not lead to major alterations in the spectrum of DEN-induced DNA mutations.
HBx-induced hepatocyte proliferation.Since previous
[image:4.612.55.552.83.456.2]stud-ies have shown that HBx may stimulate cell cycle progression and proliferation in cell culture (4, 30), we hypothesized that a similar effect of HBx in vivo might explain the 70% increase in the incidence of DEN-induced preneoplastic lesions in ATX mice. To determine the possible effect of HBx on hepatocel-lular proliferation, livers of ATX and wild-type animals were examined for the expression of PCNA, a marker of cellular proliferation (25). A significant increase in the percentage of TABLE 1. Determination of the MF in DEN-treated transgenic mice
Age
(days) Genotype AnimalIDa Total no. of PFU(105)b No. of mutantPFUc (10MF⫺4) Mean MFSD (10⫺4)⫾
30 ATX 2101 4.72 121 2.57 2.01⫾0.67
2108 2.12 55 2.59
2133 4.56 102 2.24
2134 6.52 102 1.57
2136 3.39 36 1.06
WTd 2084 3.13 76 2.43 1.83⫾0.38
2085 4.12 69 1.67
2096 4.20 65 1.55
2097 5.85 116 1.98
2098 4.78 72 1.51
90 ATX 2055 3.58 139 3.88 2.39⫾1.33
2058 2.95 58 1.97
2128 3.02 40 1.32
WT 2052 3.87 82 2.12 1.79⫾0.70
2056 3.13 71 2.27
2127 4.23 42 0.99
240 ATX 2059 2.54 87 3.43 2.49⫾0.75
2061 2.80 70 2.50
2062 2.77 95 3.43
2070 2.07 47 2.27
2082 4.01 104 2.59
2137 5.94 95 1.60
2139 3.87 63 1.63
WT 2060 3.58 94 2.62 1.99⫾0.46
2063 5.28 104 1.97
2065 2.57 39 1.52
2067 2.49 56 2.25
2132 3.87 61 1.58
WT (untreated)e 0.306⫾0.28
aID, identification.
bCalculated from dilutions incubated at 37°C.
cIsolated mutants replated and incubated at 24°C to verify phenotype. dWT, wild type.
eMF value reported for untreated WT mice at 240 days old by Madden et al. (35).
on November 9, 2019 by guest
http://jvi.asm.org/
PCNA-positive hepatocytes was observed in 14-day-old ATX mice versus results for wild-type mice (Fig. 3A) (allPvalues were ⬍0.004). This difference was apparent for both DEN-treated and unDEN-treated mice and was confirmed independently by the determination of steady-state levels of PCNA in mouse livers by Western blot analysis (Fig. 3B). Hepatocellular pro-liferation had diminished considerably by 30 and 240 days of age, with no measurable effect of HBx observed (data not shown). These results indicate that the contribution of HBx-induced cellular proliferation is likely limited to the early stages of DEN-induced carcinogenesis.
To establish that the increased levels of PCNA found in ATX mouse livers correlated with an increase in hepatocellu-lar replication, the incorporation of BrdU into hepatocelluhepatocellu-lar DNA was measured in 14-day-old mice. Significantly more BrdU-positive hepatocytes were detected in DEN-treated ATX than in treated wild-type mouse liver tissue sections (Fig. 3C and D) (P ⫽ 0.03). Together, these data clearly demon-strate that under certain conditions, HBx expression can pro-mote hepatocellular proliferation in vivo.
DISCUSSION
Previous work with humans and woodchuck X-transgenic mice has established a cancer cofactor role for the HBx (or WHx) protein (13, 54, 58). The purpose of the present study was to investigate the molecular basis of the role of the HBx cofactor in DEN-mediated carcinogenesis. Using a novel dou-ble-transgenic mouse model that allows determination of the DNA MF in vivo, we show that HBx expression is associated with a twofold increase in altered hepatic foci, a modest (23%) increase in the DNA MF, and a two- to threefold increase in hepatocellular proliferation in young mice (measured by PCNA staining and BrdU incorporation). Together, these re-sults are consistent with the idea that HBx functions as a tumor promoter in the DEN model of liver carcinogenesis.
