0022-538X/83/090452-11$02.OO/O
Copyright © 1983, American Society for Microbiology
Identification
of Four
Complementary
RNA
Species
in
Akabane
Virus-Infected
Cells
ASITK.PATTNAIKANDG. ABRAHAM*
School of Science,Griffith University,Nathan, 4111,Australia
Received 8 March1983/Accepted 26May1983
The analysis ofRNA extracted from purified Akabane virusdemonstratedthe
presence of three size classes of single-stranded RNAs with sedimentation coefficients of 31S (large, L), 26S (medium, M), and 13S (small, S). Molecular
weights of theseRNAspecies were estimated to be 2.15 x
106,
1.5 x106,
and 0.48x 106 for the L, M, and S RNAs, respectively. Hybridization analysis involving
viral genomic RNA and RNA fromvirus-infectedcellsresulted in the
identifica-tion of four virus-specific cRNA species in infected cells. These cRNAs were
found tobenonpolyadenylated by their inabilityto bind to
oligodeoxythymidyl-ate-cellulose. KineticanalysisofcRNAsynthesis in infected cells at various times
postinfection suggested that cRNA synthesis could be detected as early as 2 h
postinfection and that maximal synthesis occurred at 4 to 6 hpostinfection. The
RNAssynthesized in infectedcells could be partially resolved by sucrose density
gradient centrifugation. TheRNA fraction thatcosedimented with the S segment
of viral genomic RNA yielded two duplex RNA species when hybridized with
viral genomicRNA,suggestingthepresence of two small cRNA species. Specific
hybridization with individual viralgenomic RNAs confirmed that two species of
cRNA are coded by the S RNA segment. Analysis of cRNA synthesis in the
presenceof the protein synthesis inhibitors cycloheximideandpuromycin
indicat-ed that cycloheximide completely inhibited virus-specific RNA synthesis early
and late in infection, whereas a very low level of synthesis occurred in the
presence of puromycin. The inhibitory effects of these drugs were found to be reversible when the drugs were washed from the cells. It is concluded that continuedprotein synthesis is required forcRNA synthesis to proceed in Akabane
virus-infected cells.
Akabane virus is a member of the genus
Bunyavirus and family Bunyaviridae (7). All
bunyaviruses sofar characterized contain three
unique segments ofsingle-stranded RNA (large
[LI, medium
[Ml,
and small [S]) of negativepolaritywithatotalmolecularweightof
approx-imately 4 x 106 to 6 x 106 (3). These RNAs
reside within the virion as three helical and
circular nucleocapsids, being complexed with
multiple copies of a nucleocapsid protein (N)
and afewcopies ofalargeprotein(L), believed
tobe atranscriptase component(5). Two
exter-nalglycoproteins(G1 andG2)form thespikeson
the viralenvelope;G1 protein containsasitefor
binding to cellular receptors and also functions
astheviralhemagglutinin,whereasthe function
ofG2 still remains to be demonstrated (15). A recentreport(25)alsosuggests the presenceofa
fifth virion-associated polypeptide of molecular
weight16,000 (16K). In addition to these
struc-tural proteins, the viral genome also codes for
several nonstructural proteins (12).
Recombina-tion studies (4, 13,14)showthat the small viral
RNA contains coding information for the N
protein, whereas the MRNA segmentcontains
theinformation for both G1 and G2.Itis
there-fore inferred that theLproteinis codedbythe L
RNAsegment.
Although more than five virus-specific
pro-teins have been identified, the mechanism of
bunyavirus gene expression in infected cells
remains to be explained. It is not known how many mRNAs mediate the expression of three
genome segments into more than five
virus-specificpolypeptides. Using duplexRNA
analy-ses, Cash et al. (9) identified the presence of
three cRNA species in snowshoe hare virus-infected cells but could only detect messenger activity in the smallest of the species. By
frac-tionatingRNAfrom Uukuniemivirus(amember
of the familyBunyaviridae)-infected cells in
su-crose gradients, Ulmanen et al. (26) detected
fourvirus-specificRNAs, twoofwhich directed
the synthesis of virus-specific polypeptides in
vitro. The mRNA species that cosedimented
withthe M RNA segmentdirected thesynthesis
452
on November 10, 2019 by guest
http://jvi.asm.org/
ofa11OK-molecular-weight protein (foundtobe theprecursor toGl andG2).Inaddition, a 12S
RNA species directed the synthesis of the N
protein anda30K nonstructuralprotein.
This communicationreportstheidentification of four nonpolyadenylated cRNA species
syn-thesized in Akabane virus-infected cells and the
sensitivity of theirsynthesistotheprotein
syn-thesisinhibitors cycloheximide andpuromycin.
MATERIALS ANDMETHODS
Cellsand virus stock. Akabane virus(strain R7947)
wasobtained from the Queensland Institute of Medical
Research, Brisbane, Australia, and was cloned by
plaque isolation twice before use. Vero cells were
grown in minimal essential medium containing 5%
fetalcalfserum. Virus stocks wereusually prepared by infecting confluent monolayers of Vero cells in roller bottles at amultiplicity of infection (MOI) of
0.01 PFUpercell. High-titer stockswereprepared by directly pelleting virus from the clarified culture fluids andresuspending it in phosphate-buffered saline con-taining 0.2% bovine serum albumin and 0.001% DEAE-dextran.
Growth ofvfrusandpreparationof vRNA. Subcon-fluentmonolayercultures of Vero cells in roller bottles
wereinfected with Akabane virusatanMOI of 0.001
PFU percell. After 1 hof virus adsorption at room
temperature,inoculawereremoved andreplaced with virus growth medium and incubatedat37°C for about 35 h. Virus wasconcentrated from clarified
superna-tant by polyethylene glycol precipitation (22) or by direct pelleting and purification by centrifugation in potassium tartrate-glycerol gradients (22).After repel-leting the virus, viral genomic RNA (vRNA) was extracted with sodium dodecyl sulfate (SDS) and phenol (1).
