Vol. 49, No. 2 JOURNAL OFVIROLOGY. Feb. 1984. p. 371-378
0022-538X/84/0'0371-08$02).00/0
Copyright t 1984. American Society for Microbiology
Mapping of
a
Gene Coding for
a
Major Late Structural Polypeptide
on
the Vaccinia Virus
Genome
RICCARDO
WITTEK,'*
MARKUSHANGGI,1
ANDGERHARD HILLER2Institit de BiologieAnimale, Universit de Laulsanne, Bititment de Biologie, CH-IOIS Laiusanntie, Swvitzerland,1 and Max
Planck
Institlitefor Biophysital
Chemistry,
D-3400Glittingen, Feder-al
Republic
o(
Ge2,nanv
Received 2 August 1983/Accepted 13 September 1983
Cell-free translation of total RNAisolated from vaccinia virus-infected cells late in infection results ina
complex mixture of polypeptides. A monospecific antibody directed against one of the major structural
proteins of the virus particle immunoprecipitated a single polypeptide with a molecular weight of 11,000
(11K) from this mixture. Immunoprecipitation was therefore used to identify the structural polypeptide
amongthe in vitro translation products ofRNApurified by hybridization selectiontorestriction fragments
of the vaccinia virus genome. This allowed us to map the mRNA coding for the 11K polypeptide to the
extreme left-hand end of the HinidIII E fragment. Detailed transcriptional mapping of this region ofthe genomebynucleaseS1 analysis revealed thepresenceofalate RNAtranscribed from the rightward-reading
strand. Its 5' end mapped at ca. 130 base pairs tothe left ofthe HinidIII site at thejunction between the HindIll Fand Efragments. Themapposition ofthis RNAcoincided precisely with themap position of the
late message codingfor the 11K polypeptide.
Vaccinia virus is a complex animal virus with a large double-stranded DNA genome of ca. 180 kilobase pairs (reviewed in reference 26). Expression of thislarge amount
of genetic informationoccursin atemporally tightly
regulat-ed fashion (reviewed in reference 13). A first class of genes (early genes) is transcribed shortlyafterinfectionbytheviral
RNA polymerase. After viral DNA replication, a second
class (late genes) is expressed. The molecular basis forthe
switch from early to late gene expression is not known. To learn more about the fine structure and regulation of vaccinia virus genes, detailedtranscriptionmapsof selected
regions of the genome encoding predominantly early RNAs
have been established (1; reviewed in reference 26). Based
onthese maps,fourearlygenes have beenDNA sequenced.
and their putative promoter regions upstream of the initia-tion site oftranscription were found to lack the consensus
sequencestypical ofmosteucaryotic promoters(21, 22, 24). Very little is known about vaccinia virus late genes. Extensive symmetrical transcription lateininfection and the
enormous length heterogeneity of late RNA transcripts (reviewed in reference 26) render late genes less accessible
to analysis. However, a detailed fine structure analysis of late genes is an important prerequisite for understanding generegulation in vaccinia virus.
In this communication we describe the mapping of a
vaccinia virus late gene encoding a polypeptide with a molecular weight of 11,000
(ilK).
This structural polypep-tide representsasignificant fraction of the total proteinmassof thevirion. Bycombininghybridizationselectionof RNA,
in vitro translation. and immunoprecipitation with nuclease S1 mapping, wewere able to map precisely the gene on the vaccinia virus DNA.
MATERIALS AND METHODS
Virusesand cells. Thevaccinia virus strains WR (obtained from Bernard Moss, NationalInstitutes of Health, Bethesda, Md.) and IHD (obtained from Keith Dumbell, St. Mary's Hospital Medical School, London) were used in this study.
*Corresponding author.
HeLa cells were grown in suspension in Eagle minimal essential medium(Spinner modification) supplementedwith
10%horse serum. Infection of cells and purification ofvirus
have been described (10, 12). Rabbit kidney cells (RK-13) were grown asmonolayer culturesin Eagle minimal essential medium supplemented with 5% fetal calf serum. Cells were infected with 10 PFU of vaccinia virus per cell and then maintained in medium containing 2% fetal calf serum.