We originally predicted that HBx expression in the presence
of exogenous DNA damage would lead to unrepaired DNA and an increased DNA MF. This hypothesis was based on the observation that HBx inhibits the repair of DNA damage in cell culture (2, 20, 23, 35, 44). Although we were able to measure an HBx-associated elevation in the MF at each time point in this study, the increase measured (23%) was very small. One interpretation of this result is that the cofactor role of HBx in this model is restricted to promoting the growth of DEN-altered hepatocytes, as was previously suggested (13). However, our data showing an HBx-associated increase in hep-atocellular proliferation suggest that HBx might additionally enhance the initiation of carcinogenesis. The major DEN ad-duct O6-ethylguanine (O6-EtG) has a half-life of 20 h (48).
HBx-induced cell division that occurs prior to repair of the O6-EtG lesions would lead to a mutation at the position of the
unrepaired adducts, specifically at a guanine residue. Indeed, we are able to measure a modest increase in the MF in ATX mice at all time points and also observed an increase specifi-cally in G (or C) to A (or T) transitions. These latter results indicate that HBx may enhance the initiation of carcinogenesis by promoting the division of hepatocytes that contain unre-paired DNA damage.
The results of the present study are consistent with current models of known tumor promoters. Analysis of the tumor promoter 12-myristate 13-acetate (TPA) has yielded results that are strikingly similar to those observed for HBx. The two-to fivefold induction of mitwo-tosis in TPA-treated keratinocytes (37) is similar to the two- to threefold induction of hepatocyte proliferation measured in ATX livers. In addition, the modest elevation in the MF in carcinogen-treated Big Blue mice ad-ditionally treated with TPA (64%) (37) is comparable to the HBx-associated 23%-increased MF found in DEN-treated mice in the present study. Finally, neither TPA nor HBx alone has any affect on the MF (35, 37); their effect is apparent only in the presence of DNA damage. Thus, the influence of HBx measured in the present study is very similar to that measured for the well-known tumor promoter TPA.
The mechanism by which HBx increases hepatocyte turn-over remains unknown. This effect of HBx was measurable only in neonatal mice, when hepatocyte numbers are increas-ing and growth factors are abundant. It is interestincreas-ing to con-sider that many of the growth-stimulatory factors present in neonatal liver are also induced during liver cell regeneration (19, 59). It seems likely that a similar growth-promoting effect of HBx during immune-mediated cycles of liver cell death and regeneration (reviewed in reference 53) would contribute to the pathology of chronic HBV infection in humans.
[image:5.612.53.294.84.238.2]The expression of HBx in many transgenic mouse lines is not associated with any detrimental effects (6, 15, 21, 38, 42, 47). However, the ability of HBx to induce hepatocyte proliferation has been reported for a line of X-transgenic mice that are susceptible to HCC (31). Those mice also demonstrate a sim-ilar two- to threefold increase in cell proliferation. It remains unknown whether the ability of HBx to increase hepatocyte replication is responsible for the HCC in those mice. Other variables to consider when comparing those mice with the ATX mice used in the present study include genetic variations between mouse lineages, the level at which HBx is being ex-pressed, and possible environmental cofactors unique to indi-vidual animal colonies.
TABLE 2. cIImutation spectrum in ATX and wild-type animals
Mutation type Occurrence in
a:
ATXb WTc Controld
Transitions
G/C3A/T 11 6 27
A/T3G/C 5 8 2
Transversions
G/C3T/A 4 4 8
G/C3C/G 0 1 6
A/T3T/A 9 9 1
A/T3C/G 2 2 0
Othere 1 1 6
Total isolates 32 31 50
aTotal no. of mutants bearing the specified mutation.
bMutantcIIisolates from three 240-day-old DEN-treated ATX animals. cMutant cIIisolates from three 240-day-old DEN-treated wild-type (WT)
animals.
dControl, mutation spectrum of mutantlacIisolates from untreated mouse
liver tissue according to the work of Ross and Leavitt (49).
eIncluded are deletions, additions, and jackpot mutations (mutations that occur
early in the development of the mouse and are amplified by cellular replication).
on November 9, 2019 by guest
http://jvi.asm.org/
A delicate balance between cellular proliferation and apo-ptosis is necessary for normal liver homeostasis. Indeed, a proapoptotic property of HBx has been demonstrated in sev-eral studies (26, 43, 56). However, it is difficult to reconcile an induction of apoptosis by HBx with the increase in the devel-opment of liver foci demonstrated in the present study. Rather, the prevention of apoptosis would lead to the survival of cells that contain DNA mutations. DNA-damaging agents, such as DEN, are known to induce apoptosis (62). However, we ob-served an HBx-associated increased in PCNA-positive cells in both the presence and the absence of DEN treatment. This result indicates that the increased rate of cellular turnover measured in ATX livers is not merely a compensatory response to increased cell death in DEN-treated ATX mice.