32P-labeled vRNAwasprepared similarlyfrom virus
growninphosphate-free mediumcontaining1 mCi of
32Pi per ml. The vRNA segments were resolved by
electrophoresis ina3%polyacrylamide gel (120Vfor 16 h) containing 6 M urea and90 mM Tris-EDTA-boratebuffer, pH8.3. The individual RNA segments
were detected by autoradiography, excised, eluted from the gel (in 10 mM Tris-hydrochloride, pH 7.5, 1 mMEDTA,0.5%SDS,0.5 MNaCI),and precipitat-ed with ethanol.
Preparation of RNA from infected cells. Confluent monolayersof Vero cellswereinfected with Akabane virusatausualMOIofapproximately7. After 1 h of virusadsorption at room temperature, inocula were
removed and the monolayerwaswashed with warm phosphate-buffered saline and incubated in the pres-ence of Hanks balanced salt solution supplemented with15 mMHEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonicacid) adjustedtopH7.4. Actinomy-cin D (2 sLg/ml), [5-3H]uridine (25 to 50 pCi/ml), cycloheximide (100 pLg/ml),andpuromycin (100iLg/ml) wereaddedatappropriate times postinfection accord-ingtotheprotocolofeachexperiment. After comple-tion of the labeling period, cell monolayers were
washedwith cold phosphate-buffered saline and dis-solved in buffercontaining 0.5%SDS, and the RNA
wasextractedwithphenolandprecipitatedwith
etha-nol. Polyadenylated and nonpolyadenylated RNAs were separated by oligodeoxythymidylate [oligo(dT)]-cellulosechromatography (1).
RNA-RNA hybridization and electrophoresis. La-beled cRNAsfrom infected cells were hybridized with anexcessof unlabeled vRNA (17). The hybrid mole-cules thus formed were digested with single strand-specific nuclease Si (1,000 U/ml) at 37°C for 2 h and the residual double-stranded RNAs (dsRNAs) recov-eredby ethanol precipitation were analyzed by elec-trophoresis on a 4% polyacrylamide gel at 40V for 15 to 20 h (17). Thegel was processed for fluorography (8), and the radioactive bands were detected by expos-ing the dried gel to X-ray film at -70°C (19).
To determine the excess of unlabeled vRNA re-quired by hybridization, fixed amounts of [3H]cRNA from the virus-infected cells were hybridized with increasing amounts of unlabeled vRNA. The trichloro-aceticacid-precipitable radioactivity in portions of the hybridized RNAs before and after S1 nuclease diges-tion was determined (Table 1). This analysis indicated that saturation levels of hybridization were achieved by usingatleast6FxgofpartiallypurifiedvRNA.
Sucrosedensity gradient analysis.Labeled RNAs for sucrose density gradient analysis were dissolved in water, denatured by heating at 100°C for 1 min,
immediately chilled, and adjusted to 10 mM Tris-hydrochloride (pH 7.5)-100mMNaCl,1 mM EDTA-0.1% SDS. RNA samples were then analyzed by sedimentation through 15 to 30%o (wt/vol) sucrose density gradients (prepared in the same buffer) in a BeckmanSW41 rotor (25,000 rpm, 16 h,20°C). Frac-tions (0.4 ml) werecollected from the bottom of the gradient, and trichloroacetic acid-precipitable radioac-tivity in portions of each fraction was determined. Where appropriate, RNAs were recovered from the gradient by ethanol precipitation of the pooled peak fractions.
Materials.Cycloheximide, puromycin, and nuclease
Siwereobtained fromSigmaChemicalCo., oligo(dT)-cellulose from Collaborative Research Inc., and
[5-3H]-uridine from the Radiochemical Centre, Amer-sham, England. ActinomycinDwas agiftfrom Merck
Sharp & Dohme, Australia. [3H]uridine-labeled
[image:2.492.257.451.529.655.2]dsRNAfrom Wallal virus was agift from P. J. Walker, Queensland Institute of Medical Research, Brisbane, Australia.
TABLE 1. Determination of thesaturatinglevelof unlabeled vRNArequiredforhybridizationwith
[3H]cRNA
Amt(.g)of unlabeled vRNA
hybridizedwith[3HJcRNA %Radioactivity resistantto from 2 x106cells Si nucleasedigestiona
0 32
1 41
2 50
4 67
6 82
8 83
10 82
aDetermined by precipitating aportion of the
hy-bridizedRNAsbefore and afterS1 nucleasedigestion.
on November 10, 2019 by guest
http://jvi.asm.org/
454
RESULTS
Sucrose density gradient analysis of vRNA.
[3H]uridine-labeled RNA was extracted from
purified virus, and the denatured productswere
analyzed by sedimentation inanSDS-containing sucrose density gradient. A profile of the
acid-precipitable radioactivity in the gradient frac-tions revealed three discrete peaks (Fig. la) representing RNA species with sedimentation coefficients of 31, 26, and 13 for theL, M,and S RNAs, respectively. Molecular weights of2.15
x 106, 1.5 x 106 and 0.48 x 106 for these three
RNA species were calculated. The single-stranded nature of the RNA was shownby its sensitivity to prior digestion with pancreatic RNase (Fig. la). These data confirmed that Akabane virus RNA shows the characteristic properties of RNA fromamember ofthe
Bunya-viridae.
Identification of cRNA species in infected cells. RNA species synthesized in mock-infected or
Akabane virus-infected cells were labeled with [3H]uridine from 2to 8 h postinfection (p.i.) in thepresenceofactinomycinD. Total RNAwas
extracted andanalyzed by density centrifugation in SDS-containing sucrose density gradients
(Fig. lb). Incorporation of radioactivity into mock-infected cell RNA was considerably less
than into infected cellRNA andwas
concentrat-edatthetopof the gradient. Two of the major peaks of radioactivity in the infected cell RNA sample corresponded with the sedimentation positions of the M and S vRNA segments(Fig. la), whereas a shoulder of radioactive material
(fractions 6to10) correspondedtothe sedimen-tationposition of the L vRNA segment.
