CopyrightX 1991,American Society forMicrobiology
In
Vitro mRNA Degradation System To Study
the
Virion
Host
Shutoff
Function of Herpes Simplex Virus
CHARLES R.
KRIKORIAN'
ANDG. SULLIVAN READ2*DepartmentofMicrobiology andProgram in Molecular Biology, Stritch School of Medicine, Loyola Universityof
Chicago, Maywood, Illinois
60153,1
and Divisionof Cell Biology and Biophysics, School of BasicLife Sciences, University of Missouri-Kansas City, KansasCity, Missouri 64110_24992Received 15May 1990/Accepted 25 September 1990
The virion host shutoff (vhs) gene of herpes simplex virus encodes a virion polypeptide that induces degradation of host mRNAs at early times and rapidturnover of viralmRNAsthroughout infection. Tobetter
investigate the vhs function, an in vitro mRNA degradation system was developed, consisting of cytoplasmic
extracts
fromHeLacells infected with wild-type herpes simplex virus type 1ora mutantencodingadefective vhspolypeptide. Host and viral mRNAs were degraded rapidlyinextracts from cellsproductively infectedwithwild-type
herpes simplex virus type 1 but notinextracts from mock-infected cells orcells infected with themutant vhsl. Incontrast, 28S rRNA was stable in all three kinds ofextract. Accelerated turnover ofhost
mRNAs was alsoobserved in extracts from cells infected with wild-type virusinthepresenceofdactinomycin, indicating that the activity was induced by a structural componentofthe infecting virions. The in vitro vhs
activity was inactivated by heat orproteinaseKdigestion butwas insensitive to brief treatmentoftheextracts
withmicrococcal nuclease.Itwas notinhibitedbyplacental RNase inhibitor,it exhibited astrongdependence
upon addedMg2+,it wasactive at concentrations ofK+ up to200mM,and itdid notrequire the components of anenergy-generatingsystem. Insummary, the in vitro mRNA degradation systemappearstoaccurately reproduce the vhs-mediated decay of host and viralmRNAsandshould be useful forstudiesof themechanism
of vhs action.
In cells infected with herpes simplex virus (HSV), the
shutoff ofhost macromolecular synthesis and the cascade
regulation of viral gene expression are achieved through a
complex set oftranscriptional and posttranscriptional
con-trols (26, 45, 59, 66, 74, 76, 77). Ofthe posttranscriptional
regulatorymechanisms,the most thoroughly studied to date has been theregulationof host and viral mRNAstabilitiesby theproduct ofthe HSV virion hostshutoff(vhs) gene. The
vhs gene encodes apolypeptidethatis astructural compo-nentof virionsand atearly times afterinfectioncauses the
shutoff of most host cell protein synthesis by inducing degradation of cellularmRNAs(16-22,28, 42, 43, 55, 58, 66, 69, 71,72). Since copies ofthe vhsproteinarepresent within the infecting virion, virion host shutoff is not dependent uponpriorde novoviral protein synthesis (16-22, 42, 43, 55,
66, 69). The activity of the vhs protein is not limited, however, topreexistingcellular mRNAs. After the onset of
viraltranscription, the vhs protein induces rapidturnoverof
viralmRNAsbelonging to all kinetic classes (32, 44, 45, 69).
Thus, virus mutants that encode a defective vhs protein are
defective in virion host shutoff and produce viral mRNAs with significantly longer half-lives than those of mRNAs produced by the wild-type virus (32, 44, 45, 69). The vhs
polypeptide, therefore, appears to provide a nonselective mRNA degradation function that, when coupled with
spe-cific transcriptional controls, plays an important role in
regulatingthe steady-state levels and patterns of
accumula-tion ofboth viral and cellular mRNAs (32, 44, 45).
An ever-increasing body of data has made it clear that controlofmRNA stability plays an important role in
regu-lating the expression of many mammalian genes (5, 60).
Exposure of cellsto avariety of hormones and other stimuli
*Correspondingauthor.
increases the levels ofspecific mRNAs, at least in part,by increasing their cytoplasmic half-lives. Glucocorticoids
en-hance the stability of human growth hormone mRNA (46),
whereas estrogen specifically stabilizes vitellogenin mRNA
(8)andprolactininduces anincreasein thehalf-life ofcasein
message(25).Conversely,stimuli thatincreasethe cytoplas-mic concentration of iron destabilize transferrin receptor mRNA, apparently by reducing the affinity of a specific
mRNA binding protein for cis-acting sequences located
within the 3' untranslatedregion ofthe message(40,41,64).
Thestabilityof
3-tubulin
mRNAisregulated bythe concen-tration ofunpolymerizedP-tubulin
in the cytoplasm. Thus,treatment of cells with cytochalasin B, which disrupts mi-crotubules and raises the concentration of
unpolymerized
tubulins,induces rapiddegradation of
,3-tubulin
mRNA(23,78). Similarly, in vivo and in vitro studies suggest that an
increase in the level of freehistones,whichaccompaniesthe
cessation of DNA synthesis at the end of the S phase,
induces rapiddestabilization ofhistonemRNAs(24, 27, 38,
47,51, 52, 61-63).Finally,the presenceofspecificcis-acting
sequences within the 3', and sometimes 5', untranslated regions ofthe mRNAs encoding a number oflymphokines
andproto-oncogene products causes these messages to be very short lived (11,29, 53,54, 67, 68, 75).Abrogationof the normal controlofthestabilities of these mRNAscanleadto
celltransformation, altered growthproperties, or abnormal
differentiation (1, 10, 12, 13, 15, 36, 37, 50, 67).
A number of studies suggest that the degradation of mRNAs
normally
occurs by 3'-to-5' exonuclease digestion.Studies from J. Ross's laboratory with extracts from unin-fected K562 cells have shown that the in vitrodegradationof c-myc mRNAinvolvesprogressive shorteningof thepoly(A) tail, followed by 3'-to-5' degradation of the body of the message(6,7). That this mechanismisgenerally followedfor
112
on November 10, 2019 by guest
http://jvi.asm.org/
the decay of polyadenylated messages in vivo is suggested by the finding of Wilson and Treisman that sequences located in the 3'
untranslated
region of c-fos mRNA, which accelerate its rate ofturnover,
also cause more rapid short-ening of the poly(A) tails of c-fos messages (75). 3'-to-5'degradation also occurs for nonpolyadenylated messages. Ross and co-workers have shown, both in vitro and in
vivo,
that the decay of nonpolyadenylated histone mRNAs pro-ceeds by cleavage of the last 4 to 13 nucleotides from the 3' end of the mRNA, followed by rapid3'-to-5' degradation of the remainder of the message (51, 52, 61-63).At present, only a limited amount has been reported
concerning
the development of in vitro mRNA degradation systems. The most extensive studies to date have come from Ross's laboratory involving extracts from uninfected K562 erythroleukemia cells (3, 4, 6, 7, 51, 52, 60-63). In this system, in vitro degradation of histone mRNAs is mediated by a polysome-bound exonuclease that can be isolated in an active, soluble form by washing the polysomes with buffers containing 0.3 M KCl (61, 62). Although the RNase that degrades histone messages is normally polysome associated, in vitro regulation of the rate of histone mRNA decay by the concentration of free histones also requires a factor that is present in the S130 high-speed supernatant obtained after pelleting the polysomes from a cytoplasmic extract (51). Similar results have been obtained for the decay of some polyadenylated messages. In vitro poly(A) shortening and3'-to-5'
degradation of c-myc message can be observed upon incubation of isolated polysomes, indicating that the relevant nucleases are polysome associated (6). However, c-myc decay is greatly accelerated by a labile factor that is found in theS130postpolysomal supernatant fraction from cytoplas-mic extracts (7). This factor specifically accelerates the decay rates of c-myc and c-myb mRNAs and is inactivated by treatment with micrococcal nuclease, suggesting that it may be a ribonucleoprotein (7). However, in spite of this impressive progress, as yet little is known concerning the identity of the nucleases involved in mRNAturnover and the factors that control their activities.The vhs-mediated control of mRNA stabilities in HSV-infected cells provides a particularly attractive model system for studying the factors that control message half-lives in mammalian cells. In particular, the vhs protein is one of the few trans-acting regulators of mRNA stability that have been identified to date. In addition, mutants encoding defective vhs proteins have been isolated and characterized (17, 55), allowing a combination of genetic and biochemical tech-niques to be applied to the problem. Mapping of the mutation carried by the mutant vhsl to the UL41 open reading frame of HSV type 1 (HSV-1) has allowed identification of the vhs gene (34, 39), and recent characterization of the structural polypeptides encoded by a vhs deletion mutant has allowed identification of the vhs polypeptide within purified virions (55a). Clearly, elucidation of the mechanism of vhs-mediated control of mRNA half-lives would be greatly facilitated by the availability of an in vitro mRNA degradation system that accurately reflects the vhs activity observed in vivo. In this paper we report the development and preliminary character-ization of such a system from HSV-1-infected HeLa cells.