Labeling of cellsandpreparationof celllysatesfor
immuno-precipitation. For in vivo labeling of polypeptides. the growth medium of infected cell monolayers was removed and replaced with medium containing 1/10 of the regular methionine concentration and 10 pCi/ml of[355]methionine (specific activity, 800 Ci/mmol). Afterbeing labeledfor1 h, the cells were washed with phosphate-buffered saline and
lysed by the addition of0.1 ml per
106
cells of 50mMTris-hydrochloride (pH 7.5)-150mM NaCI-1 mM
phenylmethyl-sulfonyl fluoride-0.1 mM 2-mercaptoethanol-1% Triton
X-100-0.5% sodium deoxycholate. The lysate wastransferred toacentrifuge tube, and insoluble material wasremovedby
centrifugationat10,000 xgfor 15min at4°C. Samplesof the cleared lysate were used for immunoprecipitation (see
be-low) or analyzed directly by sodium dodecyl sulfate
(SDS)-polyacrylamidegelelectrophoresis after theaddition, per 30 ,ul of lysate, of 10 pL ofasolution containing 0.3125 M Tris-hydrochloride (pH 6.8). 10% 2-mercaptoethanol, 50% glyc-erol, 0.015% bromophenol blue, and 10 pL1 of 20% SDS and then heating the sample in aboilingwater bathfor 3 min.
RNAextraction.Totalcytoplasmic RNA waspurified from infected HeLa cells by sedimentation through a CsCI
cush-ion(4,7). Early RNA was isolated at 4 h after infection from cells maintained in medium containing either 100 pug of cycloheximide per ml (cycloheximide RNA) or 40 pL1 of
cytosine arabinoside (CAR) per ml (CAR RNA) (4). Late
RNAwasextractedat 6 hafterinfection fromcells thatwere
not treated witheither inhibitor (4).
Total RNAfrom infectedRK-13 monolayers wasisolated by a procedure communicated by Hans Koblet (University of Berne, Berne, Switzerland). The procedure is described for one culture flask with a
175-cm2
growth area. Infected cells were washed once with ice-cold phosphate-buffered371
on November 10, 2019 by guest
http://jvi.asm.org/
372 WirTEK, HANGGI, ANDHILLER
saline and then lysed by the addition of 9 ml of 0.02 M sodium acetate-8 M guanidinium hydrochloride-0.1 M 2-mercaptoethanol (pH 5). The viscous solution was trans-ferred to a Dounce homogenizer and homogenized with 10 strokes of a tight-fitting pestle. RNA was then precipitated selectively bythe addition of 4.5 ml of absolute ethanol, and the solution was allowed to stand at -20°C for at least 2 h. After centrifugation, the pellet was washed twice with absolute ethanol and then dissolved in 4.5 ml of 50 mM sodium acetate (pH 5)-10 mM EDTA-1% SDS-200 xLg of proteinase K per ml. The solution was incubated at 37°C for 1 h. The RNA was then extracted several times withphenol, followed by several extractions with chloroform, and then precipitated with ethanol.
Hybridization selection of RNA. Approximately 20 .tg of recombinant plasmid DNA carrying cloned vaccinia DNA restriction fragments was cleaved withHinidIlI,denatured in alkali, and immobilized on nitrocellulose filters as described (5). Alternatively, DNArestriction fragments were separat-edby agarose gel electrophoresis and transferred to nitrocel-lulose sheets by blotting (19). Total cytoplasmic RNA (200
jig)
washybridized to 10[tg
ofimmobilized DNA essentiallyas described (5), except that all hybridizations were per-formedat42°C for6to 18 h. Afterhybridization, filters were washed three times at room temperature with 5-ml portions of10 mMTris-hydrochloride (pH 7.6)-0.1% SDS and three times with 10 mM Tris-hydrochloride (pH 7.6). Filters were thenwashed twice for 15 min each with80% formamide-0.4 M NaCI-0.04 M sodium-PIPES [sodium piperazine-N,N'-bis(2-ethanesulfonic acid)] (pH 6.4)-0.001 MEDTA at420C.
RNA was eluted from the filters with 90% formamide-0.04
*'iYR i1 LJ
;ielj Si e I.,SrA R ,.i e
t i XL "I
4_ - 3 u)
do*
FIG. 1. lmmunoprecipitation of the 11K polypeptidefrom
vac-ciniavirus-(strainWRorIHD)infected ells. Cellsweremaintained inmediumcontaining CARor inmedium without the inhibitor(late)
and labeled for 1 h with[35S]methionine at3 and 4h, respectively, after infection.Total celllysateswereanalyzedeitherdirectly (lanes t)orafterimmunoprecipitation (lanes i)with the anti-ilKantibody. A fluorograph of a 20% polyacrylamide gel is shown. Lane m,
molecularweights(x103) of polypeptides usedassize markers.
FIG. 2. Immunoprecipitation of in vitro translation products. Late RNA was isolated from vaccinia virus- (strain IHD orWR) infected cells and translated in vitro. Samples of the incubation mixturewereanalyzed eitherdirectly(lanes t) orafter immunopreci-pitation(lanes i)or mock-immunoprecipitation (lanes-). A fluoro-graph of a 20% polyacrylamide gel is shown. Lane m, molecular weightmarkers.