To demonstratethevirus-specific and
comple-mentary nature ofthe RNA synthesis, we
hy-bridized [3H]uridine-labeled RNAs from either mock- or virus-infected cells with unlabeled
vRNA.Preliminary experiments had shown that saturation levels ofhybridizationwereachieved
instandardizedexperiments using approximate-ly 7 pug of crude vRNA extracted from partialapproximate-ly purified virus. After removal of unhybridized
[image:3.492.50.243.61.562.2]BOTTOM FRACTION NUMBER TOP
FIG. 1. (a)Sucrosedensitygradient centrifugation ofRNA frompurifiedAkabanevirus.3H-labeledRNA from purified virus was denatured and sedimented
througha15to30% SDS-sucrosedensitygradient as
described inthetext. Fractions(0.4 ml)were
collect-ed, andtrichloroacetic acid-precipitable radioactivity in eachfraction wasdetermined. Symbols:.0, RNA
from purified virus; 0, RNA from purified virus
digested withpancreatic RNase(20 ,ug/ml in 10 mM Tris-hydrochloride [pH 7.5],0.3 MNaClat37°Cfor 30 min). Arrows indicate the sedimentation position of 3H-labeled 35S poliovirus RNA and 28S and 18S
rRNAs. (b) Analysis of RNA from Akabane
virus-infectedormock-infectedcells. The virus-infectedor
mock-infected cells were labeled with [3H]uridine from 2to8 hp.i.in thepresenceof 2,ugofactinomycin Dperml(addedat0 hp.i.). Total labeled RNAwas
extracted from each cultureandanalyzed by centrifu-gation on15 to 30% SDS-sucrose densitygradients. Fractions(0.4 ml)werecollected and the
trichloroace-ticacid-precipitable radioactivity inportions of each fractionwasdetermined. Symbols: 0,RNA from the
virus-infected cells; 0, RNA from mock-infected
cells. w
0
z
ar
r-J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
A B C D E F G b
<1
_n _lU
-*
<d2
A B C
nr-,
Da
2
<3 < 4
m-.'fl 10,I
o-u.no
MO sw-O "urn
3 . J4
[image:4.492.68.433.76.388.2]4 *ow
FIG. 2. Polyacrylamide gel electrophoresis of virus-specific dsRNA hybrids. (a) Total RNA from mock-infected (lane A)orvirus-infected (lanesB andC) cellslabeled with [3H]uridine at 2 to8 hp.i. wereeither hybridized with unlabeled vRNAs(lanes A and B) or self-annealed (lane C),digestedwith nuclease Si, and
analyzedbyelectrophoresison a4%polyacrylamide gelasdescribedin thetext.Lane Drepresents the dsRNAs produced byhybridizationof[3H]uridine-labeled vRNAswith unlabeled RNAsfrom virus-infected cellsat8 h
p.i. Afterelectrophoresis, the radioactive bands were detected byfluorography. Arrowheads with arbitrary
numbers represent thefourvirus-specificdsRNAhybrids.(b)Infectedormock-infected cultureswereincubated inpresenceof actinomycinD at0 hp.i. and labeled with[3H]uridinefor 2-hperiodsatvarioustimesstartingat0 hp.i.The RNAswerethenhybridizedwithunlabeled vRNAs, and theduplexesformedwereanalyzedasabove. Duplexes in lanes B to G werederived from infected cells labeledat0,2, 4, 6,8,and 10 hp.i., respectively. Lane Acorrespondsto amock-infected culture labeled similarly.
RNAs by digestion with Si nuclease, the
dou-ble-strandedhybridmolecules were resolvedby
polyacrylamide gel electrophoresis (Fig. 2a).
Four radioactive bands representing four
dsRNA hybrids (arbitrarily numbered 1 to 4) were detected in extracts from virus-infected
cells (lane B) but not from mock-infected cells
(lane A). When a portion of3H-labeled RNA
from infected cells was self-annealed, digested
with nuclease Si, and analyzed similarly, the
samefourhybridRNAs weredetected(Fig. 2a,
laneC). This result indicatedthat RNA of both
polarities was synthesized in infectedcells
dur-ing the period 2 to 8 h p.i. To confirm that
hybridization to vRNA was occurring in these
experiments, thereverse procedureof
hybridiz-ing[3H]uridine-labeledvRNAtounlabeledRNA
from infected cells was done (Fig. 2a, lane D).
FourhybridRNAswith thesame
electrophoret-icmobilitiesasseenin lane B were detected. All
fourcRNAspeciesininfected cellswere shown
to be cytoplasmic rather than nuclear in their
location (data notshown).
Codingorigin of cRNA species. The results in
Fig. 2demonstrated the presence of four cRNA species in the virus-infected cells. To identify
thecoding origins ofthese cRNAs, vRNA
uni-formly labeled with 32p was prepared, and the
three single-stranded RNA segments were
re-solvedbypolyacrylamidegelelectrophoresis(as
VOL.47,1983
v0 -* 1
W*
S.
lw on November 10, 2019 by guest
http://jvi.asm.org/
456 PATTNAIK AND ABRAHAM
A
B
CD
4-
U-FIG. 3. Coding origin of the induced (
cies.Uniformlylabeled (32P]vRNA(totaloi
segments)washybridizedwithan excesso cRNAs from virus-infected cells and ar
electrophoresis as in Fig. 2 and detecte radiography. Hybrids were produced [32P]vRNA (lane A), L RNA segment (l RNAsegment(lane C),and S RNAsegmel
[3H]uridinewasadded for 2-h periodsatvarious times from 0 to 10 h p.i. Labeled RNAs were
extracted,
hybridized
withan excess ofvRNA,
and analyzed (Fig. 2b)asdescribedabove (Fig.
< 2 2a). Two of the virus-specific RNAs were
de-tected as early as 2 h p.i. (lane B) by this
procedure. By 4 h p.i. all four duplex RNAs
were readily detected, and RNA synthesis
reached a peak at 4 to 6 h p.i. After 8 h, the synthesis of the largesthybrid species fell below detectable levels, whereas the synthesis of the other three hybrids continued but in reduced
amounts.These dataindicate thattemporal
con-trol of the synthesis of cRNA species of Aka-bane virus occurs during infection. In similar
experimental conditions, the time of maximal
< 3 virus-specific protein synthesis coincided with < 4 the time ofmaximal cRNAsynthesis (data not
shown).