MATERIALS AND METHODS
Cells and virus. HeLa S3 and Vero cells were purchased from the American Type Culture Collection and grown at
37°C
in Eagle minimum essential medium (MEM; GIBCO) supplemented with antibiotics and 10% (vol/vol) calf serum(45). Stocks of wild-typeHSV-1 strainKOSand the mutant vhsl were prepared by infectionofVero cellmonolayers as previously described (45). vhsl grows well at all
tempera-tures from 34 to 39°C and exhibits adefective virion host shutoff function at alltemperatures(55). In these
studies,
all infections wereperformedat34°C.HeLa S3 cellswereusedfor the preparation of all invitro mRNAdegradationextracts and were infected and maintained as described
previously
forVerocells (31, 45). Inallexperiments, virus wasallowed to adsorb for 1 hin MEMcontaining 5%(vol/vol) calfserum. The inocula werethenaspirated,and thecellswereoverlaid
with fresh MEM plus 2% calfserum. Mock-infected cells
were treated inthe same way asinfected cells, except that they were exposed to lysates of uninfected Vero cells prepared in the samewayasthe virus stocks were
prepared
from infected cells.Plasmids. The plasmid pHcGAP contains a1.2-kb cDNA
insert encoding a portion ofhuman
glyceraldehyde-3-phos-phate dehydrogenase (GAPD) (73) and was obtained from
the American Type Culture Collection. The
plasmid,
pHSV106, whichcontains a 3.4-kb BamHIfragment
encod-ing the HSV-1 thymidine kinase, was purchased from Be-thesdaResearchLaboratories. Theplasmid pXlrll containsa 4.6-kb fragmentofXenopus laevis rDNAinsertedintothe EcoRI site of colicin El (14) and was provided by Jeff
Doering. The inserted fragment in pXlrll hybridizes with
28S rRNA. pXlrll was maintained in Escherichia coli
HB101, whereas pHcGAPandpHSV106weremaintained in E. coli DH5 alpha (Bethesda Research
Laboratories).
Plas-midDNAs were prepared by CsCl density gradientcentrif-ugation asdescribed previously (45, 56, 57).
In vitro mRNAdegradation extracts. Cytoplasmic extracts
for studyinginvitromRNAdegradation were
prepared
from infectedormock-infected HeLa S3 cellsbyamodification oftheprocedureofBrown andcolleagues forthe
preparation
ofin vitro translation extracts (9). At various times after
infection ormock infection, HeLacells werewashed twice with ice-cold wash buffer consisting of0.15 M sucrose, 33 mM NH4Cl, 7 mM KCl, 4.5 mM magnesium acetate
[Mg(OAc)2], and 30mM HEPES
(N-2-hydroxyethylpipera-zine-N'-2-ethanesulfonic
acid) (pH7.4). Thecellswere thenpermeabilized by the addition of 300
jxg
oflysolecithin
(L-a-lysophosphatidyl
choline; Sigma)per mlin wash bufferdirectly tothe monolayers for 60 s. After the
permeabiliza-tion buffer was removed, the cells from one 100-mm dishwere scraped into 200,ul ofstandard reaction buffer
[0.1
M HEPES (pH 7.4), 0.2 M NH4Cl, 20 mMMg(OAc)2,
7 mMKCl, 1 mM dithiothreitol, 1 mM ATP
(dipotassium
salt),
1 mM GTP (sodium salt), 40 ,uM eachofthe 20 aminoacids,
0.1 mMS-adenosylmethionine,
1 mMspermidine,
10 mM creatine phosphate (dipotassium salt), 40 U of creatinekinase per ml, and 100 U of placental RNase inhibitor (RNasin; Promega Corp, Madison,
Wis.)].
The cells werethen disrupted by 10 passages through a
25-gauge
needle.The nuclei were removed by low-speed
centrifugation,
and the supernatant was stored on ice.To initiate mRNA degradation reactions,
cytoplasmic
supernatants were transferred to a 30°C water bath. At
various times after the start of incubation,
samples
were removed, addedto an equal volume ofureabuffer(7
Murea, 10 mM Tris hydrochloride [pH 7.9], 0.35 MNaCl,
10 mMEDTA, and 1% sodium dodecyl
sulfate),
extracted twicewith phenol-chloroform (1:1,
vol/vol)
andtwicewith chloro-form, and precipitated fromethanol,
all as describedprevi-ously (45, 56, 57). Samples taken
immediately
before trans-ferring the reaction mixtures from4°C
to30°C
served as0-hon November 10, 2019 by guest
http://jvi.asm.org/
time points. The sampleswere subsequently analyzed forin
vitro decay of host and viral mRNAs and 28S rRNA by
Northern RNA blotting and hybridization as described
be-low.
In experimentsto optimize the concentrationof
Mg2+
ion needed for efficient in vitro mRNA degradation, extractswere prepared in the standard reaction buffer modified to
contain 2.5 mM Mg(OAc)2 and then supplemented with concentrated Mg(OAc)2 tobring the
Mg2+
concentration tothe desired values. To optimize the
K+
ion concentration,standard extracts were prepared containing 7 mM KCl and
then supplemented with concentrated KCl to bring the
K+
concentration tothe values described in the figure legends. To testthe requirement for energy-generating components,extracts were prepared in standard reaction buffer lacking
ATP, GTP, creatine phosphate, and creatine kinase. The extractswereall incubatedand analyzedasdescribedabove.
Pretreatment ofextracts with heat,proteinase K,or
micro-coccal nuclease.Totestthesensitivity of theextracts tobrief heattreatment, standard in vitrodegradation reactions were
heated at90°Cfor 10 min, cooled to
4°C,
and then analyzedfor in vitro decay of mRNAs according to the standard
protocol. To test the sensitivity ofthe extracts to
pretreat-ment with protease, standard extracts were supplemented
with proteinase K (Sigma; molecular biology grade) to a
concentration of1 mg/ml and incubated at
30°C
for 30 min.Theextractswerecooledbrieflyto
4°C
and thenincubatedat30°C andanalyzed forin vitromRNAdegradation according
to the standard protocol.
Totest the effect of pretreating the extracts with
micro-coccal nuclease, standardin vitro degradationextracts were
supplemented with micrococcal nuclease (Pharmacia) to
1,000 U/mlandCaCl2to 1 mMand thenincubatedat
30°C
for 10min.Ethyleneglycol-bis(,3-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA) was added to 2 mM, and the extracts were chilled on ice for 10 min. To each reaction
mixture was added deproteinized total cytoplasmic RNA
fromanequivalentnumber of uninfected HeLa cells, and the
mixtures were incubated at 30°C. Samples were taken at
various times and analyzed by Northern blotting for the in
vitro decay ofhost mRNAs and 28S rRNA.
Agarosegel electrophoresis, Northern blotting, and
hybrid-ization. RNA samplesweredenaturedwith glyoxal,
electro-phoresed through 1% agarose gels cast in 10 mM sodium
phosphate (pH 7.0), and transferred to Nytran membranes
(Schleicher and Schuell) by capillary blotting, all as
de-scribed previously (45). Filters containing immobilized
RNAswereprehybridizedfor 1to2h and thenhybridizedto
nick-translated probes as described previously (44, 45). In
someexperimentsprobes werestripped from the filters, and
the membranes were rehybridized with a second probe as
previously described (45). Nick-translated pHcGAP and
pHSV106were used as probes to detect the cellular GAPD
and the HSV-1 thymidine kinase mRNAs, respectively.
Nick-translated pXlrll was used to detect28S rRNA.
Quantitation ofmRNA levels. To quantitate the levels of
host and viralmRNAs and 28S rRNA, autoradiograms from
the Northern blots were scanned with a Hoeffer model
GS300scanningdensitometer. Since 28S rRNA provedtobe
stable in all types ofextracts, the levels of specific mRNAs
were normalizedtotheamountof28S rRNA in each sample
before mRNA decaycurves were plotted in all experiments
exceptthatshownin Fig.7. In Fig. 7the relativeamountsof
28S rRNA and GAPD mRNA were plotted without prior
normalization. Nick-translated pHcGAP hybridized to
GAPD mRNA;it alsocross-hybridized atalow levelto28S
rRNA. Equivalent results were obtained for the decay
curvesof GAPD mRNAregardlessofwhether theamountof
GAPD mRNA was normalized to the level of
28S
rRNA detected by cross-hybridization with pHcGAP orby
strip-ping the pHcGAPprobe from the blot and thenrehybridizingwith nick-translated pXlrll.