M sodium-PIPES (pH 6.4)-0.001 M EDTA by heating at
65°Cfor 10min and then wasalcoholprecipitatedwith 2.25
p.g
of calf liver tRNA (Boehringer, Mannheim, Federal Republic of Germany) as carrier after sodium acetate wasaddedto afinalconcentrationof 0.2 M.The precipitatewas
washed twice with 75%cethanol and dried.
In vitrotranslation. Amicrococcal nuclease-treated rabbit
reticulocyte lysate was prepared and used as described in detailby Jackson and Hunt(9). Invitrotranslationreactions contained [3S]methionine(specific activity, 800Ci/mmol)at afinalconcentration of1
pCi/>il
andtotalcytoplasmic RNA(finalconcentration, 100
pLg/ml).
RNAselectedbyhybridiza-tionwastranslated afterthedried RNApelletwasdissolved
directly in 20
RI
of the in vitro translation mixture.Immunoprecipitation. A monospecific antibody directed
againstamajorstructural
polypeptide
wasused inthisstudy.
Preparation and characterization of this
antibody
has been described in detail (8). Cleared cell lysates(see above)wereused directly forimmunoprecipitation. For
immunoprecipi-tation of polypeptides made in vitro, the translationreac-tionswere firstdiluted with10to20volumes of bufferA
(50
mM Tris-hydrochloride [pH 7.5], 150 mM NaCI, 1 mM
phenylmethylsulfonyl fluoride, 0.1 mM
2-mercaptoethanol,
1%7Triton X-100). Two tothree microliters of antiserum(or water for controls) was then added, and the mixture was
rotated end overendat4°Cforca. 15h. Forty microliters of
a 50%o protein A-Sepharose
(Pharmacia,
Uppsala,
Sweden)
slurry in bufferA wasthenadded,and mixingwascontinued for 2 h at room temperature. Theprotein A-Sepharose
washarvested by centrifugation and washed three times with 1-ml nortions of buffer A, three timeswith buffer B (same as
buffer A but containing 300 mM NaCI), and
finally
threetimeswith20mM
Tris-hydrochloride (pH 6.8)-80
mMNaCl.J. VIROL.
:- & i,. -il. -.
as
ro-wo
00
in
40
4w
-:.,z
it
W
'AMi--MkdlA..
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.340.541.54.318.2] [image:2.612.89.282.400.649.2]VOL.49,1984~~~~~~~MAPPINGOF AVACCINIA VIRUS LATE GENE 373
After the last wash,the
protein
A-Sepharose
wassuspended
in
sample
buffer(0.0625 MTris-hydrochloride [pH 6.8],
4%SDS, 2%
2-mercaptoethanol,
10%glycerol,
0.003%bromo-phenol
blue) and heated in aboiling
water bath for 3 mmn.The supernatant was recovered after
centrifugation.
Immunoprecipitated
materialwasanalyzed by
electropho-resis on 20%
SDS-polyacrylamide gels.
Afterelectrophore-sis,
gels
were treated with either En3Hance
orEnlightning
(New
England
NuclearCorp.,
Boston, Mass.)according
tothe instructions of the
manufacturer, dried,
andexposed
toprefogged
Kodak X-Omat R film at -70'C.NucleaseSI
analysis.
Nuclease Sianalysis
(3)wascarriedout as described
previously
(25). Various DNA restrictionfragments
were labeledat their 5' ends with[-y-32
PIATPand T4polynucleotide
kinase to map the 5' ends oftranscripts
(23).After
hybridization
andnuclease 51digestion,
resistantfragments
wereanalyzed by electrophoresis
on eitherneu-tral agarose
gels
orsequencing polyacrylamide gels
(16).Molecular
cloning.
An almostcomplete
library
of cloned Hindlll restrictionfragments
was obtained from Bernard Moss(National Institutes ofHealth). Restrictionfragments
derived from this recombinant DNA were subcloned inpBR322
andpropagated
in Escherichia coli K-12 HB101 cellsby
standardprocedures.
AClal restrictionfragment
ofparticular
interest (seebelow)
was cloned fromgenomic
vaccinia DNA. Bacteriacontaining
the desiredplasmid
wereidentified
by
colony hybridization
(20)withaDNAfragment
32P-labeled by
nick translation (15) as aprobe.
RESULTS
Immunoprecipitation
ofpolypeptides synthesized
invivo. Amonospecific antibody
directedagainst
a vaccinia virus structural 11Kpolypeptide
hasrecently
been described in detail (8). Since many structuralpolypeptides
are made late ininfection, thisantibody
appeared
to beparticularly
useful for themapping
ofavaccinia virus lategeneby
the strategyoutlined below. To confirm that the 11K
polypeptide
is indeedexpressed
as alateviralfunction, cellswere infected withtwostrainsof vacciniavirus,and thepolypeptides
werelabeled with
[35S5]methionine
for 1 h at 5 h after infection. Parallel infected cultures were maintained in mediumcon-taining
CAR, an inhibitor of DNAsynthesis.