The molecular weights of the RNA hybrid speciesweredeterminedby comparison with the
known values of the dsRNAofWallal orbivirus (16). Table 2 shows the molecular weights of thesehybrids, their corresponding single-strand-ed components, and the calculated coding
po-tential, after translation, of possible proteins. Capacity of cRNAs to bind to oligo(dT)-cellu-lose. Todetermine whether the cRNAspecies in Akabane virus-infected cells could beseparated intopopulations of polyadenylated or
nonpolya-denylated molecules, fractionationwas
attempt-ed by oligo(dT)-cellulose chromatography. [3H]uridine-labeled RNA was extracted from
cRNA spe- infected cells at either early or late times after
rindividual infection, and the polyadenylated RNA species
funlabeled were separated from the nonpolyadenylated
dalyzed
by RNA. When actinomycin D (2 ,ug/ml) wasin-withd
total
cludedduring
thelabeling
period
(4 to8 hp.i.),
lane B), M less than 4% oftheacid-precipitable radioactiv-nt(laneD). ity was found to bind specifically to and elutefrom oligo(dT)-cellulose. In contrast, a similar
described above). The recovered individual RNA segments were each hybridized with an
excess ofunlabeled cRNA from infected cells,
digested with Si nuclease, and analyzed by polyacrylamide gel electrophoresis. The unfrac-tionated[32P]vRNAuponhybridization produced the fourtypical dsRNA hybrids (Fig. 3, lane A). The L RNA upon hybridization produced
hy-brid1, whereas the M RNAgaverisetohybrid 2 (lanes B and C,respectively). Incontrast, the S RNAyielded bothhybrid 3 and hybrid4(lane D)
uponhybridization.
Kinetics of cRNA synthesis. To follow the kinetics of synthesis of cRNA species after infection, we infected parallel cultures at an
MOI of 7 PFU per cell and incubated in the
[image:5.492.74.220.78.405.2]presence of 2 ,ug of actinomycin D per ml.
TABLE 2. Molecular weights of Akabane virus-specificcRNAs andtheir coding potentials
Molwt(x106) Approximate HybridRNA Corresponding coding
species dsRNA0 single-stranded potential
RNA(x0)
1 3.8 1.9 210
2 2.76 1.38 153
3 0.25 0.125 14
4 0.21 0.105 12
aMolecularweights of the dsRNAs were calculated
by using the double-stranded RNAs of Wallal orbi-virus (16) as molecular weight standards.
bThecoding potential of each of the cRNAspecies wascalculated as one-ninth of their respective molecu-larweights.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.492.257.447.539.622.2]fractionation of host cell RNA labeled in the
absenceofactinomycinshowed that 35% of the
incorporated radioactivity was in a polyadeny-lated form.
Evidence that virusspecificcRNAs were pre-sent in the fraction that failed to bind to
oli-go(dT)-cellulose was provided by hybridization
experiments. Polyadenylated and
nonpolyaden-ylated RNAs from infected cells labeled either
early (0to 4 h p.i.)or late (4to 8 h p.i.) were
isolated and hybridized withan excess ofvRNA
and analyzed as described above. The
virus-specific dsRNA hybrids could only be seen in
the lanescontaining products derived from
non-polyadenylated RNAs (Fig. 4, lanes A and C).
HybridRNA 1 species couldbe detected early
afterinfection by longer exposure of the gelto
X-ray film (lane A), but was clearly present
when labelingwasdone atthe timeof maximal
RNA synthesis (lane C). The broad band of
radioactivityseeninthelowerpartof the gels is
aresidual hostcomponent asactinomycinD was
not included in this experiment. Preliminary
experiments to test the sensitivity of this
fluo-rography method showed that if virus-specific
radioactivity in the polyadenylated samples had
exceeded 2% of that in the nonpolyadenylated
samples then discrete bands would have been
visible in lanes B and D (Fig. 4). Additional
experiments were done to try to detect
virus-specific polyadenylated RNA species in
Aka-bane virus-infected cells by analyses in sucrose
density gradients. When the small amount of
polyadentylated
RNA thatcould be isolatedwas analyzed asdescribed inFig. lb,noradioactivepeaks corresponding to viral RNAs could be
detected(datanotshown). Thus, by either
meth-od ofdetection, no virus-specific
polyadenylat-ed RNA species could be found in Akabane virus-infected cells.
Virus-specificRNAsynthesis in the presence of
protein synthesis inhibitors. Results of
self-an-nealingof RNAs from infected cells(Fig.2a) and thepartialresistance of suchintracellularRNAs
to digestion with pancreatic RNase (data not shown) indicated that RNAs of both polarities
werebeing synthesizedeven atearlytimes after
infection. Ithasbeen well established (2, 20, 23)
with othernegative-strand viruses that inhibitors
ofprotein synthesis prevent the translation of
mRNA synthesized early in infection and so
restrict viralRNA synthesis to that ofonly the
primary type. In such conditions, only cRNAs
thatcorrespond to viral mRNAsare produced.
Theclassical protein synthesis inhibitors
cyclo-heximide and puromycin were used to treat
Akabane virus-infected cells from the time of
infection.
[3H]uridine
wasadded at 2 h p.i. for afurther 4 h. After extraction and denaturation,
the labeled RNAs were analyzed by sucrose
A
B
C
.