The design of the present study does not permit definitive conclusions regarding the ability of HBx to directly compro-mise DNA repair in vivo. At present, HBx has only been shown to inhibit the NER pathway (2, 20, 23, 44), one of several DNA repair pathways in the cell (61). DEN is a metabolically acti-vated mutagen that ethylates nucleophilic sites in DNA, pri-marily theO6 and O4 positions of guanine and thymine,
re-spectively (5). In addition to being repaired by the NER pathway (7),O6-EtG lesions are also removed viaO6
-alkylgua-nine DNA alkyltransferase activity in eukaryotes (5). The re-dundancy of repair pathways forO6-EtG moieties and the long
half-life ofO4-EtT lesions (11 days) may have minimized our
[image:6.612.68.540.73.468.2]ability to measure the impact of HBx expression on NER. We considered the possibility that HBx might inhibit the repair of FIG. 3. Impact of HBx on hepatocyte proliferation. (A) Percentage of PCNA-positive hepatocytes in 14-day-old DEN-treated (n⫽4) and untreated (n⫽5) ATX and in DEN-treated (n⫽5) and untreated (n⫽4) wild-type (WT) mouse liver tissue sections. Mean values were determined by counting PCNA-positive hepatocytes in five random fields of approximately 250 cells per field. Error bars represent standard deviations. (B) Detection of PCNA in ATX and wild-type mouse liver extracts by Western blotting analysis. Liver extracts were prepared from DEN-treated 14-day-old male ATX and wild-type mouse littermates. (C) Percentage of BrdU-positive hepatocytes in 14-day-old DEN-treated ATX (n⫽3) and wild-type (n⫽4) mouse liver tissue sections. Mean values were determined by counting BrdU-positive hepatocytes in five random fields of approximately 250 cells per field. Error bars represent standard deviations. (D) Representative immunohistochemical staining of incorporated BrdU in DEN-treated 14-day-old ATX and wild-type mouse liver tissue sections. BrdU-positive nuclei are black, and nonlabeled nuclei are counterstained with methyl green.
on November 9, 2019 by guest
http://jvi.asm.org/
a subset of DEN lesions and so investigated the DNA mutation spectrum for 63 lambdacIImutants. Those experiments re-vealed an increase in G (or C) to A (or T) transitions that could be explained by the HBx-induced cell division in the presence of unrepairedO6-EtG (discussed above). Studies
de-signed to measure the impact of HBx expression on the re-moval of lesions repaired exclusively by the NER pathway are ongoing.
In summary, a strong correlation exists between HBV status, exposure to environmental carcinogens, and the development of HCC (10, 57). In the present study, we demonstrate that the expression of HBx in vivo leads to a significant increase of DEN-induced hepatic lesions by a mechanism that does not include a large increase in the DNA MF, data consistent with a model in which HBx acts as a tumor promoter. In addition, we propose that HBx may enhance the initiation of DEN damage by inducing hepatocellular proliferation in cells that contain unrepaired DNA lesions. It is therefore possible that expression of HBx at the time of carcinogen exposure in hu-mans will similarly lead to enhanced carcinogenesis. Further investigation of the molecular mechanism(s) by which HBx alters the hepatocyte cell cycle will lead to a better understand-ing of the molecular basis of HBV-associated liver cancer and may reveal novel targets for intervention and treatment of HCC.
ACKNOWLEDGMENTS
This work was supported by NIH research grant CA54557. C.R.M. was supported by research training grant T32DK07664.
We thank Christopher Wagner, Thenaa Said, and Stephanie Moses for technical assistance.
REFERENCES
1.Beasley, R. P.1988. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer61:1942–1956.
2.Becker, S. A., T. H. Lee, J. S. Butel, and B. L. Slagle.1998. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol.72:266–272. 3.Benn, J., and R. J. Schneider.1994. Hepatitis B virus HBx protein activates
Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase sig-naling cascade. Proc. Natl. Acad. Sci. USA91:10350–10354.
4.Benn, J., and R. J. Schneider.1995. Hepatitis B virus HBx protein deregu-lates cell cycle checkpoint controls. Proc. Natl. Acad. Sci. USA92:11215– 11219.