Under theseconditions,
only early
polypeptides
are made. Cells treated with the inhibitor were labeled for 1 hstarting
at 3 h after infection. Afterbeing
labeled, the cellswerelysed,
and thepolypeptides
wereanalyzed
eitherdirectly (Fig.
1,lanest)orafter
immunoprecipitation
with theanti-ilKantibody
(lanes i). Cells labeled in the presence orabsence of CARyielded
thetypical
patterns (14) ofearly
and latepolypeptides,
respectively.
No band wasdetected after immunoprecipita-tion of theearly
polypeptides.
Incontrast, immunoprecipita-tion ofpolypeptides
labeled in the latephase
of infection resulted inanintense bandattheexpected position,
and this band was seen with both virus strains used. Apolypeptide
withavery similarelectrophoretic mobility
waspresentas amajor
band in thenonimmunoprecipitated samples.
Thispolypeptide
is agood
candidate for the 11K structuralpolypeptide
since theamountofimmunoprecipitated
materi-al loadedontothegel
wasequivalent
toonly
approximately
four times the amount of total cell extract present in thenonimmunoprecipitated samples
(lanes t).The 11K polypep-tideis thus made inlarge
amountslate ininfection. Theslight
difference inmobility
wasprobably
due to thedetergents
whichwere usedto solubilize the cells, which werepresent in thetotalpolypeptide samples
butwhichwerepresumably
removed
during
extensivewashing
of theimmunoprecipi-tate.
Immunoprecipitation
of in vitro translation products. The strategy formapping
the genecoding
for the 11K polypep-tide involvedimmunoprecipitation
of the in vitro translationproduct.
Todetermine whether theanti-ilKantibody
indeed detects such a target, total RNA was isolated at 7 h after infection from cells infected with either strain WRorIHDof vaccinia virus. This RNA was translated in vitro, and theproducts
wereanalyzed
directly
(Fig.
2, lanes t), afterimmunoprecipitation
(lanes i),orafter mock-immunoprecipi-tation without the addition ofantibody
(lanes-). As expect-ed, acomplex
polypeptide
pattern was obtained upon in vitro translation of total late RNA, and no bands weredetected after
mock-immunoprecipitation
ofparallel
sam-ples.
Afterimmunoprecipitation,
apolypeptide
of theex-pected
sizewasobserved. Theamountof immunoprecipitat-ed materialanalyzed
on thegel
wasequivalent
totwice theamount of total in vitro translation
products
present in thecorresponding
lanes. Asjudged
from the intensities of thebands, the 11K
polypeptide
appears to be made inlarge
amounts in
reticulocyte lysates programmed
with total late RNA.In vitro translation of RNA selected onHindlll restriction
m a b 1 2 3 E 0 1G m
200
-92.5
-69 -
I
46
-3i
I
U
30- "
143 -
411
a6
2 3
C NMK F Eo .i.JH 0 B
FIG. 3. Immunoprecipitation of in vitro translation products of RNA selected on HindllI restriction fragments. Polypeptides syn-thesized in vitroby late RNApurified by hybridization selectionto
pools of cloned Hinzdlll fragments (lanes 1. 2. and 3) and to individual Hindlll fragments (lanes E, 0. 1. and G) were immu-noprecipitatedandanalyzed by polyacrylamidegelelectrophoresis.
Total unselected RNA was translated in vitro, and the products
wereeitherimmunoprecipitated(lane b)or
mock-immunoprecipitat-ed(lane a)as controls. The molecular weights (X103)of polypep-tides used as size markers (lanes m) are indicated. The Hindlll restrictionmapof vaccinia(strainWR) DNA(6. 11) is shownatthe bottom of the figure. kb, Kilobases.
VOL.49, 1984
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.318.557.333.619.2]374 WITTEK,
HANGGI,
ANDHILLERaT1WI b 2 3 4 5
"I) -~
.~~~~~~~~- 4
FIG. 4. Gelanalysis ofimmunoprecipitated in products of RNA selected on restriction fragments fragment. The Clal restriction fragments of the -wereseparated by agarose gelelectrophoresis. tra cellulose membrane, and used to select RNA t Lanes 1 to 5 show the immunoprecipitated polyr
after in vitro translation of RNA selected on th( indicated at the bottom of the figure. The samples order of decreasing size(from left to right) of th( fragments used for hybridization selection. Note,I leftmost Cl/alfragment of 600 bp was blotted asa containing almost the entire pBR322 DNA. For immunoprecipitation; lane b, mock-immunopreci legendtoFig. 3. Lanes m,Size markers and theirn (X103). kb, Kilobase.
