0022-538X/92/116273-07$02.00/0
Copyright © 1992, American Society for Microbiology
Surface Lysine and Tyrosine
Residues Are Required for
Interaction of
the
Major
Herpes
Simplex Virus Type 1
DNA-Binding Protein with Single-Stranded
DNA
WILLIAM T.
RUYECHAN1.2*
AND JONATHAN W.OLSON2
DepartmentofMicrobiology, School of Medicine, State University of New York at Buffalo, Buffalo, NewYork14214, 1*andDepartmentofBiochemistry, Uniformed Services
University of the Health Sciences, Bethesda, Maryland 208142 Received4May 1992/Accepted 27 July 1992
Modification of the herpes simplex virus tpe 1
major
DNA-binding protein (ICP8) with reagents and conditionsspecific for arginine, lysine,andtyrosine
residues indicates thatsurfacelysine andtyrosine
residues arerequired for the interactionof thisproteinwithsingle-strandedDNA. Modification of either of thesetwo aminoacids resulted inalossand/ormodification of bindingactivity
asjudged bynitroceliulose filter assays and gel shift. Modificationspecificforarginine
residues didnotaffectbinding withinthelimitsof the assays used. Fminally, quenching of the intrinsictryptophan
fluorescence of ICP8 in thepresence ofsingle-stranded DNA either suggests involvement of this amino acid in thebinding reactionorreflectsaconformational changeinthe protein uponbinding.The herpes simplex virus type 1 (HSV-1) major DNA-binding proteincommonlydesignatedinfected-cell polypep-tide 8(ICP8) (25) hasbeen theobjectofintense study since its initialidentificationandsubsequentpurification (5,28-31, 36, 39, 40,43).Theprotein is encoded by HSVopenreading frame UL 29 (35), and early genetic evidence showed that ICP8 is absolutely required for virus replication in tissue culture (10, 52).
Conditional
lethal mutantsmapping in the ICP8 gene display a DNA-negative phenotype and abnor-malitiesin theproduction ofotherviralproteins, suggesting roles for ICP8 in DNAreplication and generegulation (10, 14, 16-19,32).Subsequentanalysis has borneoutthesepossibilities and identifiedother important functions of ICP8. Challberg (7), Olivoetal.
(37),
andWeetal.(53)
have shown that ICP8 isoneofsevenviral geneproductsnecessaryandsufficientfor
origin-dependentDNAreplication. ICP8iscurrently thought tofunction atthereplication forkas aclassic helix-destabi-lizing protein based on its preferential and cooperative bindingtosingle-stranded DNA,its abilityto stimulate the viral DNApolymerase, andrequirement for its presence in the synthesisoflongDNAstrands (20, 23, 45). Bushetal. (6), de Bruyn Kops and Knipe (12), and Knipe and Spang (28) have shown that ICP8 is involved in the formation of protein complexesatprereplicative sites and isrequiredfor the correct intranuclear localization of the viral DNA poly-merase.
Severalstudies oftemperature-sensitive and deletion
mu-tantshave indicatedarole forICP8inregulationof
expres-sion ofviral genes. ICP8 appears to be involved in down-regulation of immediate-early and early gene products and up-regulationof late geneswhich require viralDNA synthe-sis forexpression
(16-19).
The mechanism ormechanisms involved in these processesare asyetunknown,andseveral, notnecessarily mutually exclusive, possibilities exist.These include interaction ofICP8 withinput parental viral DNA, interaction of the protein with mRNA, promotion of viral*Correspondingauthor.
DNAreplication,and interaction withspecificpromoterson progeny DNAmolecules.
AthirdpropertyofICP8, alluded to above, involves the ability of this polypeptide to interact with proteins. These include a varietyof interactions with the viral DNA poly-merase (6, 8, 49, 50), stable interactions with the viral alkaline exonuclease (48), as well as potential interactions between ICP8 and cellular proteins which make up the nuclear matrix. The nature and strengths of these interac-tions areunknown and are currentlythe focus of intensive investigation.
Anunderstanding of the various functions of ICP8 there-fore will require theidentification ofregions of the protein and, eventually, specific amino acidswhich areinvolved in the interactions described above. Considerable progress in this regard has been made since the publication of the nucleotide sequence of the ICP8 gene (42). Analysisof the predicted amino acid sequence of the protein coupled with extensivegenetic and biochemical investigation has led to theidentification of severalimportantdomains within ICP8. A nuclearlocalizationsignalliesatthecarboxyterminus and is required for correct transport of ICP8 to prereplication sites in the nucleus (15, 16). A zinc finger consensus se-quence lies within residues 499 to512. Site-specific muta-genesisofcysteineresidues 499 and 502 results ina nonfunc-tional protein, and recent work has shown that ICP8 containsatightlybound zincatom(14, 21). Finally,aregion absolutely required for theinteraction of ICP8with single-stranded DNA, and which contains a possible consensus DNA-binding site shared by other prokaryotic and viral DNA-binding proteins, has been delineated. Correlation of the results from several laboratoriesplacestheboundaries of this region between residues 564 and 849 of the predicted primary sequence (14, 15, 31), with the consensusbinding motif, identified by sequence analysis, encompassing resi-dues 803to849
(51).
Basic and aromatic amino acid side chains have been shown to be involved in the interaction of
prokaryotic
single-stranded DNA-binding proteins(SSBs)
with single-stranded DNA(2, 4, 9, 13, 26, 27,33).