'-D
9
M
tr---<3
[image:6.492.282.422.72.376.2]4
FIG. 4. Electrophoretic analysis of dsRNAs formed by hybridization ofpolyadenylated or non-polyadenylated RNAs from the virus-infected cells. [3H]uridine-labeledRNAfrom the virus-infectedcells
at0 to 4 h (lanes A and B) or4to8 h(lanes C and D) werefractionated into polyadenylated and nonpolya-denylated RNAs by oligo(dT)-cellulose chromatogra-phy and hybridized to vRNA and analyzed as de-scribed inthelegend toFig. 2. Lanes A and C contain
duplexes derived from nonpolyadenylated RNAs, whereas lanes B and D contain duplexesfrom poly-adenylated fractions.
density gradientcentrifugation (Fig. 5). The
gra-dients containing mock-infected cell RNAs or
infected cell RNAs(in the absence of
cyclohexi-mideorpuromycin) produced profiles similarto
that seen inFig. lb.However,the RNAprofiles from cultures treated with either of the drugs
failed to reveal any radioactive peaks
corre-spondingtothesedimentationpositions ofthe L
andMvRNAsegments.Minorpeaks of
radioac-tivitywere seenin thesegradients,
correspond-ingtothesedimentationposition ofthe S vRNA
segment, which was also the region to which
some labeled host RNAs sedimented (see the
mock-infected profile, Fig. 5). These results
indicatedthat RNA synthesis in thepresenceof
on November 10, 2019 by guest
http://jvi.asm.org/
0
10
5-v 5 lo is 20 25 30
BOTTOM FRACTION NUMBER TOP
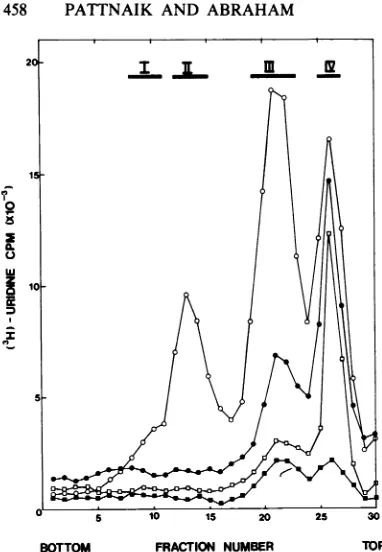
FIG. 5. Sucrosedensitygradient analysisof RNA fromvirus-infectedormock-infectedcells in the
pres-ence of cycloheximide or puromycin. Infected or mock-infected cultureswereincubated inthepresence
ofactinomycinDand eithercycloheximide (100 ,ug/ml)
orpuromycin (100 ,ug/ml)from the time ofinfection. RNAslabeled with[3H]uridinefrom2to6hp.i.were extracted andanalyzedbycentrifugationon15to30% SDS-sucrose density gradients as described in the
legend to Fig. 1. RNAfrommock-infected cells (0)
and RNA from Akabane virus-infected cells either directly (0)orin thepresenceofcycloheximide (O)or
puromycin(U).
either of these inhibitors was below detectable
levels in thisanalysis. Even if the MOI in such experiments was increased to 50 PFU percell,
no RNA synthesis was detectable in cells in
which protein synthesis had been stopped by these inhibitors (see below).
To determine whether the radiolabeled RNA species inFig. 5 were virus specific, the RNAs
infourregions of the gradients (numbered I, II,
III, and IV) wererecoveredby ethanol
precipi-tation and hybridized with an excess of
unla-beled vRNA. When theresulting dsRNAs after S1 nuclease digestion were analyzed by
poly-acrylamide gel electrophoresis, virus-specific RNAscouldbedemonstrated only in fractions I
to III of thegradient containing RNA from the control infected cells (data not shown). No vi-rus-specific productsweredetected in gradients
containing RNA from either cycloheximide- or
puromycin-treatedcells (data not shown), which
confirmed that primary RNA transcription in
such treated cellswas belowdetectable levels.
Sincetheinhibitoryeffects ofpuromycin and
cycloheximide on protein synthesis are fully
reversible, itwasofinteresttodetermine
wheth-erRNAsynthesis intreatedinfected cells would
recommence if the drugs were removed. To
make thedetection ofanyprimary transcription
in such cellsmorelikely,anMOI of50PFUper
cell was used. Cycloheximide and puromycin
were added to four cultures at the time of
infection. At 1.75 h p.i., the drugs were
thor-oughly washed from two ofthecultures over a
period of10 min. All four cultures (together with
anuntreated control culture) were labeled with
[3H]uridine
from2 to 6 h p.i. The labeled RNAfrom each culture was hybridized with vRNA
andanalyzed as described above (Fig. 6a). No
dsRNAs were seen when cycloheximide was
retained intheculture (lane B), butafaint band
corresponding to the fourth viral hybrid RNA
could be seenin the corresponding
puromycin-treated sample (lane D). When the inhibitors
werewashedfromthecultures, cRNA synthesis was restored as shown by the presence of the
typicalpatternof four dsRNAs in lanes C andE.
However,theinhibitory effect of cycloheximide
could not be completely reversed within the
labeling periodused asjudgedby acomparison
of the intensities oftheradioactive bands (lanes
A and C).
Since in the presence of cycloheximide and
puromycin primary transcription wasibelow
de-tectablelevels,anexperimentwasdonetoshow
theireffectsonsecondary(amplified)RNA
tran-scription when syntheticratesaremuchhigher.
Five cell cultures were infected (at the usual
MOIof7PFU percell) and incubated for4h to
allowRNA synthesisto reachitsmaximalrate.
Cycloheximide and puromycin were added to
two cultures each and then washed from two
culturesat5.75 hp.i.
[3H]uridine
wasaddedfora further 4 h before preparing and analyzing
samples as described forFig. 6a.