fragments. We used a combination of hybr
tion, in vitro translation, and
immunoprecil
the gene coding for the 11K polypeptide c
virus genome. For technical reasons, hybridi was performed with RNA isolated from cel strain IHD of vaccinia virus and cloned DN
strain WR. However, for nuclease Si analy
homologoushybridizations with RNAofstra
comparison, with RNAofboth strains were In thefirstexperiment, RNAfrom vaccini cells isolatedat7 hafter infection(lateRNA) hybridization selection to cloned HindIII E
fragments. To keepthe numberofsamples
small, three nitrocellulose filters, with the recombinantplasmids immobilizedoneach,N
first screening. Selected RNA was translate
the productswere immunoprecipitated with fic antibody directed against the 11K polyp, lyzed by polyacrylamide gel electrophoresiV
control, total unselected RNAfrom infected lated in vitro,and the productswere either
mq
tated with the antibody or mock-immunoprecipitatedwith-2 out the addition of antibody but otherwise with the same 2
_ manipulations as for immunoprecipitation. As expected, a polypeptide with a molecular weight of ca. 11,000 was
-S
- 2precipitated
from an in vitro translation mixture of totalRNA(lane b), but no band was detected in the absence of the antibody (lane a).
ej 46 When RNA selected by
hybridization
to thepooled
HindIII restrictionfragments N, M, K, and F (Fig. 3, lane 1) and L, J, H, and D (lane 3) was translated in vitro, no
:30
polypeptide
wasimmunoprecipitated
with the anti-ilK anti-body. Incontrast,RNA selectedon a mixtureof the HindlIl fragments E, 0, I, and G (lane 2) directed the synthesis of the 11Kpolypeptide.RNA wastherefore selected on eachof the Hindlll fragments E, 0, 1, and G individually and translated in vitro, and the polypeptides were immunopre-cipitated. Onlythe HinzdlIl Efragment selected the message- '4 Jfor the 11K
polypeptide
(lane E).In vitro translation of RNA selected on ClaI restriction fragments. Forfinermapping of the gene coding for the 11K
polypeptide, the Clal restriction sites were mapped within the HinidlIl E fragment (Fig. 4). The recombinant plasmid containing the HiidIlll E fragment was cleaved with Clal, andthefragmentswereseparated by gel electrophoresis and 2- blotted ontonitrocellulose membrane. Strips containing indi-vidual restriction fragments were cut from the membrane, andthesefragmentswere usedtoisolate RNAby
hybridiza-tionselection. Total unselectedRNA wastranslated, and the products were either immunoprecipitated (Fig. 4, lane a) or
ivitro translation
of theHindlll E
Hindlll fragment m a b 1 2 3 m
Lnsferred to
nitro-by hybridization. )eptides obtained
e Clal fragments 25
were arrangedIni
e Clal restriction _
however, that the hybrid fragment
controls (lane a. --46
ipitation) see the nolecularweights
idization selec-pitation to map
)n the vaccinia
zation selection Is infected with
[Afragments of
sis(see below),
in WRor, fora
carried out. avirus-infected
waspurified by
)NA restriction
to be processed
DNA of four wereused inthe
Id in vitro, and the
monospeci-eptide and
ana-s(Fig. 3). As a cells was
trans-
Immunoprecipi-.2 D.. -- _w
(,1>I. -- _
I
-.U
- 3
2 3
- - ;30
d. -1
FIG. 5. Immunoprecipitation of in vitro translation products of RNA selected on BamnHI restriction fragments of the Hindlll E fragment. The three BarnHI fragments of the Hindlll Efragment wereused topurify late RNA byhybridization selection. The RNA was translated in vitro, and the products were immu-noprecipitatedandanalyzedbypolyacrylamide gelelectrophoresis.
Lanes 1,2,and 3 show theproducts obtained byRNA selectedon the corresponding fragments (bottom of the figure). For controls (lane a, immunoprecipitation; lane b, mock-immunoprecipitation)
seethelegendtoFig.3. Lanesm,sizemarkers and their molecular weights(x103) kb. Kilobase.