5.Beranek, D. T.1990. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res.231:11–30. 6.Billet, O., G. Grimber, M. Levrero, K. A. Seye, P. Briand, and V. Joulin.
1995. In vivo activity of the hepatitis B virus core promoter: tissue specificity and temporal regulation. J. Virol.69:5912–5916.
7.Bronstein, S. M., J. E. Cochrane, T. R. Craft, J. A. Swenberg, and T. R. Skopek.1991. Toxicity, mutagenicity, and mutational spectra of N-ethyl-N-nitrosourea in human cell lines with different DNA repair phenotypes. Can-cer Res.51:5188–5197.
8.Buendia, M. A.1992. Hepatitis B viruses and hepatocellular carcinoma. Adv. Cancer Res.59:167–226.
9.Caselmann, W. H.1995. Transactivation of cellular gene expression by hep-atitis B viral proteins: a possible molecular mechanism of hepatocarcinogen-esis. J. Hepatol.22:34–37.
10. Chen, C. J., M. W. Yu, and Y. F. Liaw.1997. Epidemiological characteristics and risk factors of hepatocellular carcinoma. J. Gastroenterol. Hepatol.
12:S294–S308.
11. Chen, H.-S., S. Kaneko, R. Girones, R. W. Anderson, W. E. Hornbuckle, B. C. Tennant, P. J. Cote, J. L. Gerin, R. H. Purcell, and R. H. Miller.1993. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol.67:1218–1226.
12. Chomarat, P., J. M. Rice, B. L. Slagle, and C. P. Wild.1998. Hepatitis B virus-induced liver injury and altered expression of carcinogen metabolising enzymes: the role of the HBx protein. Toxicol. Lett.103:595–601. 13. Dandri, M., P. Schirmacher, and C. E. Rogler.1996. Woodchuck hepatitis
virus X protein is present in chronically infected woodchuck liver and wood-chuck hepatocellular carcinomas which are permissive for viral replication. J. Virol.70:5246–5254.
14. Doria, M., N. Klein, R. Lucito, and R. J. Schneider.1995. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J.19:4747–4757. 15. Dragani, T. A., G. Manenti, H. Farza, G. Della Porta, P. Tiollais, and C.
Pourcel.1989. Transgenic mice containing hepatitis B virus sequences are more susceptible to carcinogen-induced hepatocarcinogenesis. Carcinogen-esis11:953–956.
16. Feitelson, M. A., and L. X. Duan.1997. Hepatitis B virus x antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol.150:1141–1157.
17. Feitelson, M. A., M. Zhu, L. X. Duan, and W. T. London.1993. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene8:1109–1117.
18. Ghebranious, N., and S. Sell.1998. Hepatitis B injury, male gender, afla-toxin, and p53 expression each contribute to hepatocarcinogenesis in trans-genic mice. Hepatology27:383–391.
19. Grasl-Kraupp, B., W. Rossmanith, B. Ruttkay-Nedecky, L. Mullauer, B. Kammerer, W. Bursch, and R. Schulte-Hermann.1998. Levels of transform-ing growth factor beta and transformtransform-ing growth factor beta receptors in rat liver during growth, regression by apoptosis and neoplasia. Hepatology28:
717–726.
20. Groisman, I. J., R. Koshy, F. Henkler, J. D. Groopman, and M. A. Alaoui-Jamali.1999. Downregulation of DNA excision repair by the hepatitis B virus-x protein occurs in p53-proficient and p53-deficient cells. Carcinogen-esis20:479–483.
21. Guidotti, L. G., B. Matzke, H. Schaller, and F. V. Chisari.1995. High-level hepatitis B virus replication in transgenic mice. J. Virol.69:6158–6169. 22. Jakubczak, J. L., G. Merlino, J. E. French, W. J. Muller, B. Paul, S. Adhya,
and S. Garges.1996. Analysis of genetic instability during mammary tumor progression using a novel selection-based assay for in vivo mutations in a bacteriophage lambda transgene target. Proc. Natl. Acad. Sci. USA93:9073– 9078.
23. Jia, L., X. W. Wang, and C. C. Harris.1999. Hepatitis B virus X protein inhibits nucleotide excision repair. Int. J. Cancer80:875–879.
24. Kekule, A. S., U. Lauer, L. Weiss, B. Luber, and P. H. Hofschneider.1993. Hepatitis B virus transactivator HBx uses a tumour promoter signaling path-way. Nature361:742–745.