RESULTS
Strategy. TheHSVvirion host shutoffproteinis known to
induce rapid degradation of host and viral mRNAs in the cytoplasm (32, 44, 45, 66, 69). In an effort to develop anin vitro mRNA degradation system to study the vhs function,
we decided to compare the rates of mRNAdegradation in in vitro translation extracts from mock-infected HeLa cells and extracts from cells infected with either wild-type HSV-1 or the mutant vhsl, which has been shown to encode a
defec-tive vhs polypeptide (55). HeLacells were chosen because they are readily infected with HSV and because numerous studies have proven them to be a particularly suitable cell line for the preparation of in vitro translation extracts. Although in vivo studies indicated that vhs-induced mRNA degradation does not require efficient ongoing translation of
the mRNAs (66, 69), we decided to prepare invitro transla-tion extracts because we reasoned that they would be the best initial approximation to a functional cytoplasm.
Extracts were prepared by a procedure shown by Brown and co-workers to be suitable for the preparation of highly active in vitro translation extracts from a variety ofcultured
cells (9). This procedure involves briefly exposing the mono-layers to lysolecithin to permeabilize the cells, harvesting the cells directly into buffer containing the components required for in vitro translation, disrupting the cells by repeated passage through a 25-gauge needle, and removing the nuclei by low-speed centrifugation. This protocol was chosen because it is simple and rapid, making itpossible to prepare and analyze the activity of extracts on the same day. This enabled us to perform all experiments with freshly prepared lysates and eliminated the need to freeze in vitro degradation extracts before analysis, an obvious advantage in view of the possible detrimental effects that freezing and thawing could have upon the activities of as yet uncharac-terized proteins.
To optimize the concentration of lysolecithin needed to permeabilize HeLa cells and to verify that this procedure yielded active in vitro translation extracts, standard reaction mixtures were prepared by using a variety of lysolecithin concentrations in the permeabilization buffer. Unlabeled methionine was omitted from the reaction buffer and
[35S]methionine
was added to a concentration of 75,uCi/ml.
At various times after the start of incubation, samples were withdrawn and the amount of[35S]methionine
incorporated into trichloroacetic acid-precipitable material was deter-mined as described previously (55). Exposure of the HeLa cells to 300pug
oflysolecithin per ml was found to be optimal for in vitro translation (data not shown). For extracts pre-pared with this concentration of lysolecithin, efficient in vitro translation continued for approximately 40min,
after which the amount of incorporated label leveled to a plateau (Fig. 1).In vitro degradation of host mRNAs. The vhs function was originally identified on the basis of its ability to induce rapid degradation of host mRNAs and the concomitant shutoff of host polypeptide synthesis (16, 17, 19, 20, 55, 66). To determine whether HeLa celllysateswould be suitable for in vitro studies of the vhs function, standard in vitro translation
on November 10, 2019 by guest
http://jvi.asm.org/
1.0
-0 20 40 60 TIMEOFINCUBATION (MINUTES)
FIG. 1. In vitro translation by HeLacell extracts. An in vitro
mRNAdegradationextractwasprepared from mock-infectedHeLa
cells in standard reaction buffer containing 2.5 mM Mg(OAc)2, lacking unlabeled methionine, and supplemented with
[3S]methio-nineto75 uCi/ml.Thereaction mixturewasincubatedat30°C.At
varioustimessampleswerewithdrawn, and theamountof radioac-tivity incorporated into trichloroacetic acid-precipitable material
wasdeterminedasdescribed previously (55).
extracts were prepared from mock-infected cells and from
cells 5 h after infection with 20 PFU of either wild-type
HSV-1 or the mutant vhsl per cell. The extracts were
incubatedat30°C;atvarious timessampleswerewithdrawn
andextracted withphenol and chloroform, and the decay of specific cellular mRNAswasanalyzed by Northern blotting.
Tocontrolfor the totalamount ofcytoplasmic RNA loaded
ontoeachlaneofthegel,theblotswerealsoprobed for 28S rRNA, and the amount of mRNA was normalized to the
amount of28S rRNA before the mRNA decay curve was
plotted.
Intheseinitialexperimentswedecidedtostudy the decay
ofendogenous cellular mRNAs rather than that of added
exogenousmRNAsbecause wereasoned that the structure
ofmessengerribonucleoprotein particles(mRNPs)
reconsti-tutedon exogenousmRNAsmight differ from that of
endog-enous mRNPs. mRNP structure could easily affect mRNA
stability; in fact, HSV infection has been shownto induce changes in mRNPstructure that correlate with awild-type
virionhostshutoff function (31). Focusinguponthe decayof
endogenous mRNAs should, therefore, remove one poten-tial variable from the experiments. In addition, the results involving decay of endogenous mRNAs should provide a
base line for later attempts to study the degradation of
exogenous messages.
Thedecay ofthecellularmRNAencoding GAPDisshown inFig.2and3. GAPDmRNAwaschosenforstudybecause
it has along invivo half-life in uninfected cells (73). Thus,
any vhs-induced reduction in message stability should be
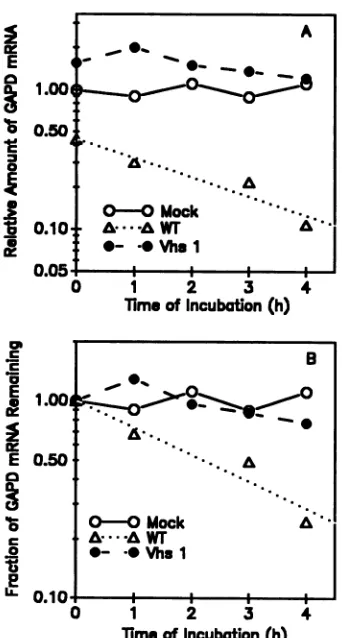
moreeasilydetectedforthismRNAthanforamessagethat is inherently unstable. GAPD mRNA was relatively stable foratleast 4 h inextractsfrom mock-infected cells (Fig. 2,
lanes 1through 5; Fig. 3). Incontrast, itdecayed rapidlyin extracts from cells infected with wild-type HSV-1, so that little detectable mRNA remained by 1 h after the start of
incubation(Fig. 2,lanes6through 9; Fig. 3).That consider-ablevhs-induced degradation had occurred invivo in wild-typeinfections before thepreparationofextractsisindicated by the fact that the intensity of the band formed by GAPD mRNA at 0 h of incubation for wild-type extracts was
reduced considerably relative to that observed at 0 h for mockorvhsl infections (compare Fig. 2,lanes1,6,and 10,
FIG. 2. Invitrodegradation of host mRNAs. Standard in vitro mRNA degradation extracts were prepared from HeLa cells S h
aftermockinfection (lanes 1through 5),orinfection with 20 PFU of wild-type HSV-1 (lanes 6 through 9)orvhsl (lanes 10through 14)
percell.Sampleswerewithdrawn fromthereactionsat0 h(lanes 1, 6, and 10), 1 h (lanes 2, 7, and 11), 2 h (lanes 3 and 12), 3 h (lanes 4, 8, and 13), or4h(lanes 5, 9, and 14). Thesampleswereextracted
twice with phenol-chloroform and twice withchloroform, and the RNAswereprecipitatedfromethanol. Samples of totalcytoplasmic
RNA were denatured with glyoxal, electrophoresed through 1% agarose gels, and transferred to Nytran membranes by capillary blottingasdescribed in thetext.Themembraneswerethenprobed
todetectGAPD mRNA and 28S rRNAasdescribed in thetext.
withFig. 3A). Incontrast tothe casefor wildtype-infected
cell extracts, inextractsfromcells infected with vhsl GAPD mRNA was every bit as stable as in extracts from mock-infected cells (Fig. 2, lanes 10 through 14; Fig. 3). In vitro
decaywasspecificformRNAs,asevidencedbythe fact that
28S rRNA was equally stable in mock-, wild type-, and
vhsl-infected cellextracts.Alltold, the rankorder ofinvitro
decayratesof GAPD mRNAwasthe sameasthatobserved in mock,wild-type, and vhsl infections in vivo.