J. VIROL.
i.3o -- 41M
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.66.303.77.356.2] [image:4.612.360.528.382.627.2]MAPPING OF AVACCINIA VIRUS LATE GENE 375
U1)
U) -D
2000-
1000-
500-
250-t 2 3
m p t e I p t e I p t e I
-0~
F
z
i
4
p t e I m
m
L
E
(U
E
m
(u
L- ~~~~~~~~~~~VIl~
2 3
4
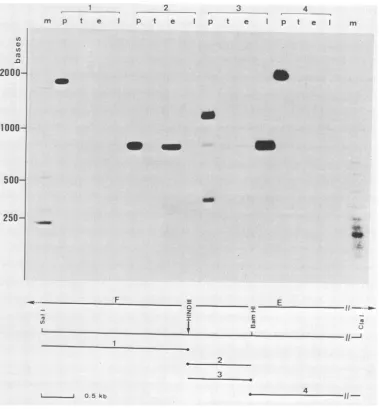
FIG. 6. Fluorographofelectrophoreticallyseparated nucleaseSi-resistantDNA-RNAhybrids. DNA fragmentsderivedfrom theHindill Eand Ffragments(see bottompartofthe figure)werelabeled at the 5' end (solid circle). Each probe( to4) was either analyzed directly (lanesp) orafter hybridizationtotRNA (lanes t) and to early (lanes e) and late (lanes 1) RNA from vaccinia virus- (strain WR) infected cells. After hybridization, samples were treated with
Si
nuclease, and resistant material was resolved byelectrophoresis on a neutral2%agarose gel.m, Molecularweightmarkers. kb. Kilobase.mock-immunoprecipitated (lane b)as acontrol. The invitro
translationproducts ofRNA selectedontheClaI restriction fragments were immunoprecipitated and analyzed on the same gel (lanes 1 to 5). Significant amounts of the 11K
polypeptidewere onlydetected when
polypeptides
madebyRNA selectedonthe leftmost ClaI fragment were
immuno-precipitated (lane 1).
In vitro translation of RNA selected on cloned BamHI
restriction fragments. To confirm this result with cloned
DNA fragments, three BarnHI restriction fragments, one of
ca. 800basepairs (bp)andtwowithaverysimilar size ofca.
7,100 bp, were isolated from the HindIll E fragment and subcloned in pBR322. Restriction site mapping showed that the800-bpfragmentmapstotheleft-handendoftheHindlIl
E fragment. The recombinant DNA was immobilized on
nitrocellulose membranesandusedtoselectRNAby
hybrid-ization. Selected RNA was translated in vitro, and the
resulting polypeptides were immunoprecipitated and
ana-lyzedbypolyacrylamide gel electrophoresis (Fig. 5, lanes 1 to 3) together with the immunoprecipitation products of in vitrotranslated, unselectedRNA (lanea) or mock-immuno-precipitated material (lane b) serving ascontrols.
Asexpected, onlythe ca. 800-bp leftmostHindIII-BarnHI
fragmentselected the mRNAcoding for the 11K polypeptide
(Fig. 5, lane 1).Thus, by combining hybridization selection,
in vitrotranslation,andimmunoprecipitation, it waspossible to mapthegenefor the 11K polypeptide to theextreme left hand end ofthe HizdIlll E fragment.
Mappingoftranscripts bynucleaseSI analysis.Tomapthe
gene more precisely and also to determine the direction of
transcription of the mRNA, nuclease SI analysis of RNA-DNA duplexes was performed. Various DNA fragments
labeled atthe 5' end were used ashybridizationprobes (Fig. 6). Each end-labeled probe was either analyzed directly (lanes p) or after hybridization with tRNA (lanes t), early
RNA(lanese),andlate RNA(lanes 1) isolated from vaccinia
VOL. 49. 1984
I --I 0 5 kb
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.123.502.77.486.2]376 WITTEK,
HANGGI,
ANDHILLERvirus-infected cells. After hybridization, single-stranded
DNA was digested with Si nuclease, and resistant material
was resolved as RNA-DNA duplexes by neutral agarosegel
electrophoresis.
With probe 1, faint bands were only detected after long
exposures ofthe gel. When probe 2 was used, an intense
band with a size very similar to that of the DNA fragment used as the probe was obtained after hybridization to early
RNA, indicatingthat an early transcript starts to therightof
the
BamHI
site and is transcribed from the leftward-readingstrand. When the same DNA fragment labeled at the
oppo-site end (probe 3) was used for hybridization, an intense
band was detected only with late RNA. The size of this
protected fragment (ca. 800 bp) was again very similar tothe size of the probe. For two reasons it is unlikely that this band
resulted from DNA-DNA reannealing followed by
incom-plete digestion with Si nuclease. First, this band was only seenafter hybridization to late RNA, not afterhybridization
to tRNA as a control or to early vaccinia RNA. Second,
probe 3 was prepared from a recombinant plasmid carrying a
BarmHlfragment which had been subcloned from aplasmid
containing the
HindlIl
E fragment. This recombinant thus contains the 800-bp fragment of vaccinia DNA fused to the375-bp fragment of pBR322 sequences from the HindlIl to theBanzHI site of the original vector. After being labeled at the
BamnHl
sites, the hybrid fragment was only partiallycleaved with Hindlll, and the entire mixture of cleaved and
uncleaved material was therefore used as the hybridization
probe (probe 3; Fig. 6, lane p). Since no reannealing of the uncleaved hybrid fragment or of the pBR322 fragment oc-curred during hybridization, the 800-bp fragment observed
after annealing to late RNA most likely resulted from
specific DNA-RNA hybridization.