Identification of these 6273on November 9, 2019 by guest
http://jvi.asm.org/
LYS,~
III ,liiI I II I I III , Io 900100011001196
100 l070ill 80l09001 0011
ARG r I" I I I
o 100 200 300 400 500 600 700 8009001000
I1100
1196TYR I .I .I .IL I.. I~L
0 100 200 300 400 500 600 700
+
TRPr, , , I 'il
0 100 200 300 400500 600700800 900 100011001196
| DNA BINING
REGION
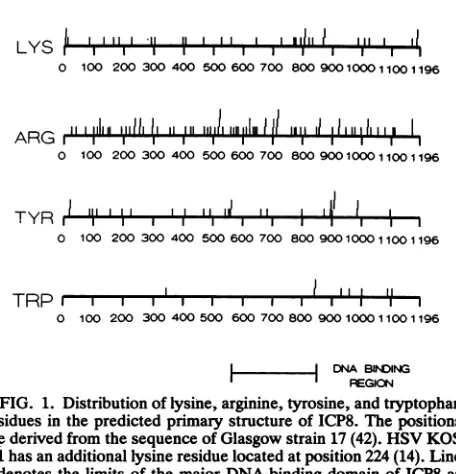
FIG. 1. Distributionof lysine, arginine,tyrosine,and tryptophan
residues inthepredicted primary structure of ICP8. The positions
arederived fromthesequenceofGlasgowstrain 17 (42). HSV KOS 1.1 hasanadditionallysineresiduelocatedatposition224(14). Line
5denotes thelimits of the major DNA-binding domain ofICP8as
defined by genetic and biochemical studies. This domain
encom-passesresidues 564to849.
residues resulted from avariety ofbiochemical and
physi-cochemicalapproaches aswell assite-specific mutagenesis.
Basedon sequence analysis, ICP8 contains 77 arginine, 33 lysine (34inHSVKOS 1.1), 27 tyrosine, and 8 tryptophan
residues (14, 42). As can be seen in Fig. 1, the arginine,
lysine, and tyrosine residues are distributed throughout the
predicted primary structure, whereas the tryptophan resi-dues, with one exception, are clustered in the
carboxy-terminal third of ICP8. Here, we report the results of
experiments designed to modify the side chains of these
amino acids in the ICP8 molecule and the effect of such
modifications on the ability of the protein to interact with
single-stranded DNA.
MATERIALS AND METHODS
Cells and viruses. Vero and U-35 cell lineswere
propa-gated in monolayer culture as described by Orberg and
Schaffer (38), using Dulbecco's modified Eagle's medium
supplementedwith 2% fetal calfserumand8% Serum Plus
(Hazleton Laboratories). Stocks of HSV-1 strain mP (24)
were propagated and titered on Vero cell monolayers as
previouslydescribed(46).
PurificationofICP8. ICP8waspurified from stably
trans-formed U-35 cells infected with HSV-1 strain mP at a
multiplicityofinfectionof 1.Phosphonoacetatewas present
at a concentration of 100 ,ug/ml tominimize productionof
late viral proteins. Purification of 10 to 20 mgof
homoge-neousICP8wasdoneaspreviously described (21,44). After
purification, ICP8wasdialyzed against buffercontaining 150 mM KCI, 10 mMTris-Cl (pH 7.6), 1 mM EDTA, 0.1 mM
dithiothreitol, 20 ,g of phenylmethylsulfonylfluorideperml,
and 50%glycerol (TEGDP) and storedat-20°Cbeforeuse.
Chemical modification of ICP8. Chemicalmodification of
ICP8was done by the protocols describedby
Bandyopad-hyay and Wu (4) in their analysis of the Escherichia coli SSB.
(i) Lysine modification. ICP8 (0.5 ml)ataconcentrationof
675 ,g/mlin150mMKCl-TEGDP-50% glycerolwasdiluted
with an
equal
volume of saturated sodium acetate in TEGDP. Aceticanhydride
(10
pl)
wasadded totheprotein,
and the resultingmixture wasincubatedonicefor30min.
A second10-p1
aliquot of aceticanhydridewasthenadded,
and the incubationwascontinued for anadditional 30min.
After the second incubation, the reaction mixture wasdialyzed
against 500 ml of TEGDPat4°C.
Asecondsample of ICP8at the same concentration was treated identically except for addition of the acetic anhydride. Binding assays, gel shifts, and sodium dodecyl sulfate-polyacrylamide gel electropho-retic(SDS-PAGE) analysiswere doneonboth the modified and control samples immediately upon removal from dialy-sis.(ii) Modification ofarginine by phenylglyoxal. A concen-trated solution of ICP8 was dialyzed against 50 mM
Tris-HCl
(pH
7.4)-150
mM KCl-20% glycerol (TGK),resulting
in afinal protein concentration of 422
p,g/ml
(3.3 x 106M).
Phenylglyoxal (10 mg/ml in TGK) was added in 300-fold molar excess to one-half of the protein, while an equal volume of TGK was added to the remainder, which acted as a control.Thereaction mixtures were incubated at25°C,
and aliquots were removed at 30, 60, 120, and 180min
and quenched on ice. Experiments were performed with the 180-min reaction products.(iii)Iodination oftyrosine. ICP8 (1 ml) at a concentration of 675,ug/ml or 1.1 mg/ml was dialyzed against 2 liters of 0.1M sodium borate (pH 9.5) for 2 h at
4°C.