The vhsproteinisastructuralcomponentofvirions and is therefore ableto induce degradation of host mRNAs in the absence ofpriorde novo viral gene expression (17, 22, 55, 66, 69). Thus, virion host shutoff is induced by
UV-inacti-vatedvirusaswellasafterinfection ofcells in thepresence
ofdactinomycin to block viraltranscription (17, 22, 55, 66, 69). To determine whether the accelerated degradation of GAPD mRNAobserved inextractsfromcells infected with
wild-type virus was induced by a virion component or
requireddenovo viralgene expression,invitrodegradation
extracts were prepared from cells 5 h after a productive
wild-type virus infectionor5 hafterinfection with 50 PFU of
wild-type virus or vhsl per cell in the presence of5 ,ug of
dactinomycinperml.Degradationof theGAPD mRNAwas
equally rapid in extractsfrom cells infected withwild-type
virus in thepresenceandabsence ofdactinomycin (Fig. 4).
In contrast, GAPD mRNA was stable for at least 5 h in extractsfromcells infected with vhsl.Thus,theaccelerated
degradation of host mRNAs that was observed in extracts
from cells infected withwild-typeHSV-1wasnotdependent
upon de novo viral gene expression and was therefore
induced bya componentof theinfecting virions.
Invitrodegradationof viral mRNAs.Althoughvhs mutants
were originallyisolated on the basis oftheir defects in the
degradation of cellular mRNAs and the shutoff of host
protein synthesis,recentstudies indicatethat the vhsprotein playsacentral roleindeterminingthe half-lives of bothviral
and cellular mRNAs within the infected cell (32, 44, 45).
3.0
2.0 0 0
75
0
0
z
a-0
co
0
9
0.0
80
_
ae0 0 0
.1
)I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.320.556.76.211.2] [image:4.612.70.286.78.213.2]E
:1
a1.00
0.50
0.10.
nnr,
A 0- _
kA..
....
....
CO- Mack
0-*VhswrAl
*- -oVhs 1
0
cm
E
1.00O
I
E 0.50
0
a
c
nIn.
1 2 3
TimoofIncubation(h) 4
a% ._-.a E
S
0,
E
2
O 1 2 3 4
[image:5.612.95.266.72.391.2]Timeof Incubation(h)
FIG. 3. Quantitation of in vitro decay of host mRNAs. The
autoradiogram shown in Fig. 2 was scanned, and the amount of
GAPD mRNAin each lane was normalized tothe amount of 28S
rRNA. In panel A the relative amounts of GAPD mRNA in
mock-infected (0), wild-type virus-infected (A), and vhsl-infected (@) cellextractsareplottedinarbitraryunits. InpanelB theamount
ofGAPDmRNApresentinasample fromanyofthethree typesof
extractisexpressedas afraction of theamountpresentat0hin that
kindofextract.
0.50
A-A vhs 1 + Act D
*- - Wr+ActD
O.0 WTproductive
A
A
A
;~~~~~~t
...Y -o....
i6 ..1.
0
i 2 3 4
Time of Incubation(h)
5
FIG. 4. Invitrodegradationin extractsfrom cellsinfected inthe presence ofdactinomycin. Standard in vitrodegradation extracts
wereprepared from cells 5 h afterinfection with 50 PFU ofwild-type
virus(0)orvhsl(A)percell inthepresenceof 5,ugofdactinomycin
permlor5hafter infection with 50 PFU ofwild-typeviruspercell in theabsenceofanydrugs(0).Theextractswereincubatedat30°C
fortheindicatedtimes,atwhichpointsampleswerewithdrawnand totalRNAs wereextracted andanalyzed for GAPD mRNA and 28S rRNA by Northern blotting as described in the legend to Fig. 2. Autoradiograms were scanned with a Hoeffer model GS300
scan-ningdensitometer, andtheamountofGAPDmRNAin eachsample
wasnormalizedto the amountof 28SrRNA. The amountof GAPD mRNAremainingatvarious timeswas plottedas afractionof the amountpresentat0 h.
mentstocharacterize someofthebiochemical
requirements
ofthein vitro mRNAdegradation system.
A preliminary experiment was undertaken to determine
theeffectof RNasinuponvhs-inducedmRNAdecay. Paral-leldegradation extracts wereprepared fromHeLacells5 h after infection with 20 PFU of either
wild-type
HSV-1 orvhsl percell. RNasin (100U/ml) wasincluded inhalf ofthe
reactionmixtures and omitted fromthe other half.
Regard-lessofwhether theinhibitorwaspresent,mRNAswereveryMeasurementsof the half-lives of 10 different viral mRNAs in cells infected with wild-type virus or the mutant vhsl
revealed the following: (i) in wild-type infections the
half-lives of all 10 messages, representing all kinetic classes of
viral mRNA, were very similar; (ii) the vhsl mutation
resulted in dramatic increases in the stabilities of all 10
messages (44, 45). Thus, the vhs protein inducesthelargely
nonselectivedegradation of bothviral andcellular mRNAs. To determine whether degradation of viral mRNAs was
alsoacceleratedinourin vitromessagedegradationsystem, in vitro degradation extracts were prepared from cells 5 h afterinfection withwild-typevirusorvhsl andanalyzedfor
the degradation of the mRNA encodingthe viralthymidine
kinase (TK). TK mRNA was degraded rapidly in in vitro
extracts from cells infected withwild-type HSV-1 but was
relatively stable foratleast 5 h inextractsfrom cellsinfected
withvhsl (Fig. 5). Therefore,once againthein vitroresults
paralleledthose observed invivo.
Characterizationof the invitromRNAdegradationsystem. Inallrespectsexaminedtothis point, in vitro degradation of
hostandviralmRNAsobserved in HSV-infected HeLa cell
extracts paralleled vhs-induced mRNA degradation
ob-served in vivo. We therefore undertook a series of
experi-'E
.0 E
E 0.1
0
9--- Vhs 1
0.05-4
0 1 2 3 4 5
Time ofIncubation(h)
FIG. 5. In vitrodegradation ofviral mRNAs. Standard in vitro mRNA degradation extracts were prepared from cells 5 h after infection with 20 PFU ofwild-typeHSV-1(0)orvhsl(-)percell.
The extracts were incubated at 30°C for the indicated times, at
which point samples were withdrawn and total RNAs were
ex-tracted andanalyzed for TK mRNA and 28SrRNA by Northern
blottingasdescribed in thelegendtoFig.2. Autoradiogramswere
scannedwitha Hoeffer model GS300scanning densitometer, and
theamount of TK mRNA in each sample was normalized to the
amount of 28S rRNA. The amount of TK mRNA remaining at
varioustimeswasplottedas afraction oftheamountpresentat0 h. Errorbars indicate the standard errors of the means determined
fromreplicate experiments. B
..
0-A..
O-O Mock A'.
a@
-AWT
0- -OVhs1
v.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.329.548.76.212.2] [image:5.612.332.548.480.612.2]0~
.'-.a E
e
E
CZ
0 U
a
L,
1.00'
0.50
0.10*
n nr..
1 2 3 4 5
[image:6.612.66.284.77.215.2]TimeofIncubation(h)
FIG. 6. Effect of heat and proteinase K pretreatment upon
vhs-induced in vitro degradation. Standard in vitromRNA
degrada-tion extracts were prepared from cells 5 h after infection with 20
PFU ofwild-type HSV-1percell. Extractswerepretreatedeither by
the addition ofproteinase K and digestion for 30minorby heating
to90°C for 10min(0),after which theextractswerereturnedto4°C. Control unpretreated extracts were left at 4°C until the start of incubation (0). The extracts were incubated at30°C for the indi-cated times. Samples were withdrawn and analyzed for GAPD
mRNA and 28S rRNA by Northern blotting, and the amount of GAPDmiRNAin each samplewasnormalizedtotheamountof 28S rRNA.
stableinextractsfrom cellsinfected with vhsl, whereas they decayed rapidly in extracts from cells infected with
wild-type virus (datanotshown). In vhsl-infected cells extracts,
there was slightly more mRNA decay in the absence of RNasin than when itwasincluded in the reaction mix. We
attribute this to nonspecific RNases that are inhibited by
RNasin. Because it didnotinhibit vhs-induced degradation and itsinclusion yielded slightly cleaner results, RNasinwas
included as a component in the standard in vitro reaction
bufferandwasused inall of theotherexperiments described
in thisreport.
Thenext setof experimentswasundertakentodetermine whether the in vitro vhs activity could be inactivated by pretreating the extracts with heat or proteinase K. Three
parallel in vitro degradation extracts were prepared from
HeLa cells 5 h after infection with 20 PFU ofwild-type
HSV-1 percell. One extract was heatedto 90°Cfor 10min
and then chilledonice. A secondextractwas supplemented
withproteinase K and digested at30°C for 30min, and the
third was left untreated. All three extracts werethen
ana-lyzed for invitro vhsactivity. Pretreatment of the extracts
by either heating or proteinase K digestion completely abolished vhs-mediatedinvitro degradation(Fig. 6). These results areconsistent with the involvement ofone or more
heat-labile proteinsinvhs-mediated message turnover.