Finally, a
BamHI-ClaI
fragment of ca. 2,100 bp labeled atthe
BamHI
site was used as a probe (probe 4). No bandswere observed when this probe was hybridized to late RNA. With early RNA,a faint band corresponding to ca. 1,000 bp
was seen on longer exposures of the gel (not shown). This band could represent the 5' portion of the early transcript also detected with probe 2.
The Si analysis (Fig. 6) allowed us to identify a late RNA that maps to the left-hand end of the
HindlIl
E fragment. The map position of this RNA thus agrees very well with the map position obtained by hybridization selection and in vitro translation of the late message coding for the11K polypep-tide. Furthermore, the initiation site of the RNA is located very close to theHindIll
site at the junction between theHindlIl
F andHindlll
Efragments,
and transcription occursfrom the rightward-reading strand. However, with the DNA fragments used as probes forS1 analysis,it was not possible to more precisely map the 5' end of the transcript. There-fore, a
Clal
fragment spanning the HindlIl site was cloned from genomic vaccinia DNA in pBR322. The clone was identified by colony hybridization by using as a probe the vaccinia DNA fragment from theHindlIl
site to the firstClalsite at the left-hand end of the
HinidlIl
E fragment. Two probes derived from this fragment were used for further nucleaseS1 analysis to map more precisely the 5' end of the late RNA (Fig. 7). The first probe (probe 1) consisted of the entireClal
fragment labeled at the 5' ends. Second, aTaiqI
fragment of ca. 800 bp from the leftClal site to the firstTaqI
site at 165 bp to the right of theHindlIl
site was isolated from the recombinant DNA and also labeled at the 5' ends (probe 2). Samples of each probe were analyzed either after cleavage withHindIll
(lanes g) or uncleaved (lanes f). The smallHindIII-TaqI
fragment of 165 bases} t.' c d e t cg a
2
b) ci d e 9 ni
- -622
_ -527
_ -404
_ -309
- -272 EW -238
3 -.2i7
- additconoi --I~~~~~~~~~~~~~..c t i
,-2
L 0 h [:
FIG. 7. Fluorograph of electrophoretically separated nuclease
Si-resistantDNAfragments. Two fragments (labeled1and 2on the figure) derived from a cloned Clal fragment spanning the Hindlll site at thejunction between the HindIII E and F fragment were labeled at the 5' ends and used as probes for nuclease Si analysis. Eachprobe was analyzed either uncleaved (lanes f) or aftercleavage with Hindll (lanes g). Hybridizations were performed to tRNA (lanes a) as controls and to cycloheximide RNA (lanes b), CAR RNA (lanes c), and late RNA (lanes d) isolated fromcells infected with strain WR. Each probe was also hybridized to late RNA isolated from cells infected with strain IHD of vacciniavirus (lanes e). Afterhybridization, samples were treated with SI nuclease, and resistant DNAfragments were resolved by electrophoresis ona4% polyacrylamide sequencing gel. Lane m, sizes (in bases) of DNA fragments used as markers. kb, Kilobase.
obtained upon cleavage of probe 2 with HindIll is not
shown. Hybridizations were performed with the probes that
were not digested with HindIll. Each probe was hybridized
to tRNA as a control (lanes a), to early RNA isolated from
cells infected with strain WR of vaccinia virus and treated with eithercycloheximide (lanes b)or CAR (lanesc), and to
late RNA (lanes d). For acomparison, each probe was also hybridized to late RNA isolated from cells infected with
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.318.560.72.493.2]MAPPING OFAVACCINIA VIRUS LATE GENE 377
strain IHD (lanes e). With cycloheximide RNA hybridized to probe 1, an Si-protected fragment of ca. 500 bases was detected (lane b). A much fainter band was also seen with CAR RNA (lane c) after longer exposures ofthegel. Similar bands were obtained after hybridization of early RNA to probe2, indicating that an early RNA is transcribed from the leftward-reading strand since thetwoprobes haveacommon left-hand terminus. When probe 1 was hybridized to either WR or IHD late RNA, SI-protected fragments ofca. 720 baseswereobtained. (The intensities of the bands cannot be directly compared since hybridizations with the late RNAs wereperformed on different occasions with different prepa-rationsof the probe.) If the observedfragment resulted from
hybridization of the late RNA transcribed from the
right-ward-reading strand, the 5' end would map at 130 bases to the left of the HindIII site, and a much shorter fragment
should be obtained with probe 2. Indeed, afragment of 300 bases was detected after hybridization of late RNA from both virus strainstoprobe2(lanes d and e). With this probe. the 5'end of the late RNAmappedat135 basestothe left of the HindIll site, which is in excellent agreement with the mapposition obtained with probe 1.
The map position of the late mRNA coding for the 11K
polypeptide is shown in Fig. 8.