The resulting 2.2 ml (307 or 500pg
of ICP8 per ml, respectively) was split into two 1.0-ml fractions. A 15- or 30-fold molar excess of K13 (0.05M 12 in 0.24 M KI) was added to one of the fractions, and an equal volume of deionized water was added to the control. Both mixtures were incubated on ice for 20min, and the reaction was stopped by the addition of 15pl
of 1 M NaHSO3 to each mixture. The individual reaction mixtures were then dialyzedovernight against TEGDP.Nitrocellulose filter binding assays. Modified and control protein solutions were diluted 1:10 with TEGDP. Increasing amounts of protein were then mixed with 16 ng of
3H-labelled heat-denatured bacteriophage lambda DNA (5.3 x 106 dpm/pg) in TE buffer (0.01 M Tris, 0.001 M EDTA[ph
7.61).
The final reaction volume was 100pl.
The mixtures were then incubated on ice for 10 min. After incubation,80-pl
aliquots were withdrawn and filtered throughnitrocel-lulose discs. Filtration, washing, drying, and scintillation counting were done as previously described (44 46).
Agarose gelshift assays. Various amounts of modified and control ICP8 were mixed with 300 or 500 ng of single-stranded M13 DNA (Boehringer) and incubated on ice for 30 min. The final volume was 25
pl,
and buffer and salt conditionswereidentical to those in a previous study by this laboratory (44). After incubation, the samples were applied to 0.5%horizontalagarose gels and electrophoresed at 60V for3 h. The gel was then stained with ethidium bromide in the presence of 1 M NaCl, and the DNA bands were visualizedwith short-wavelength UVlight.SDS-PAGE. Equalvolumes of both modified and control ICP8 were diluted 1:2 with SDS loading buffer, boiled for 2 min,and run onSDS-9.5% polyacrylamide gels. After elec-trophoresis, proteins were visualized by staining with Coomassie brilliant blue.
Protein quantification. Protein was quantified by using either a Bio-Rad protein assay kit or the method of Lowry (34a) after precipitation of protein with 5% trichloroacetic acid andredissolution in 1 N NaOH (21).
Fluorescence measurements. Fluorescence measurements and titration data were obtained with aPerkin-Elmer 650-40
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.68.296.67.304.2]spectrofluorimeter. Measurements were corrected for dilu-tion, and inner filter effects resulting from addition of single-stranded DNA wereminimal because of the low concentra-tions used. Excitiation and emission slit widths were set at 5 nm.
RESULTS
Allthe modificationreactions were done in aqueous media under mild conditionswhich were specific for the side chain inquestion. The molar ratio of reagent to amino acid was kept low to further ensure that the side chains being modified werethe most highly accessible to the reagents used. Thus, theconditionsused were designed to identify a minimum set of amino acids critical for the interaction of ICP8 with single-stranded DNA and which most likely lie on the surface of the protein or in hydrophilic clefts in its tertiary structure.
Reaction with acetic anhydride. ICP8 was modified as described above by reaction with acetic anhydride, which, under these conditions, results in selective acylation of the epsilon aminogroupoflysineresidues(4). The molar ratio of aceticanhydridetototallysine residueswas5/1. The ability ofthe modified ICP8 tointeract with single-strandedDNA was evaluated by a nitrocellulose filter binding assay with heat-denatured DNA and by agarose gel shift with single-stranded M13 DNA. Theuse of both of these assays was requiredtodeterminewhether themodifications were having an effect specifically on cooperative DNA binding. The agarose gel shift assay for SSBs was first described by Lohman et al.(34)and can be used to assesswhether binding is cooperative ornoncooperative. Cooperative binding re-sults in broaddistributionsatsubsaturating ratios(visualized assmears) indicativeof the nonrandom interaction of protein with nucleic acid. Noncooperative binding, on the other hand, results in the decreased mobility ofarelatively coher-entband of DNA. The nitrocellulose filterbinding assayis considerablymoresensitivethanthegelshift assay and thus canbe usedtodetect interactions which may be too weak to beobservedbygelshift.
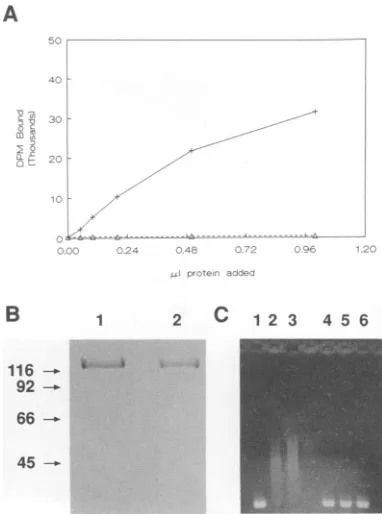
Modification experimentswere repeated three times, and typical results are presented in Fig. 2. Modification with aceticanhydride completely eliminatedtheability of ICP8 to interact with heat-denaturedornaturalsingle-strandedDNA infilterbindingandgelshift assays, respectively, compared with the controlsample (Fig.2AandC).The integrity of the proteinwas checked aftermodificationby SDS-gel electro-phoresis, andno difference between modified and unmodi-fied protein was observed (Fig. 2B). Thus, these results suggest that surface lysine residues are required for the interaction of ICP8 withsingle-stranded DNA.