Recently Brewer and Ross showed that a factor that is
presentinthe postpolysomal supernatant fraction from the
cytoplasmof K562 cells and thatspecificallyaccelerates the
decay ofc-myc and c-myb mRNAs is inactivated by brief
digestion with micrococcal nuclease (7). To determine whether similar micrococcal nuclease pretreatment of
ex-tracts from HSV-1-infected cells would inactivate the in
vitro vhs activity, standard in vitro degradation extracts
were prepared from HeLacells 5 h after mock infection or
infection with 20 PFU of wild-type HSV-1 per cell. The extracts were supplemented with micrococcal nuclease and
CaCl2and thenpreincubated at30°Cfor10min. EGTAwas
thenaddedtochelate theCa2+and inactivate the micrococ-cal nuclease, and the extracts were chilled briefly on ice.
1.001
I
0
0.10
c
0 .
0-1 2 3
TimeofIncubation (h)
[image:6.612.325.546.79.214.2]4 5
FIG. 7. Decay ofexogenous GAPD mRNA and 28S rRNA in
micrococcal nuclease-treated in vitro degradation extracts. Stan-dard in vitromRNA degradationextractswerepreparedfrom cells 5 h after mock infection or infection with 20 PFU ofwild-type
HSV-1percell.Theextractswerepretreatedasdescribed in thetext
with micrococcal nuclease in the presenceofaddedCa2". EGTA wasaddedtochelate the Ca2+,and deproteinized total cytoplasmic RNAfromanequivalent number of cellswasaddedtoeachextract.
The extracts were incubated at 30°C for the indicated times.
Sampleswere withdrawnandanalyzed for GAPD mRNA and 28S rRNA by Northern blotting. The relativeamountof 28S rRNA in mock-infected(0) and wild-type virus-infected (0) cellextractsis plottedas afraction of theamountpresent at0 h. Incontrast toother figures in thispaper,therelativeamountof GAPDmRNAdetected in mock-infected(A) and wild-type virus-infected(A) cellextractsis plotted without being normalized to the amount of 28S rRNA
presentin thesample.
Micrococcal nuclease treatmentisroutinelyusedto deplete
in vitro translation extracts ofendogenous mRNAs andto rendertranslation dependent uponexogenouslyadded
mes-sages(49). Therefore,toprovideatargetfor thevhs-induced
degradative activity, after micrococcal nuclease pretreat-ment the extracts were supplemented with deproteinized
total cytoplasmic RNA fromanequivalent numberof
unin-fected HeLa cells. Theextractswerethen incubatedat30°C
andanalyzedfor in vitrodecayofexogenous GAPDmRNA and 28S rRNA.
Figure7shows thedecayofexogenousGAPD mRNAand total 28S rRNA. Three conclusions canbe drawn from the
resultsofthisexperiment. First,pretreatmentof theextract from wild-type virus-infected cells with micrococcal
nucle-asedidnotinhibittherapiddegradationofexogenousGAPD mRNA. That thisdegradationwasduetothe vhsactivityand
wasnotthe resultsof residual micrococcal nucleaseactivity
is indicatedbythe fact GAPD mRNAwasrelativelystablein the micrococcal nuclease-treated extracts from mock-in-fected cells. Second, the fact thatexogenousGAPD mRNA
wasdegradedin the wildtype-infectedcellextractssuggests that the invitro mRNAdegradationsystem willbe usefulfor
studying the decay of both exogenous and endogenous
mRNAs. Third, 28S rRNAwasstable in extracts fromboth mock-infected and wild-type virus-infected cells. Since
ini-tiallythe totalamountof 28SrRNA was a50:50 mixtureof
endogenous and deproteinized exogenous 28S rRNA, the results indicate thatinwildtype-infectedcellextracts
depro-teinizedexogenousGAPDmRNAwasdegradedmuchmore
rapidlythandeproteinizedexogenous28S rRNA. Thisresult is an additional indication that the RNase activity seen in wildtype-infectedcellextractswasspecificfor mRNAsand
wasnotthe resultofanonspecific RNase.
The next setofexperimentswere undertaken toexamine
0-0 WT Control
*- - WT + Heat
A. A WT +ProteinaseK
%_---
4
_...,.._....!9
0-0 WT-: 28SrRNA 0-@Mock: 28S rRNA
A . *W:GAPD mRNA
A -A Mock:GAPD mRNA
on November 10, 2019 by guest
http://jvi.asm.org/
0P
._
.a
E
z
E
0
c
0
0
1.00I
0.50
0.104
1 2 3 4
[image:7.612.333.549.77.207.2]TimeofIncubation(h)
FIG. 8. Mg2+dependence of vhs-induced mRNA degradation. In vitro mRNAdegradationextractswereprepared from cells 5 h after
infection with 20 PFU ofwild-type HSV-1per cell. The extracts
wereprepared in standard reaction buffer modifiedto contain 2.5 mM (0), 10 mM (0), 20 mM (A), or40 mM (A) Mg(OAc)2. The
extracts were incubated at30°C for the indicated times. Samples
werewithdrawnandanalyzedfor viral TK mRNA and 28S rRNA by
Northernblotting, and theamountof TK mRNA in each samplewas
normalizedtotheamountof28S rRNA.
the Mg2+ dependence of the in vitro degradation system.
Parallel in vitro degradation extracts were prepared from
HeLa cells 5 h after infection with 20 PFU of wild-type
HSV-1 per cell. Individual reaction mixtures were
supple-mented with concentrated Mg(OAc)2 to bring the Mg2+
concentrationtothe desired value.The extracts were
incu-batedfor5 handanalyzed for the decayof theviral mRNA
encodingTK. Invitro degradation of TK mRNA showed a
strongdependenceupontheconcentration ofMg2+ion(Fig. 8). Although a significant amount of mRNA degradation
could beobserved atanMg2+concentration of10mM(Fig. 8) or5mM (datanot shown), increasing the Mg2+
concen-trationto20mMorhighersignificantlyincreased the degra-dation ratein wild-type extracts (Fig. 8). That theeffect of raising theMg2+ concentrationwas not simply the result of inducing nonspecific changes in mRNP structure or the
activation of nonspecific nucleases is indicated by the fact
thatasignificant differenceinthe mRNA decayratesinwild type- and vhsl-infected cell extracts was observed at an
Mg2+ concentration of 20 mM (see Fig. 10); 20 mM was
chosen as the optimal Mg2+ concentration for subsequent experiments.
A similar experiment was undertaken to determine the
effect of varying the K+ concentration upon the rate of vhs-induced decay ofTK mRNA (Fig. 9). Parallel in vitro degradation reactions were prepared from HeLa cells 5 h
afterinfection with 20PFU ofwild-type HSV-1 percell. In
thesereactions theMg2+ concentrationwasheldconstant at
20 mM, whereas the concentration of K+ ion was varied
from 7 to500 mM. Efficient degradation ofTK mRNAwas
observed at K+ concentrations from7to200mM,whereas increasing theK+ concentration to 500 mM severely
inhib-itedthedegradation reaction (Fig.9).Wechose 7 mMK+as
optimal.
The final experimentwas designed to determine whether
vhs-induced in vitro mRNA degradation was dependent
upon the components ofanenergy-generating system. Par-allelinvitro degradationextractswereprepared from HeLa
cells at 5 hafter infection with 20 PFU ofeither wild-type
HSV-1 or vhsl. Half of the reactions contained all of the
components ofthe standard reaction, whereas ATP, GTP,
.' *~~~~~~~-- 100 mM
A...A200 mM
c A--A- 500 mM
E
A A
Z 1.00 .A
E A
0.10
0~~~~~~~~
C
0.10
0 1 2 3 4 5
Timeof Incubation(h)
FIG. 9. K+ dependence ofvhs-induced mRNAdegradation. In vitromRNAdegradationextractswerepreparedfrom cells 5 h after
infection with 20 PFU ofwild-type HSV-1 per cell. The extracts
werepreparedin standardreaction buffer modifiedtocontain 7 mM (0), 100mM (0),200 mM(A), or500 mM(A) KCl.The extracts
were incubated at 30°C for the indicated times. Samples were
withdrawn and analyzed for viral TK mRNA and 28S rRNA by Northernblotting,and theamounitof TK mRNAin eachsamplewas
normalizedtotheamountof 28S rRNA.
creatine phosphate, and creatinephosphokinasewere omit-ted fromthe other half. Efficient vhs-induced degradation of TK mRNA occurred in thepresence(Fig. 10A) and absence
(Fig. 10B) of the components ofanenergy-generating
sys-tem.