25. Kelman, Z.1997. PCNA: structure, functions and interactions. Oncogene
14:629–640.
26. Kim, H., H. Lee, and Y. Yun.1998. X-gene product of hepatitis B virus induces apoptosis in liver cells. J. Biol. Chem.273:381–385.
27. Kirby, G. M., I. Chemin, R. Montesano, F. V. Chisari, M. A. Lang, and C. P. Wild.1994. Induction of specific cytochrome P450s involved in aflatoxin B1 metabolism in hepatitis B virus transgenic mice. Mol. Carcinog.11:74–80. 28. Klein, N. P., and R. J. Schneider.1997. Activation of Src family kinases by
hepatitis B virus HBx protein and coupled signaling to Ras. Mol. Cell. Biol.
17:6427–6436.
29. Kohler, S. W., G. S. Provost, P. L. Kretz, A. Fieck, and J. M. Short.1990. An in vivo assay using transgenic mice to analyze spontaneous and induced mutations at the nucleic acid level. Strategies3:19–21.
30. Koike, K., K. Moriya, H. Yotsuyanagi, S. Iino, and K. Kurokawa.1994. Induction of cell cycle progression by hepatitis B virus HBx gene expression in quiescent mouse fibroblasts. J. Clin. Investig.94:44–49.
31. Koike, K., K. Moriya, H. Yotsuyanagi, Y. Shintani, H. Fujie, T. Tsutsumi, and S. Kimura.1998. Compensatory apoptosis in preneoplastic liver of a transgenic mouse model for viral hepatocarcinogenesis. Cancer Lett.134:
181–186.
32. Lee, T.-H., S. J. Elledge, and J. S. Butel.1995. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol.69:1107– 1114.
33. Lee, T.-H., M. F. Finegold, R.-F. Shen, J. L. DeMayo, S. L. C. Woo, and J. S. Butel.1990. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J. Virol.64:5939–5947.
34. Lee, Y. H., and Y. Yun.1998. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J. Biol. Chem.273:25510–25515.
35. Madden, C. R., M. F. Finegold, and B. L. Slagle.2000. Expression of hep-atitis B virus X protein does not alter the accumulation of spontaneous mutations in transgenic mice. J. Virol.74:5266–5272.
36. McGinley, J. N., K. K. Knott, and H. J. Thompson.2000. Effect of fixation and epitope retrieval on BrdU indices in mammary carcinomas. J. Histo-chem. CytoHisto-chem.48:355–362.
37. Miller, M. L., K. Vasunia, G. Talaska, A. Andringa, J. De Boer, and K. Dixon.2000. The tumor promoter TPA enhances benzo[a]pyrene and ben-zo[a]pyrene diolepoxide mutagenesis in Big Blue mouse skin. Environ. Mol. Mutagen.35:319–327.
38. Miller, R. H., S. Kaneko, C. T. Chung, R. Girones, and R. H. Purcell.1989. Compact organization of the hepatitis B virus genome. Hepatology9:322– 327.
39. Natoli, G., M. L. Avantaggiati, P. Chirillo, P. L. Puri, A. Ianni, C. Balsano, and M. Levrero.1994. Ras- and Raf-dependent activation of c-Jun transcrip-tional activity by the hepatitis B virus transactivator pX. Oncogene9:2837– 2843.
on November 9, 2019 by guest
http://jvi.asm.org/
40.Nomura, T., Y. Lin, D. Dorjsuren, S. Ohno, T. Yamashita, and S. Murakami.
1999. Human hepatitis B virus X protein is detectable in nuclei of transfected cells, and is active for transactivation. Biochim. Biophys. Acta1453:330–340. 41. Parkin, D. M., P. Pisani, and J. Ferlay.1999. Global cancer statistics. CA
Cancer J. Clin.49:33–64.
42. Perfumo, S., L. Amicone, S. Colloca, M. Giorgo, L. Pozzi, and M. Tripodi.
1992. Recognized efficiency of the hepatitis B virus polyadenylation signal is tissue specific in transgenic mice. J. Virol.66:6819–6823.
43. Pollicino, T., O. Terradillos, H. Lecoeur, M. L. Gougeon, and M. A. Buendia.
1998. Pro-apoptotic effect of the hepatitis B virus X gene. Biomed. Pharma-cother.52:363–368.
44. Prost, S., J. M. Ford, C. Taylor, J. Doig, and D. J. Harrison.1998. Hepatitis B x protein inhibits p53-dependent DNA repair in primary mouse hepato-cytes. J. Biol. Chem.273:33327–33332.