Reaction with phenylglyoxal. ICP8 was subjected to reac-tion withphenylglyoxalasdescribed in Materials and Meth-ods. Theproteinwasmodifiedat amolar ratio of phenylgly-oxal to total arginine of 5/1. Derivitization of accessible arginineresidues didnotaffect themobilityof theproteinon SDS-polyacrylamide gels, nor were any differences,within experimental error, seen in filter binding experimentswith modified and controlproteins (Fig. 3A andB).Inthis case, agarose gel shifts were used to assesswhether the binding observed in the nitrocellulosefilter assayswas cooperative ornoncooperative.Ascanbeseenfrom the data inFig. 3C, both modified and control protein-DNA complexes were shifted up in broad, diffusebands, indicating that coopera-tivebindingwas not significantly affectedby phenyglyoxal modification.
A
B
1 2 C 1 2 3 4 5 6116- -
-
92-66
-45
-FIG. 2. Modification of ICP8 with aceticanhydride. (A)
Nitro-cellulose filter bindingassays with modified (A) and control (+)
protein. (B) SDS-PAGE of control (lane 1) and modified (lane 2)
ICP8. Numbers on leftshow size in kilodaltons. (C) Agarose gel
shiftassaysof ICP8binding activity withsingle-strandedM13 DNA.
Lanes 1 to3, 0, 5, and10plofcontrolprotein. Lanes4to6, 0, 5,and
10pAl of modified protein. The concentration of both protein samples
was-300 p.g/ml.
Quenching of tryptophan fluorescence. Tryptophan resi-dues have beenimplicatedin the interaction of both the T4 gene32proteinand E. coli SSBwithsingle-strandednucleic acids(13, 27). Numerous attemptstomodifysome orall of the eight tryptophan residues present in ICP8 by using N-bromosuccinimide(4) resultedincleavageof the polypep-tide backbone basedonSDS-PAGEanalysisof the modified protein (data not shown). As an alternative approach to investigating possible involvement oftryptophanresidues in the interaction of ICP8 with single-stranded DNA, we ex-amined the effect of the presence ofsingle-strandedDNAon the fluorescence spectrum of theprotein. Thefluorescence excitation andemission maxima of ICP8are280and 336 nm, respectively (22). These parameters indicate that the
fluo-rescence spectrumof ICP8 is derived from tryptophanand
thatthese tryptophanresidues arepresent in eithera more hydrophobic or heterogeneous environment than those in the E. coliSSBproteinorT4 gene 32product,both of which haveemission maxima at340to345 nm (13, 26, 33, 34).
Titrationof ICP8 withsingle-stranded
bacteriophage
M13 DNA resulted in a quenchingof theprotein's fluorescence intensity as shown in Fig. 4. The maximumquenching
observed inanumber ofexperimentsrepresented
adecrease of 35 to40% of the fluorescence intensityobserved in the absence of added DNA. These results suggest that tryp-tophan residues aredirectly
involved in the interaction of ICP8 withsingle-strandedDNA or,alternatively,
arepresent in a conformationally active region of theprotein
which modifies the local environment of thetryptophan
residues upon binding. It is possible that thequenching
seen is aon November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.347.538.78.335.2]A
B
1 2 C 1 23 4 5 6w;.;;^-Iw
92
-66
--45 -_
A
B
C
116
-4
92
-
66-45 -.
FIG. 3. Modification of ICP8 withphenylglyoxal. (A)
Nitrocel-lulose filter binding assays with modified (A) and control (+)
protein. (B) SDS-PAGE of control (lane 1) and modified (lane 2)
ICP8. Numbers on left show size inkilodaltons. (C) Agarose gel
shift assayswith 0.3 ,ug ofsingle-strandedM13 DNA. Lanes 1 to3,
0,5, and 10 ,ul of controlprotein. Lanes 4 to6,0, 5, and 10 p.1of
modifiedprotein. The concentrations of bothprotein sampleswere
-400p.g/ml.
combination of both of these phenomena since two of the tryptophanresidues lie within thecurrently mappedlimits of the single-stranded DNA-binding region and five of the remaining six are in the carboxy-terminal portion of the molecule, which isconformationallyactive upon interaction of ICP8 withsingle-strandedDNA (see Discussion).
Iodination of tyrosine residues. Tyrosine residues were modifiedby iodinationatmolarratios of 15/1 and30/1ofKI3
90
80
70[
60
0.00 0.42 0.83 1.25 1.67 2.08 2.50
MicrogramsM13 DNA
FIG. 4. Quenching ofintrinsic fluorescence of ICP8 by single-stranded DNA.ICP8 (1 ml)ataconcentration of 24,ug/mlin 150 mM
KCI-0.01 M Tris-HCI-0.001 M EDTA was titrated with
single-stranded DNAbytheaddition of 2-pJaliquotsofa70-p.g/ml solution
ofsingle-strandedM13 DNA. Excitationwasdone at280nm,and
emissionwas monitored at 336nm. The solution was allowed to
equilibratefor5min after addition of the aliquot before the
fluores-cenceemissionwasread. Allmeasurementsweredoneat25°C.