DISCUSSION
In this reportwedescribe anin vitro mRNA degradation
system, consisting of cytoplasmic extracts from HSV-1-infected HeLa cells, that appears to accurately reproduce the degradation of host and viral mRNAs induced by the wild-type vhs protein in vivo. This conclusion is supported by several important parallels between the in vitro data and in vivo observations. First, host messages were degraded
rapidly inextractsprepared fromcellsproductively infected
withwild-typeHSV-1 butnotinextractsfrommock-infected cells or cells infected with the mutant vhsl. Second, the
accelerated turnover of host mRNAs occurred in extracts
from cells infected with wild-type virus in the presence of
dactinomycin, indicatingthat itwasinduced byacomponent
ofthe infecting virions andwasnotdependentupondenovo
viral gene expression. Third, accelerated turnover ofviral mRNAs was observed in extracts from cells productively infected with wild-type HSV-1 but not in extracts from
vhsl-infected cells. In each of the above cases, the most
important observation supportingthe fidelityof the invitro
systemwasthestrikingdifference between the mRNAdecay
ratesinextractsfrom cells infected withwild-typevirusand in extracts from vhsl-infected cells. This observation indi-cates that the accelerated in vitro degradation of mRNAs
was dependent upon infection of the cells with virions
containingafunctional vhspolypeptideandwasnot simply
theconsequenceofanonspecificRNaseliberatedduringcell
fractionation or induced as a nonspecific consequence of
viralinfection. Finally, although the wild-type vhsfunction
induced accelerated in vitro turnover of both viral and
cellularmRNAs, endogenous 28S rRNAwasequally stable
in extracts from mock-infected cells and in extracts from cells infected with eitherwild-typevirusorthe vhslmutant. Thislends further supportto the conclusion that the
degra-dative activity seen in wild type-infected cell extracts was
specific for mRNAs and was not due to a contaminating
Mg2+ Effects 0-0 2.5mM
*- -0 10 mm
A/ A 20 mM
A 40 mM
*
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.79.296.79.213.2]vhs-INDUCED mRNA DECAY IN VITRO 119
z z
I
mE
0
10.50
0.50
0.10~
z
z
E
m
I-0
1 2 3 4 5
TiMEOFINCUBATION(H)
0 1 2 3 4 5
liME OFINCUBAllON (H)
FIG. 10. Dependence of vhs-induced mRNA degradation upon the components ofan energy-generating system. In vitro mRNA
degradation extracts were prepared from cells 5 h after infection
with 20 PFU of wild-type HSV-1 (0) or vhsl (A) per cell. The
extracts were prepared eitherin standard reaction buffer (A) orin
standardreaction buffer fromwhich ATP,GTP,creatinephosphate, and creatine phosphokinase had been omitted (B). The extracts
were incubated at 30°C for the indicated times. Samples were
withdrawn and analyzed for viral TK mRNA and 28S rRNA by
Northernblotting, and theamountofTK mRNAineachsamplewas
normalizedtotheamountof 28S rRNA.
nonspecific RNase that should havebeen presentin allthree kinds ofextract.
Analysis of the crude in vitro system showed thatone or
morefactors necessary forin vitro vhs activity was
inacti-vatedby heating the extracts to90°C or bybriefproteinase Kdigestion.These dataareconsistent with theinvolvement
ofone or moreheat-labile proteins in vhs-induced
degrada-tion. In contrast, pretreatment of wild-type extracts with micrococcal nuclease did notinhibit the subsequent degra-dation of added exogenous mRNA, indicating that the
fac-torsrequiredfor in vitro vhs activity are apparently
insensi-tivetomicrococcal nucleasetreatments knownto inactivate
anumberof small RNAs andribonucleoproteins (30)as well as afactor that accelerates in vitro decay ofc-mycandc-myb
mRNAs (7). Furthermore, the finding that exogenous
mRNAs were rapidly degraded in extracts from cells
in-fected with wild-type virus but were relatively stable in
extracts frommock-infected cells suggests that the in vitro
degradation system will be suitable for studying the
vhs-induced decay ofbothexogenous and endogenousmRNAs.
Itisalso worthnotingthatin theseexperimentsthesourceof
exogenousmRNAwastotaldeproteinizedcytoplasmic RNA
containingamixtureof mRNAand rRNAs.Thus,theresults
shown in Fig. 7 indicate that added deproteinized mRNA
wasdegradedmorerapidly thanaddeddeproteinized rRNAs
in wild-type extracts. This adds further support to the
conclusion that the in vitro vhs activity observed in wild
type-infected cell extracts wasspecific formRNAs anddid
notsimply resultfroma contaminating nonspecific RNase.
Preliminary biochemical characterization ofthe in vitro mRNA degradation system from HSV-infected cells
indi-cates that it is similar in a number of respects to in vitro mRNAdegradationsystemsfromuninfected cells described
previouslybyRossetal. (3, 4, 6, 7, 51, 52, 60-63) and others
(48,
70).
Inparticular,
the vhs-induced mRNAdegradation
activity
was not inhibitedby
the placental RNase inhibitor RNasin and was dependent upon added divalent cation.Efficient vhs-induced degradation occurred at K+ ion
con-centrations ofup to 200 mM but was inhibited by 500 mM
K+,
and mRNA degradative activity was not dependentupon the addition of ATP, GTP, creatine phosphate, or
creatine phosphokinase. In each of these respects, the
vhs-induced RNase activity was similar to that ofthe
exo-nuclease shownbyRoss and co-workers toinduce degrada-tion of histone mRNAs in extracts fromK562 erythroleuke-miacells (62).
Althoughbothrequiredaddeddivalent cation, the in vitro vhs activity reported here and the exonuclease described by
Ross (62)differ somewhat in the nature of their dependence upon added
Mg2+.
Whereas the exonuclease that degradeshistone mRNAs exhibited abroad optimum ranging from 5 to20mM
Mg2+
(62), the vhsactivityobserved in theextractsdescribed here was more strongly dependent upon added
Mg2 . Thus, although a striking difference was observed
between mRNA decay rates in extracts from wild-type
virus-infected and vhsl-infected cells at Mg2+ concentra-tions ranging from 2.5 to 20 mM (Fig. 2 through 5, 8, and 10; unpublished data), the rate of mRNA degradation in wild-type extracts increased continuously as the Mg2+
concen-trationwasraised to20or40 mM. At present, the reason for this difference in the Mg2+ dependence of the two in vitro systemsis unclear. It mayreflect anincreased Mg2+
depen-denceof one of the proteins involved in vhs-induced degra-dation. Alternatively, higherMg2+concentrations may favor aconformational change in mRNP structure that renders the mRNA more susceptible to vhs-induced degradation.
Besidesprovidingthe groundwork forfutureexperiments,
the preliminary biochemical characterization of the vhs-induced RNase allows it to be distinguished from several previously characterized RNases that are commonly found in cell extracts. Pancreatic RNase is resistant to boiling but is inhibited by RNasin (48). The fact that the vhs activity is sensitive to heating to 90°C but is not inhibited by RNasin therefore indicates that it does not involve a pancreatic-type RNase. A nucleolar exonuclease has been described that, like the vhs-induced RNase, is dependent uponadded Mg2+ (35). However, unlike the vhs-induced enzyme, the nucleo-lar RNase is inactive at K+ ion concentrations greater than 90 mM (35). A lysosomal acid RNase has been described (65). However, unlike the vhs-induced enzyme, it does not
require added Mg2+.
In several previously characterized in vitro degradation systems, the RNases responsible for mRNA turnover were found to be polysome associated. This was the casefor the nucleasesresponsible for invitro degradation of histone and c-myc mRNAs, although in both cases the decay rate was greatly accelerated by soluble factors present ina postpoly-somal supernatant fraction of the cytoplasm (7, 51). In addition, Brawerman and co-workers haverecentlyreported an RNase that is associated with polysomes as well as free mRNPs in a variety ofmammalian cells (2). At present, the
A. WITH ENERGYCOMPONENTS
A- -
-A\
°-
-A~-A--A-VHS 1\
O~~~~~~~~
B. WITOUT ENERGYCOMPONENS
~~~~~~A-
--A- -AVHS 1
VOL. 65, 1991
1.00
0.50.
0.10.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.67.288.75.348.2]subcellular localization of the proteins required for vhs-inducedmRNA degradation is unknown.