Results observed(Fig.6b) indicated that both
cycloheximide and puromycin had a dramatic
inhibitory effect on secondary RNA
transcrip-tion also(lanes B and D) and that thisinhibition
couldbe readilyreversed bywashingthedrugs
from the cellcultures(lanes C and E). However, theeffectofpuromycin onsecondary
transcrip-tion was not complete, as afaint band of
radio-activity correspondingto hybridRNA 4 canbe
seen in lane D, Fig. 6b. The sensitivity ofthe
fluorography procedure used couldjust detect
bands containing 2% of the radioactivity of
control bands(lane A),andsoanestimateofthe
effect ofpuromycin on RNA synthesis in this
experimentis that itisat least90% complete.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.492.48.239.55.331.2]A
-e ~- ,
B C D E a
A B C D E b
<1
_ <2 gEE
<3
_- 4
FIG. 6. Effect of cycloheximide and puromycin on RNA synthesis in virus-infected cells. (a) Cultures infected with Akabane virus at a multiplicity of infection of 50 PFU per cell were treated with either
cycloheximide(100,ug/ml)orpuromycin (100,ug/ml)at0hp.i.At 1.75 hp.i., drugsfromone setof cultureswere washed out and thecellswerelabeled with[3H]uridinefrom 2to6hp.i.The RNAswereextracted, hybridized
with unlabeled vRNAs, and analyzed by polyacrylamide gel electrophoresis. Duplexes were derivedfrom infected cells (lane A), in the presence ofcycloheximideorpuromycin (lanes BandD,respectively), orwhen cycloheximide and puromycin were washed out (lanes C and E, respectively). (b) Similaranalysis of the duplexes when cell cultures were infected at an MOI of7 PFU percell were treated withcycloheximide or
puromycinat4hp.i.,and thedrugswerewashedout at5.75 hp.i.RNAwaslabeledwith[3H]uridinefrom6 to 10 hp.i. Duplexeswerefrom infected cells(laneA) in the presence ofcycloheximideorpuromycin (lanesB andD,
respectively)orwhencycloheximide andpuromycinwerewashedout(lanesC and E,respectively).
DISCUSSION
Analysis ofthe RNA contained in Akabane
virus particles indicated that it was typical of
that found for other bunyaviruses in that the
genome RNA was single stranded and in three
segments, although the measured sizes of these
segments wereslightlysmaller thanthose found
for other members of the family (5). However,
Akabane virus was found to differ from the
generalpropertiesshownby negative-strand
vi-ruses in that no polyadenylated cRNA species
could be detected in infected cells, and
estab-lished secondary RNAtranscription was
sensi-tivetotheaction of inhibitors ofprotein
synthe-sis. In addition, four species of cRNA were
detected inAkabane virus-infectedcells, which
differs from thethreefoundpreviouslyfor
snow-shoe hare bunyavirus(9).
The synthesis of
[3H]uridine-labeled
cRNAinAkabane virus-infected cells was followed by
hybridizationwith unlabeledvRNAandanalysis
of the 3H-labeled hybrid species so formed by
gelelectrophoresis. Thiswasnecessarybecause
the usual experimental procedures used with
negative-strand virusestoanalyze only primary RNAtranscripts resulted in undetectable levels of[3H]uridine incorporation. Kinetic studies of cRNAsynthesis atvarious times afterinfection
at an MOI of7 PFUper cell (Fig. 2b) showed
that detectablesynthesiswasestablishedas
ear-lyas 2h p.i. Atthis time, only RNA hybrids 2
and 4 were visible(Fig. 2b,lane B). Synthesisof cRNAs increased as the infection proceeded,
reaching amaximal rateat4 to 6 h p.i. At this
time, all four hybrid species representing the
fourcRNA species were detected readily (lane
D). The synthesis ofthe cRNAspecies leading
to the formation of hybrid 1 was the first to
decrease to below detectable levels (lane E),
whereas synthesis ofthe cRNA speciesleading
totheformation ofhybrid3persisted alongwith
gI-4
<1 <2
w
M
<3
< 4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.492.57.446.73.336.2]460 PATTNAIK AND ABRAHAM
hybrids2and 4untilafter 10 h p.i. These results
show that although all four cRNA species
reached their time of maximalsynthesis together
(4 to 6hp.i.),there waslimited temporal control
overthe synthesis of individual cRNA species.
Toconfirm that the fourcRNA species being
labeled in these experiments truly represented
viral cRNA species, the alternative experiment
inwhich[3H]uridine-labeledvRNAwas
hybrid-ized with unlabeled infected cell RNAs was
done andanalyzed similarly. Againfourspecies
of double-stranded hybrid molecules that
showed the same electrophoretic mobilities as
those in theconverseexperiment(Fig. 2a, lanes
BandD) werefound. This result contrasted with
the results of Cash et al. (9) who found only
three such hybrid species in analyses of
snow-shoe harevirus-infected cells. Although the
sin-gle-strand-specific S1 nuclease was used
rou-tinely inourexperimentstodigest unhybridized
(single-stranded) molecules, the same four
hy-brid RNA species could be produced if the
RNases T1 or T2 were used instead for the
digestionstep(datanot shown).
MolecularweightsofthehybriddsRNAs and
the corresponding cRNAs were determined by
comparison with the genome RNAs of the
de-scribedorbivirus, Wallal (16). Thederived sizes
of the cRNAspecies (Table1)weresmallerthan
the values measured for the vRNAs, indicating
that none represented full-length transcripts of
thegenomeRNAs. Thecodingpotentialsof the
cRNAs(calculatedasone-ninthof the molecular
weight of the cRNA)werefoundtobe
approxi-mately210 x 103, 153 x
103,
or14x 103daltonsrespectivelyfor the L, M,orStranscripts (Table
1). Either of the cRNAs represented in hybrid
species 1 and 2appear tohavesufficient coding
information for either the L protein (121 x 10
daltons)ortheG1 andG2proteins together(140
X 103 daltons, total). In contrast, the cRNAs presentinhybridspecies 3 and4do notappear
tobelargeenoughtocode for either the LorG1
plus G2 proteins. Within the approximations
used for calculating the coding potentials of
these cRNAs, it seems possible that one of
cRNA species present in either hybrid 3 or 4
codes for the N protein. This conclusion has
been supported by preliminary in vitro
transla-tion ofthe RNA species in region III, Fig. 5,
which hasresulted in thetranslation ofonly the
polypeptidecorrespondingtotheNprotein
(un-published results). HybridRNAspecies3and4
are similar in size and may have overlapping
sequences since hybridization shows that they
areboth derived from segment S ofviral RNA
(Fig. 3). Ifso,thisinformation is consistentwith
thereportofBishop and co-workers (6)that the S segments of bunyaviruses have sequences
that allow for the potential expression oftwo
gene products by using overlapping reading
frames. Additional nonstructural proteins found
in variousbunyavirus-infectedcells (12, 25, 26),
including Akabane virus(21),could conceivably
betranslated from the remainingcRNAspecies.