FIG. 5. Modification of ICP8by iodination. (A) Nitrocellulose
ifiterbinding assays with modified(A\)and control(+) proteins.(B)
SDS-PAGE of modified ICP8.Asinpreviousfigures,themodified
and control proteins showed identical migration rates (data not
shown). Numbersonleft show size in kilodaltons. (C)Agarosegel
shift assays of ICP8binding activity with 0.5 ,ug ofsingle-stranded
M13 DNA. Lanes1 to4,0, 2, 5, and 10,uJofcontrolprotein.Lanes
5to8, 0,2, 5, and 10 ,ul of modifiedprotein. The concentrations of
bothprotein sampleswere-800,ug/ml.
to ICP8. These ratios correspond to ratios ofKI3 to total tyrosines of 0.5/1 and 1/1, respectively. The same assays were performed on the iodinated and control proteins as were described above for ICP8 modified by reaction with aceticanhydrideandphenylglyoxal. Theresultspresentedin Fig.5arethose obtainedin the15/1 molarratioexperiments and show that iodination under these conditions did not result in cleavage ofthe protein. The filter binding assay results(Fig. 5A) indicate thatsomeresidualbinding remains aftermodification.This low level ofbindingwasconsistently observed andamounts to10to15% ofthebindingseenwith controlproteinbasedon acomparisonofpointstakenfrom the linear portions ofeach binding curve. Modification of ICP8 with a 30-fold molar ratio of K13 yielded identical results, includingthe levelsof residualbindingin the nitro-cellulose filter assay.
Fluorescencetitrationexperimentswith the modified ICP8 resultedinamaximumquenchingof8%comparedwith36% for the unmodified protein in the presence of increasing
amountsof DNA. These results suggest that 22to23%of the
protein retaineditsDNA-binding properties after modifica-tion, which is in reasonably good although not complete agreementwith the filterbindingdata.
Agarosegel assayswith concentrations ofprotein similar to those used in the acetic anhydride and phenylglyoxal experiments
(300
to400,ug/ml)showednoclear evidence of a shift resultingfrom eithercooperative ornoncooperative.L .7
2
c
0
,, p
0
ae
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.79.271.74.327.2] [image:4.612.327.543.76.348.2] [image:4.612.80.275.524.650.2]bindingin themodified proteins (datanotshown). However, using concentrations of -800,ugofICP8 per ml and running thegels overnight resulted in shifts of the DNA in the lanes containing modified protein (Fig. 5C). The shifts observed were considerably smaller than those seen with equivalent amounts of the control protein as would be expected if a substantial fraction of the protein was inactivated. However, the DNA bands were also less diffuse, suggesting that the cooperativityof binding was also being affected (see Discus-sion). Thus, iodination significantly affects the ability of ICP8 to bind to single-stranded DNA, indicating that ty-rosine residuesarerequired for the interaction.
DISCUSSION
We found that lysine and tyrosine residues accessible to modification are required for the interaction of ICP8 with single-stranded DNA and that tryptophan fluorescence is quenched uponbinding. Positively charged lysine and argi-nine side chains have been implicated in a number of protein-single-stranded nucleic acid interactions and are believed to be primarilyinvolved incharge-charge interac-tions with the phosphate backbone, although theyare also capable of participating in hydrogen bonding (9, 26, 33). Thus, it is not surprising that modification of lysine side chains results inaloss ofDNA-binding activity.
The lack of any observed diminution in binding activity resulting from arginine modification is rather unexpected basedonthe above arguments. These results could be dueto argininesnotbeinginvolved in the interactionorbecause the arginines essential for interaction are not accessible under our experimental conditions. Such inaccessibility could be due to either a local hydrophobic environment for critical arginines which precludes reaction under aqueous condi-tions or the bulky nature of the diphenylglyoxal adduct, which may not be compatible with the three-dimensional location of suchside chains.Thus,thestudiesreportedhere do not necessarily eliminate the
possibility
thatarginine
residuesarerequired and/or directlyinvolved in the interac-tion of ICP8 withsingle-strandedDNA.Aromatictyrosineandtryptophanresiduesareinvolved in theinteractionof the E. coliSSBandthe T4 gene32
protein
(2,4, 13, 27,47). Ithas beensuggestedthattheseresidues, inaregular array,formhydrophobic pockets into whichthe heterocyclic rings of the DNA bind(26).
Two tryptophan residues(Trp-844
andTrp-846)
arepresentwithin the puta-tive single-strandedDNA-bindingmotifidentified by Wang and Hall (51), and one of them(Trp-846) occupies
acon-servedpositionin thepredicted primary sequenceofICP8, the varicella-zoster virus ORF29 product, the
Epstein-Barr
virus BALF 2product,and thesimian Colbum cytomegalo-virus DB129protein(1, 3, 11, 14, 42). Thequenchingof the tryptophanfluorescence observed in thisstudy
isconsistent with thepossibility thatafraction of thetryptophanresidues undergo a change in environment upon the interaction of ICP8withsingle-stranded
DNA,eitherthrough
direct inter-action with hydrophobic bases or through conformational changes in the protein induced uponbinding.