45. Pugh, T. D., J. H. King, H. Koen, D. Nychka, J. Chover, G. Wahba, Y.-H. He, and S. Goldfarb.1983. Reliable stereological method for estimating the number of microscopic hepatocellular foci from their transections. Cancer Res.43:1261–1268.
46. Qadri, I., J. W. Conaway, R. C. Conaway, J. Schaack, and A. Siddiqui.1996. Hepatitis B virus transactivator protein, HBx, associates with the compo-nents of TFIIH and stimulates the DNA helicase activity of TFIIH. Proc. Natl. Acad. Sci. USA93:10578–10583.
47. Reifenberg, K., J. Lohler, H.-P. Pudollek, E. Schmitteckert, G. Spindler, J. Kock, and H.-J. Schlicht.1997. Long-term expression of the hepatitis B virus core-e- and X-proteins does not cause pathologic changes in transgenic mice. J. Hepatol.26:119–130.
48. Richardson, F. C., M. C. Dyroff, J. A. Boucheron, and J. A. Swenberg.1985. Differential repair of O4-alkylthymidine following exposure to methylating
and ethylating hepatocarcinogens. Carcinogenesis6:625–629.
49. Ross, J. A., and S. A. Leavitt.1998. Induction of mutations by 2-acetylamin-ofluorene in lacI transgenic B6C3F1 mouse liver. Mutagenesis13:173–179. 50. Rossner, M. T.1992. Hepatitis B virus X-gene product: a promiscuous
transcriptional activator. J. Med. Virol.36:101–117.
51. Sambrook, J., E. F. Fritsch, and T. Maniatis.1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
52. Sitterlin, D., T. H. Lee, S. Prigent, P. Tiollais, J. S. Butel, and C. Transy.
1997. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and re-stricted to transactivation-proficient X-insertion mutants. J. Virol.71:6194– 6199.
53. Slagle, B. L., S. A. Becker, and J. S. Butel.1994. Hepatitis viruses and liver cancer, p. 149–171.InA. Minson, J. Neil, and M. McCrae (ed.), Viruses and cancer, vol. 51. University of Cambridge, Cambridge, England.
54. Slagle, B. L., T.-H. Lee, D. Medina, M. J. Finegold, and J. S. Butel.1996. Increased sensitivity to the hepatocarcinogen diethylnitrosamine in trans-genic mice carrying the hepatitis B virus X gene. Mol. Carcinog.15:261–269. 55. Slagle, B. L., T. H. Lee, and J. S. Butel.1992. Hepatitis B virus and
hepa-tocellular carcinoma. Prog. Med. Virol.39:167–203.
56. Su, F., and R. J. Schneider.1997. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor␣. Proc. Natl. Acad. Sci. USA94:8744–8749.
57. Sun, Z., P. Lu, M. H. Gail, D. Pee, Q. Zhang, L. Ming, J. Wang, Y. Wu, G. Liu, and Y. Zhu.1999. Increased risk of hepatocellular carcinoma in male hepatitis B surface antigen carriers with chronic hepatitis who have detect-able urinary aflatoxin metabolite M1. Hepatology30:379–383.
58. Terradillos, O., O. Billet, C. A. Renard, R. Levy, T. Molina, P. Briand, and M. A. Buendia.1997. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene14:395–404.
59. Tomiya, T., I. Ogata, and K. Fujiwara.1998. Transforming growth factor alpha levels in liver and blood correlate better than hepatocyte growth factor with hepatocyte proliferation during liver regeneration. Am. J. Pathol.153:
955–961.
60. Wang, X. W., K. Forrester, H. Yeh, M. A. Feitelson, J. R. Gu, and C. C. Harris.1994. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. USA91:2230–2234.
61. Wood, R. D.1996. DNA repair in eukaryotes. Annu. Rev. Biochem.65:135– 167.
62. Wyllie, A. H., C. O. Bellamy, V. J. Bubb, A. R. Clarke, S. Corbet, L. Curtis, D. J. Harrison, M. L. Hooper, N. Toft, S. Webb, and C. C. Bird.1999. Apoptosis and carcinogenesis. Br. J. Cancer80(Suppl. 1):34–37.
63. Zoulim, F., J. Saputelli, and C. Seeger.1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol.68:2026–2030.