None of the RNAs synthesized in Akabane
virus-infected cells wasfound to be
polyadeny-lated, basedontheirinabilityto bindspecifically
tooligo(dT)-cellulose. The RNA synthesizedat
earlytimes after infection and presumably
con-taining the products of primary transcription
failed to bind to oligo(dT)-cellulose in amounts
sufficienttoanalyze asbeing virus specific (Fig.
4). In contrast, RNA from the fraction which
failedtobindtooligo(dT)-cellulose could readily
bedemonstratedto contain virus-specific
mole-cules. Similar results were obtained if RNA
synthesized atlater times (undoubtedly
includ-ingtheproductsof secondary transcription)was
used for the analysis (Fig. 4). If the MOI in
experiments of this type was increased to
ap-proximately 50 PFU per cell to increase the
probability of detecting primary RNA
tran-scripts, the results were unchanged (data not
shown). It could be argued that the level of
polyadenylated RNA synthesis was toolow to
detect in theseexperiments. However,thedata
in Fig. 4 combined with measurements ofthe
sensitivity of the fluorography detection
proce-dureindicate that the synthesis of virus-specific
nonpolyadenylated RNA was at least 30 times
higher than thatof polyadenylatedRNAin
Aka-bane virus-infected cells. Such a situation is
unique among the negative-strand viruses in
general, but is consistent with work reported previously for another bunyavirus, Uukuniemi
virus (26). However, it conflicts withtheresults
of Cash et al. (9), who were able to show that
both polyadenylated and nonpolyadenylated
RNAfractions from snowshoe hare
virus-infect-edcells could be translated in vitroto produce
thenucleocapsidprotein N. Preliminary studies
in our laboratory with in vitro translation
sys-tems suggestmRNAactivity is associated only withnonpolyadenylatedRNAin Akabane
virus-infected cells, thus supporting our contention
that in this system nopolyadenylatedRNAs can be detected.
It is accepted that the first new synthetic
event in cells infected with a negative-strand
virus isthe production ofmRNA by the
virus-associatedRNApolymerase. Thisprimary
tran-scription is independent of protein synthesis.
However, the change to secondary (amplified)
transcriptionis dependenton theexpression of
viralproteins, and this transitioncanbe
prevent-ed by the use of inhibitors ofprotein synthesis (10, 11, 28). Thus, cycloheximide and puromy-cin were used to treat Akabane virus-infected
cells from the time of infection to examine
on November 10, 2019 by guest
http://jvi.asm.org/
primary RNAtranscription products. When
ana-lyzedeither bysedimentation inSDS-containing
sucrose density gradients or by hybridization
plus gelelectrophoresis, novirus-specific RNA
productscould be detected (Fig. 5and6).
Simi-lar results were obtained if the inhibitors were
not added untilthe time of maximal RNA
syn-thesis, when secondary transcription was
un-doubtedly occurring. This was a surprising
re-sult as secondary transcription with other
negative-strandviruses hasnotbeen reportedto
bedependentuponcontinuing protein synthesis
(23, 24). Similar effects were observed if an
alternate protein synthesis inhibitor, emetine,
was used (data not shown). Studies on the
primary transcription of bunyaviruses in the
presence ofcycloheximide orpuromycin have
been reported previously. In snowshoe hare
virus-infected cellsat ahigh MOI, low levels of
RNAsynthesis could be detected by
hybridiza-tion in thepresenceofcycloheximidebutnotin
thepresence ofpuromycin (27). Similar results
alsowereobtainedbyusingmutantsof this virus
(27). RNA synthesis in Bunyamwera
virus-in-fected cells in thepresenceofcycloheximide has
been examined(18) andagain[3H]uridine
incor-poration levels were only marginally above
background levels, and the products were not
showntobecomplementary tovRNA.Thus, in
bothof thesereportsand the resultspresentedin
thiscommunication, primaryRNAtranscription
by bunyaviruses in the absence ofprotein
syn-thesis hasnotbeen demonstrated conclusively.
Thesynthesis of protein andRNAin Akabane
virus-infected cells appearedtobelinked.Since
theinhibitory effects of cycloheximideand
puro-mycin on protein synthesis can be readily
re-versedby thewashing of treated cells, itwasof
interest to see whether secondary RNA
tran-scription was affected similarly. The results in
Fig. 6aandb show thatsecondarytranscription
can be readily established after the removal of
either inhibitor (Fig. 6a) and can recommence
andapproach formersyntheticratesif
interrupt-ed by the action of these drugs (Fig. 6b).
Al-though it is not known whether the effects of
cycloheximideandpuromycinonAkabaneviral
RNA synthesis are direct orindirect, it seems
that the latteralternative ispossible because of
thecorrespondencebetween theeffects on
pro-teinand RNA synthesis and the reversible
na-ture ofeach (Fig. 6b). The conclusion follows
thatsecondaryRNAtranscription (and perhaps
primarytranscription as well) in Akabane
virus-infectedcells isdependentoncontinuingprotein
synthesis.
ACKNOWLEDGMENTS
WethankCecilia Devlin for excellent technical assistance. This work was supported by grants from the National
Health and Medical Research Council ofAustraliaand the Australian Meat ResearchCommittee.
LITERATURE CITED
1. Abraham, G. 1979. Theeffect of ultraviolet radiationon
the primary transcription of influenza virus messenger RNAs.Virology 97:177-182.
2. Bean,W.J.,andR. W.Simpson. 1973.Primary transcrip-tionof the influenza virus genome in permissive cells. Virology56:646-651.
3. Bishop, D. H. L. 1978.Geneticpotentialofbunyaviruses. Curr.Top. Microbiol. Immunol. 86:1-33.
4. Bishop, D. H. L., B. J. Beaty, and R.E. Shope. 1980. Recombinationand genecoding assignmentsof bunyavi-rusesandarenaviruses.Ann.N.Y.Acad.Sci.354:84-106. 5. Bishop, D. H. L., C. H. Calisher, J. Casals, M. P. Chumakov, S.Y.Gaidamovich, C. Hannoun, D. K. Lvov, I. D. Marshall, N. Oker-Blom, R. F. Pettersson, J. S. Porterfleld, P.K. Russell, R.E. Shope, and E.G. Westaway.1980.Bunyaviridae.Intervirology 14:125-143. 6. Bishop, D. H. L., K. G. Gould,H. Akashi, and C.M. Clerx-van Haaster. 1982. The complete sequence and coding content of snowshoe hare bunyavirus small (S) viral RNA. Nucleic Acids Res. 10:3703-3713.