Theprecise
nature of the effect and the identification of
specific
tryp-tophans involved mustawait a combination of
site-specific
mutagenesis in which each of thetryptophan
residues is modifiedindividually aswellasfurtherchemical and physi-cochemicalmapping
studies.Corroborating
evidence for aconformational change in ICP8 upon interaction of the protein with single-stranded DNA comes from
continuing
work inourlaboratorywhich has shown that thesignal
fromcysteine side chains, labeled with fluorescent maleimide derivatives, and mappingin the carboxy-half of ICP8, un-dergoes quenching in the presence of single-stranded DNA (41).
This study showed the interaction of ICP8 to be highly sensitivetoreaction ofthe protein withKI3,which leads to the iodination of the phenolic ring in tyrosine. The results reported are intriguing with regard to several points. First, the ratio of
KI3/ICP8
represents -0.5 to 1.0 mol ofK13per moloftyrosine.This suggests that the tyrosine(s) modified is highly accessibleto the aqueous environment. Second, no tyrosinesarepresent intheconsensusbinding site identified byWang and Hall(51), the proposed aromatic array being made up of phenylalanine and tryptophan residues. Five tyrosine residues, however,arepresent in themajor DNA-binding domainencompassingresidues 564 to 849, and one (Tyr-801)occurstwoamino acidsbeforethe Wang and Hall (51) binding motif.Finally, while iodination appearedtosignificantly diminish binding, incontrast tothe results obtained for lysine modi-fication, residual signalswere seenin thefilterbinding and fluorescencetitrationassays. The mostdirectinterpretation of these results would be that 10 to 20% of the ICP8 remained unmodified, while the remainderwas completely inactivated.Thegel shiftresults shown inFig.SC, however, indicate that the situation is morecomplex. Assuming that -20%of theICP8remainedunmodified,lanes 2 and 8 ofFig. SCshould bedirectly comparable with regardtotheamount of active ICP8 present and the DNA shifts should be identical. This is not the case. The DNAband in lane 8 is shiftedhigherthan the DNAband in lane 2. Moreover,the DNA band in lane 8 is more coherent and shows less smearing than the bandin lane2,
indicating
that thebinding
of the iodinated protein is less cooperative than that ob-served inthe control lanes.Similarly,lesscooperativeshifts arealso seen in lanes 6 and 7. Whilewe cannotruleoutthepossibility
thatafraction of the ICP8 losesallDNA-binding
activityuponiodination,these results suggest that the roleof the modified tyrosine residues involves thecooperative
as well as the intrinsic or noncooperative interaction ofICP8 with single-strandedDNA.In summary, the work
presented
above resulted in the identification of several amino acid side chains which are required for, or affected by, the interaction of ICP8 with single-stranded DNA.The methodutilized, chemical modi-fication of the nativepolypeptide,
hasproved
useful in similar studieswith otherSSBs,
and the information gener-ated should allow a more logical and confident choice of targets forsite-specific mutagenesis
studies aimed at fine mapping criticalregions and residues of theprotein.
Since theputativeDNA-binding
regionencompassing
residues564 to849contains ninelysine,
fivetyrosine,
andtwotryptophan
residues, it seems reasonable to propose thesespecific
amino acidsasthe initial targets of such ananalysis.
ACKNOWLEDGMENTS
This workwassupported byPublic Health Service grantAI22468 fromthe NationalInstituteofAllergyand Infectious Diseases.
The assistance of Helen Ling and Kathy Dudas is gratefully acknowledged.
REFERENCES
1. Anders,D.G.1990. Nucleotide sequence ofacytomegalovirus
single-stranded DNA-binding protein gene: comparison with
alpha- andgammaherpesvirus counterparts revealsconserved
segments. J.Gen. Virol. 71:2451-2456.
on November 9, 2019 by guest
http://jvi.asm.org/
2. Anderson, R. A., and J. E. Coleman. 1975. Physicochemical
properties ofDNAbinding proteins:gene32protein ofT4and
Escherichia coliunwinding protein.Biochemistry 14:5485-5491.
3. Baer, R.,A. T.Bankier, M. D. Biggin,P. L. Deininger, P.J.
Farrel,T.J.Gibson, G. Hatfull,G. S.Hudson,S. C.Satchwell,
C.Seguin,P.S.Tuffnell,and B.G.Barrel.1984.DNA sequence
andexpression of the B95-8 Epstein-Barr virusgenome.Nature
(London)310:207-211.
4. Bandyopadhyay,P.K., andC.-W.Wu.1978. Fluorescenceand
chemical studies on the interaction of Escherichia coli
DNA-binding protein with single-stranded DNA. Biochemistry 17:
4078-4075.
5. Bayliss,G.J.,H.S.Marsden,andJ. Hay. 1975.Herpes simplex
virusproteins: DNA-binding proteinsin infected cells and in the
virusstructure. Virology68:124-134.
6. Bush, M., D. R. Yager, M.Gao, K.Weisshart, A. J. Marcy,
D. M. Coen, and D. M. Knipe. 1991. Correct intranuclear
localization ofherpes simplex DNA polymerase requires the
viral ICP8DNA-binding protein.J.Virol. 65:1082-1089.
7. Challberg,M. D. 1986. Amethodforidentifying the viralgenes
required for herpesvirus DNA replication. Proc. Natl. Acad.
Sci. USA 83:9094-9098.
8. Chiou,H.C.,S. K.Weller,and D. M.Cohen. 1985. Mutations
in the herpes simplex virus major DNA-binding proteingene
leading to altered sensitivity to DNA polymerase inhibitors.
Virology145:213-226.