Inpreliminaryexperiments, invitrodegradation extracts
from mock-infected, wild-type HSV-1-infected, or vhsl-infectedHeLacells wereseparated bycentrifugation into a polysome pellet and a postpolysomal supernatant. Polyso-mal mRNAs from extracts ofcells infected with wild-type virus or vhsl were equally stable upon suspension and
incubationof the polysomesin standardreaction buffer or in the postpolysomal supernatant from mock-infected cells
(31a). However, the addition of thehigh-speed supernatant fromextracts ofcellsinfected withwild-typevirus to any of
the three types ofpolysomeresultedinrapid degradation of
thepolysomalmRNAs. These dataindicate thatone or more
factors required for vhs-induced mRNA degradation are found inthepostpolysomal supernatantfraction of extracts fromcells infected with wild-type virus. Whetheradditional
polysome- ormRNP-associatedfactors are alsorequiredfor
vhsactivityis currentlyunderinvestigation.
A central unanswered question concerning the vhs
func-tion is whether the vhsprotein isitself an RNase or,instead, activates a cellular nuclease, perhaps one involved in the normal turnover of mRNAs in uninfected cells. To date,
attempts todemonstrateanRNaseactivityinpreparations of disrupted virions have beenunsuccessful (31a). The reason forthismaysimply be thatmethods ofvirion disruption that
preservetheRNase activity have yet to befound. Alterna-tively,oneor morecellularmacromolecules mayberequired for vhsactivity. Thesimilarity ofthebiochemical character-istics of the vhs-induced activityto those of in vitro degra-dation systems from uninfectedcells (62) is consistent with
this second possibility. Resolution of this question and elucidation of the detailed mechanism ofvhs-induced
mes-sageturnover will require further fractionation and
charac-terization of the in vitro mRNAdegradation system. ACKNOWLEDGMENTS
We thank Annette Schmidt and Kim Knight for many helpful discussions and critical appraisals of the data presented in this report. We are also indebted to Christine Sorenson, Phil Hart, and JeffRoss for helpful discussions and for communicating their data prior to publication.
This work was supported by grant AI21501 from the National Institutes of Health.
REFERENCES
1. Adams, J. M., A. W. Harris, C. A. Pinkert, L. M. Corcoran, W. S.Alexander, S. Cory, R. D. Palmiter, and R. L. Brinster. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature (Lon-don) 318:533-538.
2. Bandyopadhyay, R., M. Coutts, A. Krowczynska, and G. Brawerman. 1990. Nuclease activity associated with mamma-lian mRNA in its native state: possible basis for selectivity in mRNA decay. Mol. Cell. Biol. 10:2060-2069.
3. Bernstein, P., S. W. Peltz, and J. Ross. 1989. The poly(A)-poly(A)-binding protein complex is a major determinant of mRNA stability in vitro. Mol. Cell. Biol. 9:659-670.
4. Bernstein, P., and J. Ross. 1989. Poly(A), poly(A) binding protein and the regulation of mRNA stability. Trends Biochem. Sci. 14:373-377.
5. Brawerman, G. 1989. mRNA decay: finding the right targets. Cell 57:9-10.
6. Brewer, G., and J. Ross. 1988. Poly(A) shortening and degrada-tion of the 3' A+U-rich sequences of human c-myc mRNA in a cell-free system. Mol. Cell. Biol. 8:1697-1708.
7. Brewer, G., and J. Ross. 1989. Regulation of c-myc mRNA stability in vitro by a labile destabilizer with an essential nucleic
acid component. Mol.Cell. Biol. 9:1996-2006.
8. Brock, M. L., and D. J. Shapiro. 1983. Estrogen stabilizes vitellogenin mRNA against cytoplasmic degradation. Cell34: 207-214.
9. Brown, G. D., R. W. Peluso, S. A.Moyer, and R. W.Moyer.
1983. A simple method for the preparation ofextracts from animalcellswhich catalyze efficientin vitroproteinsynthesis.J. Biol. Chem. 23:14309-14314.
10. Campisi, J., H. E. Gray, A. B. Pardee, M. Dean, and G. E. Sonenshein. 1984. Cell-cycle control of c-myc but not c-ras
expression is lost following chemical transformation. Cell 36: 241-247.
11. Caput,D., B. Beutler, K. Hartog, R.Thayer, S.Brown-Shimer, and A. Cerami. 1986. Identification ofa common nucleotide sequence in the 3'-untranslated region of mRNA molecules specifyinginflammatorymediators. Proc.Natl. Acad. Sci. USA 83:1670-1674.
12. Coppola, J. A., and M. D. Cole. 1986. Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell dif-ferentiation butnotcommitment.Nature(London)320:760-763. 13. Dani, C., L. M. Blanchard, M. Piechaczyk, S. ElSabouty, L. Marty, and P. Jeanteur. 1984. Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. USA 81:7046-7050.
14. Dawid, I. B., and P. K. Wellauer. 1976. Areinvestigationof the 5' to 3' polarity in 40S ribosomalRNA precursor ofXenopus laevis. Cell 8:443-448.
15. Dony, C., M. Kessel, and P. Gruss. 1985. Post-transcriptional
controlofmycand p53expressionduringdifferentiation of the embryonal carcinoma cell line F9. Nature (London) 316:636-639.
16. Fenwick, M. L. 1984. The effects ofherpesviruses oncellular macromolecular synthesis, p. 359-390. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology, vol. 19. PlenumPublishingCorp., New York.
17. Fenwick, M. L., and J. Clark. 1982. Earlyanddelayedshut-off of hostprotein synthesis in cells infectedwith herpessimplex
virus. J. Gen. Virol. 61:121-125.
18. Fenwick,M.L.,andJ.Clark. 1983.Theeffect ofcycloheximide
ontheaccumulationandstabilityoffunctionalalpha-mRNAin cells infectedwithherpes simplexvirus.J.Gen.Virol. 64:1955-1963.
19. Fenwick, M. L., and M. M. McMenamin. 1984. Early virion-associated suppression of cellular protein synthesis by herpes
simplex virus isaccompanied by inactivation ofmRNA.J.Gen. Virol. 65:1225-1228.
20. Fenwick, M. L., L. S. Morse, and B. Roizman. 1979.Anatomy of herpessimplex virusDNA. XI.Apparentclusteringof functions effecting rapid inhibition of host DNAandprotein synthesis.J. Virol. 29:825-827.
21. Fenwick, M. L., and S. A. Owen. 1988. On the control of immediate early (alpha) mRNA survival in cells infected with herpes simplex virus. J.Gen. Virol. 69:2869-2877.
22. Fenwick, M. L., and M. J. Walker. 1978. Suppression of synthesis of cellular macromoleculesbyherpessimplexvirus.J. Gen. Virol. 41:37-51.
23. Gay,D.A.,T.J.Yen, J.T. Y.Lau,and D. W. Cleveland. 1987. Sequencesthatconferbeta-tubulinautoregulationthrough mod-ulated mRNA stability reside within exon 1 ofa beta-tubulin mRNA. Cell50:671-679.
24. Graves, R. A., N. B.Pandey, N. Chodchoy,andW.F.Marzluff. 1987. Translation is required for regulation of histone mRNA degradation. Cell 48:615-626.
25. Guyette, W. A., R. J.Matusik,andJ.M.Rosen. 1979. Prolactin-mediated transcriptional and post-transcriptional control of casein geneexpression. Cell 17:1013-1023.
26. Harris-Hamilton,E., and S. L. Bachenheimer. 1985. Accumula-tion of herpes simplex virus type 1 RNAs of different kinetic classes in thecytoplasm of infectedcells. J. Virol. 53:144-151. 27. Heintz, N., H. L. Sive, and R. G. Roeder. 1983. Regulation of
human histone gene expression: kinetics ofaccumulation and changesin therateofsynthesisand in thehalf-lives of individual histone mRNAs during the HeLa cell cycle. Mol. Cell. Biol.
on November 10, 2019 by guest
http://jvi.asm.org/
3:539-550.
28.
Hill,
T. M., R. K. Sinden, and J. R. Sadler. 1983. Herpes simplex types 1 and 2 induce shutoff of host protein synthesis in Friend erythroleukemia cells. J. Virol. 45:241-250.29. Jones, T. R., and M. D. Cole. 1987. Rapid cytoplasmicturnover
of c-myc mRNA: requirement of the 3' untranslated sequences. Mol. Cell. Biol. 7:4513-4521.
30. Kass, S., K. Tyc, J. A. Steitz, and B. Sollner-Webb. 1990. The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell 60:897-908.
31. Krikorian, C. R., and G. S. Read. 1989. Proteins associated with mRNA in cells infected with herpes simplex virus. Biochem. Biophys. Res. Commun. 164:355-361.
31a.Krikorian, C. R., and G. S. Read. Unpublished data.
32. Kwong, A. D., and N. Frenkel. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 84:1926-1930. 33. Kwong, A. D., and N. Frenkel. 1989. The herpes simplex virus
virion host shutoff function. J. Virol. 63:4834-4839.