7. Bishop, D. H. L., andR.E.Shope. 1979.Bunyaviridae,p. 1-156. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology, vol. 14. Plenum Publishing Corp., New York.
8. Bonner,W.M.,and R. A.Laskey.1974. Afilmdetection method for tritium-labeled proteinsandnucleic acids in polyacrylamide gels. Eur. J. Biochem. 46:83-88. 9. Cash, P.,A.C.Vezz,J. R.Gentsch,and D. H. L.Bishop.
1979.Genomecomplexitiesof the three mRNAspeciesof snowshoe harebunyavirus and invitro translation of S mRNA toviral Npolypeptide.J. Virol.31:685-694. 10. Choppin,P.W., and R.W.Compans. 1975.Reproduction
ofparamyxoviruses, p. 95-178. In H. Fraenkel-Conrat and R.R.Wagner(ed.),Comprehensivevirology, vol.4. PlenumPublishing Corp.,New York.
11. Compans,R.W., andP. W.Choppin.1975.Reproduction oforthomyxoviruses,p.179-252.InH.Fraenkel-Conrat and R. R.Wagner (ed.),Comprehensivevirology, vol.4. PlenumPublishing Corp., New York.
12. Fuller, F., andD. H. L. Bishop. 1982. Identificationof virus-coded nonstructural polypeptides in bunyavirus-infected cells. J. Virol.41:643-648.
13. Gentsch, J. R.,and D. H. L. Bishop. 1978. Small viral RNA segmentofbunyavirusescodesfor viral nucleocap-sidprotein.J.Virol. 28:417-419.
14. Gentsch,J.R., and D.H. L.Bishop. 1979.Aviral RNA segmentofbunyavirusescodes fortwoglycoproteins,Gl and G2. J. Virol. 30:767-770.
15. Gonzalez-Scarano, F.,R. E.Shope,C.E.Calisher,andN. Nathanson. 1982.Characterisation of monoclonal antibod-ies against Gl and Nproteinsof La Crosse and Tahyna, twoCaliforniaserogroupbunyaviruses. Virology 120:42-53.
16.Gorman,B.M.,J.Taylor,P.J. Walker,and P. R.Young. 1978. Isolation of recombinants between related orbivi-ruses.J. Gen.Virol. 41:333-342.
17. Hay,A.J.,B.Lomniczi,A. R.Bellamy,andJ. J.Skehel. 1977.Transcription oftheinfluenza virus genome. Virolo-gy 83:337-355.
18. Kascsak,R.J.,andM.J. Lyons.1977.Bunyamwera virus I.Themolecularcomplexity of the virion RNA. Virology 82:37-47.
19. Laskey,R. A.,and A. D. Mils. 1975. Quantitative film detection of3Hand14Cinpolyacrylamide gels by fluorog-raphy.Eur.J.Biochem. 56:335-341.
20. Marcus, I., D. H. Engelhardt, J. M. Hurt, and M.J. Sekellick. 1971. Interferon action: inhibition of vesicular stomatitis virusRNAsynthesisinducedby virion-bound polymerase. Science 174:593-598.
47,
on November 10, 2019 by guest
http://jvi.asm.org/
21. McPhee, D. A., and A. J. Della-Porta.1980.Biochemical and serological comparisons of Australian bunyaviruses belongingtotheSimbuserogroup, p.93-101. In D. H.L. Bishop and R. W. Compans (ed.), The replication of negative strand viruses, Proceedings of the4th Interna-tional Symposium on Negative Strand Viruses, Virgin Islands. Elsevier/North-Holland, New York.
22. Obijeski,J. F., D. H. L.Bishop,F. A. Murphy, and E. L. Palmer. 1976. Structural proteins of La Crosse virus.J. Virol. 19:985-997.
23. Robinson, W. S. 1971.Sendai virus RNA synthesis and nucleocapsidformation in thepresenceofcycloheximide. Virology 44:494-502.
24. Scholtissek, C., and R. Rott. 1970. Synthesis in vivo of influenza virus plus and minus strand RNA and its prefer-ential inhibitionby antibiotics. Virology 40:989-9%.
25. Short, N. J.,A.D.Meek, and L.Dalgarno.1982.Seven infection-specific polypeptides in BHK cells infected with Bunyamweravirus. J. Virol. 43:840-843.
26. Ulmanen,I., P. Seppala, andR.F. Pettersson. 1981. In vitro translation of Uukuniemivirus-specific RNAs: iden-tification ofanon-structural protein anda precursorto
membraneglycoproteins. J. Virol. 37:72-79.
27. Vezza, A.C.,P. M.Repik,P.Cash,and D. H. L.Bishop. 1979. In vivo transcription and protein synthesis capabili-tiesof bunyaviruses: wild-type snowshoe hare virus and its temperature-sensitivegroupI,groupII,andgroupI/II
mutants.J. Virol. 31:426-436.
28. Wagner, R. R. 1975. Reproduction of rhabdoviruses,p.
1-93. In H.Fraenkel-Conrat and R. R. Wagner (ed.), Com-prehensive virology, vol. 4, Plenum Publishing Corp., New York.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/




![FIG. 4.formedatdenylatedduplexesadenylatedwerewhereasphyscribedpolyadenylated[3H]uridine-labeled 0 Electrophoreticanalysisof dsRNAs by hybridization of polyadenylated or non- RNAs from the virus-infected cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1443557.96758/6.492.282.422.72.376/formedatdenylatedduplexesadenylatedwerewhereasphyscribedpolyadenylated-uridine-labeled-electrophoreticanalysisof-dsrnas-hybridization-polyadenylated-infected.webp)