9. Coleman, J. E., and J. L. Oakley. 1980. Physical chemical
studies of the structure and function of DNA binding (helix
destabilizing) proteins. Crit.Rev. Biochem. 7:247-289.
10. Conley, A. J.,D. M.Knipe,P.C.Jones,and B. Roizman.1981.
Moleculargeneticsofherpes simplexvirus.VII.
Characteriza-tion of a temperature-sensitive mutant produced by in vitro
mutagenesis and defectivein DNAsynthesis and accumulation
ofpolypeptides.J. Virol.37:413-428.
11. Davison, A. J., and J. E. Scott. 1986. The complete DNA
sequenceof varicella-zoster virus. J. Gen. Virol. 67:1759-1816.
12. deBruyn Kops,A.,and D. M.Knipe.1988.FormationofDNA
replication structures in herpes virus-infected cells requires a
viralDNAbinding protein. Cell 55:857-868.
13. Doan, L. T., J.-J. Toulme, and C. Helene. 1984.Involvement of tryptophylresiduesinthebinding of model peptides andgene32
protein from phageT4tosingle-strandedDNA. Aspectroscopic
method for detection oftryptophaninthevicinity of nucleic acid
bases.Biochemistry23:1202-1210.
14. Gao, M., J. Bouchey,K.Curtin,and D. M.Knipe.1988.Genetic
identification of a portion of the herpes simplex virus ICP8
protein requiredforDNA-binding. Virology163:319-329.
15. Gao, M.,andD. M.Knipe.1989.Genetic evidence formultiple
nuclear functions of the herpes simplex virus ICP8
DNA-binding protein.J. Virol.63:5258-5267.
16. Gao, M., and D. M. Knipe. 1991. Potential role for herpes
simplexvirus ICP8replicationproteininstimulation of lategene
expression.J.Virol. 64:2666-2675.
17. Godowski,P.J.,and D. M.Knipe.1983. Mutations in themajor
DNA-binding proteingeneofherpessimplexvirustype 1result
in increased levelsof viral gene expression.J. Virol. 47:478-486.
18. Godowski, P. J., and D. M. Knipe. 1985. Identification ofa
herpes simplexvirus function that represses late gene
expres-sion fromparentalviral genomes. J. Virol.55:357-365.
19. Godowski,P.J.,and D. M.Knipe.1986.Transcriptional control
of herpesvirus gene expression: gene functions required for
positive and negative regulation. Proc. Natl. Acad. Sci. USA
83:256-260.
20. Gottlieb, J., A.I. Marcy, D. M. Coen,and M. D. Challberg.
1990. Theherpes simplexvirus UL42 geneproduct:asubunitof
DNA polymerase that functions to increase processivity. J.
Virol. 64:5976-5987.
21. Gupte,S.S.,J. W.Olson,and W. T.Ruyechan. 1991. The major
herpes simplex virus type-1 DNA-binding protein is a zinc
metalloprotein.J.Biol. Chem.266:11413-11416.
22. Gupte, S.S.,and W. T.Ruyechan. Unpublisheddata.
23. Hernandez,T.R., andI. R. Lehman. 1990.Functional
interac-tion between the herpessimplex-1DNApolymerase and UL42
protein. J. Biol. Chem. 265:11227-11232.
24. Hoggan, M. D., and B. Roizman. 1959. The isolation and
propertiesofavariant ofherpes simplex producing
multinucle-atedgiant cells inmonolayer culturesinthe presenceof
anti-body.Am.J. Hyg. 70:208-219.
25. Honess, R. W., and B. Roizman.1974. Regulation of herpesvirus
macromolecularsynthesis. I.Cascaderegulation of the
synthe-sis of threegroupsof viralproteins.J.Virol. 14:8-19.
26. Karpel, R. L. 1990. T4 bacteriophage gene 32 protein, p. 103-130. In A.Revzin(ed.),The biologyof non-specific
DNA-protein interactions. CRC Press,Inc., BocaRaton,Fla.
27. Khamis, M. I., J. R. Casas-Finet, A. H. Maki, J. B. Murphy, and J. W. Chase. 1987. Investigation of the role of individual
tryptophan residues in the binding ofE. coli single-stranded
binding protein to single-stranded polynucleotides. J. Biol.
Chem. 262:10938-10945.
28. Knipe, D. M., and A. E. Spang. 1982. Definition of a series of stagesin the association oftwoherpesviral proteins with the cell
nucleus. J.Virol. 43:314-324.
29. Leinbach, S. S., and J. F. Casto. 1983. Identification and
characterization of deoxyribonucleoprotein complex containing
themajor DNA-binding protein of herpes simplexvirus type 1.
Virology 131:274-286.
30. Leinbach,S.S., J. F. Casto, and T. K. Pickett.1984.
Deoxyri-bonucleoprotein complexes andDNAsynthesis of herpes
sim-plex virustype 1.Virology 137:287-296.
31. Leinbach, S. S., and L. S. Heath. 1988. A carboxy-terminal
peptide of the DNA-binding protein ICP8 of herpes simplex
virus contains a single-stranded DNA-binding site. Virology
166:10-16.
32. Littler, D., D. Purifoy, A. Minson, and K. L. Powell. 1983.
Herpessimplex virus non-structural proteins. III. Function of
themajor DNA-binding protein.J. Gen. Virol. 64:983-995.