34. Kwong, A. D., J. A. Kruper, and N. Frenkel. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912-921. 35. Lasater, L. S., and D. C. Eichler. 1984. Isolation and properties ofa single-strand 5' to 3' exoribonuclease from Ehrlich ascites tumor cell nucleoli. Biochemistry 23:4367-4373.
36. Leder, A., P. K. Pattengale, A. Kuo, T. A. Stewart, and P. Leder. 1986. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell 45:485-495.
37. Lee, W. M., M. Schwab, D. Westaway, and H. E. Varmus. 1985. Augmented expression of normal c-myc is sufficient for cotrans-formation of rat embryocellswitha mutant ras gene. Mol. Cell. Biol. 5:3345-3356.
38. Luscher, B., C. Stauber, R. Schindler, and D. Schumperli. 1985. Faithful cell-cycle regulation of a recombinant mouse histone H4 gene is controlled by sequences in the 3' terminal part of the gene. Proc. Natl. Acad. Sci. USA 82:4389-4393.
39. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.
40. Mullner, E. W., and L. C. Kuhn. 1988. A stem-loop in the 3' untranslated region mediates iron-dependent regulation of trans-ferrin receptor mRNA stability in the cytoplasm. Cell 53:815-825.
41. Mullner, E. W., B. Neupert, and L. D. Kuhn. 1989. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell 58:373-382. 42. Nishioka,Y., and S. Silverstein. 1977. Degradation of cellular
mRNA during infection with herpes simplex virus. Proc. Natl. Acad. Sci. USA74:2370-2374.
43. Nishioka, Y., and S. Silverstein. 1978. Requirements of protein synthesis for the degradation of host mRNA in Friend erythro-leukemia cells infected with herpes simplex virus type 1. J. Virol.27:619-627.
44. Oroskar, A. A., and G. S. Read. 1987. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 61:604-606.
45. Oroskar, A. A., and G. S. Read. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 63:1897-1906.
46. Paek, I., and R. Axel. 1987. Glucocorticoids enhance stability of human growth hormone mRNA. Mol. Cell. Biol. 7:1496-1507. 47. Pandey, N. B., and W. F. Marzluff. 1987. The stem-loop
structure at the 3' end of histone mRNA is necessary and sufficient for regulation of histone mRNA stability. Mol. Cell. Biol. 7:4557-4559.
48. Pei, R., and K. Calame. 1988. Differential stability of c-myc mRNAs ina cell-free system. Mol. Cell. Biol. 8:2860-2868. 49. Pelham, H. R. B., and R. J. Jackson. 1976. An efficient
mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem. 67:247-256.
50.
Pellegrini,
S., and C. Basilico. 1986. Rat fibroblasts expressinghigh levels of human c-myc transcripts are anchorage-indepen-dent and tumorigenic. J. Cell. Physiol. 126:107-114.
51. Peltz,S. S., and J. Ross. 1987. Autogenous regulation of histone mRNA decay by histone proteins in a cell-free system. Mol. Cell. Biol. 7:4345-4356.
52. Peltz, S. W., G. Brewer, G. Kobs, and J. Ross. 1987. Substrate specificity of the exonuclease activity that degrades H4 histone mRNA. J. Biol. Chem. 19:9382-9388.
53. Piecharczyk, M., J.-Q. Yang, J.-M.Blanchard, P. Jeanteur,and K. B. Marcu. 1985. Post-transcriptional mechanisms are respon-sible for accumulation of truncated c-myc RNAs in murine plasma celltumors. Cell 42:589-597.
54. Rabbitts, P. H., A. Forster, M. A.Stinson, and T. H. Rabbitts. 1985. Truncation of exon 1 from the c-myc gene results in prolonged c-myc mRNA stability. EMBO J. 4:3727-3733. 55. Read, G. S., and N. Frenkel. 1983. Herpes simplex virus
mutantsdefective in thevirion-associated shutoff of host
poly-peptide synthesis and exhibiting abnormal synthesis ofalpha
(immediate-early) polypeptides. J.Virol. 46:498-512.
55a.Read,G. S., and K.Knight. Submitted forpublication.
56. Read, G.S., J. A.Sharp,and W. C. Summers. 1984. In vitroand in vivo transcription initiation sites on the TK-encoding BamHI Q fragment of HSV-1 DNA. Virology 138:368-372.
57. Read, G. S., and W. C. Summers. 1982. In vitrotranscription of the thymidine kinase gene of herpes simplex virus. Proc. Natl. Acad. Sci. USA 79:5215-5219.
58. Roizman, B., G.S.Borman, andM. Rousta.1965. Macromolec-ular synthesis in cells infected with herpes simplex virus. Nature(London) 206:1374-1375.
59. Roizman, B., and A. E. Sears. 1990. Herpessimplex virusesand their replication, p. 1795-1841. In B. N. Fields and D. M.Knipe (ed.), Fields virology, 2nd ed. Raven Press. New York. 60. Ross, J. 1988. Messenger RNA turnover in eukaryotic cells.
Mol. Biol. Med.5:1-14.
61. Ross, J., and G. Kobs. 1986. H4 histone messenger RNAdecay in cell-free extracts initiates at or near the 3' terminus and proceeds 3' to 5'. J. Mol. Biol. 188:579-593.
62. Ross, J., G. Kobs, G. Brewer, and S. W. Peltz. 1987. Properties oftheexonucleaseactivity that degrades H4 histone mRNA.J. Biol. Chem. 19:9374-9381.
63. Ross, J., S. W. Peltz, G. Kobs, and G. Brewer. 1986. Histone mRNA degradation in vivo: the first detectablestepoccursat or near the 3' terminus. Mol. Cell. Biol. 6:4362-4371.
64. Rouault, T. A., M. W. Hentze, D. J. Haile, J. B. Harford, and R. D. Klausner. 1989. The iron-responsive element binding protein: a method for the affinity purification ofa regulatory RNA-binding protein. Proc. Natl. Acad. Sci. USA 86:5678-5772.
65. Saha, B. K., M. Y. Graham, and D. Schlessinger. 1979. Acid ribonuclease from HeLa cell lysosomes. J. Biol. Chem. 254: 5951-5957.
66. Schek, N., and S. L. Bachenheimer. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 55:601-610.
67. Schuler, G. D., and M. D. Cole. 1988. GM-CSFand oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell 55:1115-1122.
68. Shaw, G., and R. Kamen. 1986. A conserved AUsequence from the 3' untranslated region of GM-CSF mRNA mediates selec-tive mRNA degradation. Cell 46:659-667.
69. Strom, T., and N. Frenkel. 1987.Effects of herpessimplex virus on mRNA stability. J. Virol. 61:2198-2207.
70. Sunitha, I., and L.I. Slobin. 1987. An in vitro system derived from Friend erythroleukemia cells to study messenger RNA stability. Biochem. Biophys. Res. Commun. 144:560-568. 71. Sydiskis, R. J., and B. Roizman. 1966. Polysomes and protein
synthesis in cells infected with a DNAvirus. Science 153:76-78. 72. Sydiskis, R. J., and B. Roizman. 1967. The disaggregation of
host polysomes in productive and abortiveinfectionwithherpes simplex virus. Virology 32:678-686.
73. Tso, J. Y., X.-H. Sun, T.-H. Kao, K. S. Knight, and R. Wu. 1985. Isolation and characterization of rat andhuman
on November 10, 2019 by guest
http://jvi.asm.org/
dehyde-3-phosphate dehydrogenase cDNAs: genomic complex-ity and molecular evolution of the gene. Nucleic Acids Res.
13:2485-2502.
74. Weinheimer, S. P., and S. L. McKnight. 1987. Transcriptional and post-transcriptional controls establish the cascade of herpes simplex virus proteinsynthesis. J. Mol. Biol. 195:819-833. 75. Wilson, R., and R. Treisman. 1988. Removal of poly(A) and
consequent degradation of c-fos mRNA facilitated by 3'
AU-richsequences.Nature (London) 336:396-399.
76. Yager, D. R., and D. M.Coen. 1988. Analysis of the transcript
of the herpes simplex virus DNA polymerase gene provides
evidence thatpolymeraseexpression is inefficientatthe level of translation. J. Virol. 62:2007-2015.
77. Yager, D. R., A. I. Marcy, and D. M. Coen. 1990. Translational regulation of herpes simplex virus DNA polymerase. J. Virol. 64:2217-2225.
78. Yen, T. J., P. S. Machlin, and D. W. Cleveland. 1988. Autoreg-ulated instability of beta-tubulin mRNAs by recognition of the
nascentamino terminus ofbeta-tubulin. Nature (London) 334: 580-585.