33. Lohman, T. M., and W. Bujalowski. 1990. Escherichia coli
single-strand binding protein: multiple single-stranded DNA
binding modes and cooperativities, p. 131-170.In A. Revzin
(ed.), The biology of nonspecific DNA-protein interactions.
CRCPress, Inc.,BocaRaton, Fla.
34. Lohman, T. M., L. B. Overman, and S. Datta. 1986. Salt
dependent changes in the DNAbinding cooperativity of the
Escherichia coli single strand binding protein. J. Mol. Biol.
187:603-615.
34a.Lowry,0.H., N. J. Rosebrough, A. L. Farr, and R. J. Randall.
1951. Protein measurementwith the Folin phenol reagent. J. Biol. Chem. 193:265-275.
35. McGeoch, D.J.,M. A. Dalrymple,A. J.Davidson,A. Dolan,
M.C.Frame, D. McNab, L. J. Perry, J. E.Scott,and P.Taylor.
1988. ThecompleteDNA sequenceof thelong unique regionin
herpes simplexvirus type 1. J.Gen.Virol.69:1531-1574.
36. O'Donnel, M. E., P. Elias, B. E. Funnel, and I. R. Lehman. 1987.
Interactionbetween the DNApolymerase andsingle-stranded
DNA-binding protein (infected cell protein8) of herpes simplex
virus1. J.Biol.Chem.262:4260-4266.
37. Olivo,P.D.,N.J. Nelson,and M. D.Challberg. 1989.Herpes
simplexvirus type 1 geneproducts requiredfor DNA
replica-tion:identification andoverexpression. J.Virol. 63:196-204.
38. Orberg, P. K., and P. A. Schaffer. 1987. Expression of the
simplexvirus type 1major DNA-binding proteinICP8 in
trans-formed cell lines: complementation of deletion mutants and
inhibition ofwild-typevirus. J. Virol. 61:1136-1146.
39. Powell, K. L., E.Littler,and D.J.M.Purifoy.1981.
Nonstruc-turalproteins of herpes simplexvirus. II.Majorvirus-specific
DNA-binding protein.J. Virol.39:894-902.
40. Powell,K.L.,and D.J.M.Purifoy.1977.DNAbinding proteins
of cells infectedbyHSV-1 and HSV-2.Intervirology7:225-239.
41. Pujara, C. P., S. S. Gupte, J. W. Olson, and W. T. Ruyechan.
Unpublished data.
42. Quinn,J. P., and D. J. McGeoch. 1985. DNA sequence in the
genomeofherpes simplexvirus type 1containingthe genes for
DNApolymeraseand themajor DNA-binding protein.Nucleic
AcidsRes. 13:8143-8163.
43. Ruyechan, W. T. 1983. Themajor herpes simplexvirus
on November 9, 2019 by guest
http://jvi.asm.org/
binding protein holds single-stranded DNA in an extended
configuration.J. Virol. 46:661-666.
44. Ruyechan, W. T. 1988. N-Ethlymaleimide inhibition of the
DNA-binding activityofthe herpes simplexvirustype1major
DNA-binding protein. J. Virol. 62:810-817.
45. Ruyechan, W. T., A.Chytil,and C.M.Fisher. 1986. Invitro
characterization ofa thermolabile herpes simplex virus
DNA-binding protein. J. Virol. 59:31-36.
46. Ruyechan,W.T.,and A.C.Weir.1984.Interaction withnucleic
acids and stimulation of the viral DNA polymerase by the
herpes simplex virus type 1 major DNA-binding protein. J.
Virol. 52:727-733.
47. Shamoo, Y.,W.J. Roberts, J.E.Coleman,K. R.Williams,and
W. H.Konigsberg. 1987. Site specific mutagenesisof T4gene
32: the role of tyrosine residuesinprotein-nucleicacid
interac-tions, p. 385-397. In D. L. Oxebder (ed.), Protein structure,
folding,anddesign2. AlanR.Liss, Inc.,New York.
48. Thomas, M.S.,M.Gao,D. M.Knipe, and K. L. Powell. 1992.
Association between the herpes simplex major DNA-binding
protein and alkaline nuclease. J. Virol. 66:1152-1161.
49. Vaughn, P.J. L., L. M.Banks,D. J. M.Purifoy, and K. L.
Powell.1984. Interactions betweenherpes simplex virus
DNA-binding proteins. J. Gen. Virol. 65:2033-2041.
50. Vaughn, P. J., D. J. M. Purifoy, and K L. Powell. 1985.
DNA-binding protein associated with herpes simplex virus
DNApolymerase.J. Virol. 53:501-508.
51. Wang, Y., and J. Hall. 1990. Characterization of a major
DNA-bindingdomain in theherpes simplexvirustype1 DNA-bindingprotein(ICP8).J. Virol.64:2082-2089.
52. Weller, S. K., K. J. Lee, D.J. Sabourin,and P. A. Schaffer.
1983. Geneticanalysis of temperature-sensitive mutantswhich
define the gene for the major herpes simplex virus type 1
DNA-binding protein. J. Virol. 45:354-366.
53. Wu,C.A.,N.J. Nelson, D.J. McGeoch,and M. D.Challberg.
1988. Identification ofherpes simplex virus type 1 genes
re-quired for origin-dependent synthesis. J. Virol. 62:435-443.