DISCUSSION
In contrast to the steadily growing information onthefine
structure ofvaccinia virus early
genes.
verylittle is known about late genes. This ismainly because thefirst regions of the genome that were chosen for detailed transcriptionalmappingencoded veryfew late transcripts and probably no
major ones (reviewed in reference 26). In addition, two
unusualproperties of late RNAs (reviewedin reference 26) renderlate genes lessaccessibletoanalysis by conventional
procedures such as RNAblotting and nuclease S1 analysis
with uniformly labeled DNA as probes. First, late RNAs are extremely heterogeneous inlength,presumablyas aresultof
incorrect termination oftranscription late in infection. Sec-ond, a large fraction of late RNA is capable of forming double-stranded RNA uponself-annealing. Thus. in all
map-ping procedures involving DNA-RNA hybridizations with
RNAin solution, competing RNA-RNA annealing will take place. As a consequence, if late RNAs are purified by
hybridization selection and translated in vitro, the intensity
of a particular band may not reflect the abundance of the
corresponding message since significant amounts may be lost as RNA-RNA duplexes during selection.
Structuralpolypeptides aregood candidates for late
poly-peptides. However, such genes cannot be mapped simplyon
the basisof comigration of in vitro translation products with authentic structural polypeptides since many of these are knowntoundergoposttranslational modifications (13), most of which will not be performed by the in vitro translation system.
F Cla
E
Z s, ...
Taq additionalTaq sitesCla
I
Immunoprecipitationwith antibodies directed against
ma-jor structural polypeptidesincombination with hybridization selection and in vitro translation offers a possibility of mapping representative late genes despite the problems mentioned above.
In this study we used an antibody directed against a
structural polypeptide witha molecular weight of 11,000 to
map alate gene on thevaccinia virus DNA. This biologically interesting polypeptide is exposed on the surface ofmature
virus particles and interacts with actin-containing cytoskele-tal elements of the infected cell (8). The antibody appeared
to be particularly useful for our mapping study mainly for
two reasons. First, the antibody has been characterized in
great detail and shown by two-dimensional immunoblotting torecognize a single target and thus to be monospecific (8). Second, the 11K polypeptide contributes ca. 10%tothe total protein mass present in purified virions (13, 17). We there-fore assumed that the mRNA would be present in large
amounts in infected cells and thus facilitate mapping of the
gene. Indeed, from quantitative aspects of in vitro transla-tion and immunoprecipitatransla-tion experiments, the 11K
poly-peptideappears to be a major product made in response to
total unselected late RNA.
As expected for a structural protein, significant amounts
of the 11Kpolypeptide were immunoprecipitatedfrom cells labeled in the late phase ofinfection only and not from cells treated with CAR, in which late viral functions are not
expressed.
Bycombininghybridization selection, in vitro translation. and immunoprecipitation, we were able to map the gene
codingfor the11K structuralpolypeptidetotheextreme
left-hand end of the HindIll E fragment. A late polypeptide of similar size has previously been detected after in vitro
translation ofRNA selected on the HinidIll E fragment (2).
Finermapping ofthe genewasaccomplished by nuclease S1 analysis with 5' end labeled DNA fragmentsasprobes. The message wasfound to betranscribedfrom left torightandto
initiate within the adjacent Hindlll Ffragment atca. 130bp
totheleft of the
HinidIII
site. Because of the size heterogene-ity of late transcripts, we made no attempt to map the 3' endofthemRNA.However, from the size ofthepolypeptidethe length of the coding sequence should not exceed ca. 300 nucleotides. Interestingly,anearlyRNA istranscribed in the
opposite direction from the same regionof the genome.
Sincethe gene mapswithin aregionof the genome that is
highly conserved between poxviruses (11, 18), it is not
surprising that the 11K polypeptide is immunoprecipitated
fromcells infected with both vaccinia virus strains used in this study.
We are currently sequencing the late gene and its 5'
flanking region to compare its putative promoter sequence with corresponding sequences of early genes
(21.
22. 24).ACKNOWLEDGMENTS
Wethank Keith Dumbelland Bernard Mossfor virus strains and recombinantDNAs.John Knowlandfor preparing the rabbit reticu-locytelysate. LenArchard.Jacques-Edouard Germond. and Walter Wahli for critically reading the manuscript, and Beatrice ten Heg-gelerfor help in preparing the illustrations.
This work was supported by grant 3.048-0.81 from the Swiss National Science Foundation.
11K
I 01 kb
FIG. 8. Map position and direction of transcription of the late mRNA coding forthe 11Kstructuralpolypeptide. kb. Kilobase.
LITERATURE CITED
1. Bajszar, G., R. Wittek, J.P.Weir,and B.Moss. 1983.Vaccinia virus thymidine kinase and neighboring genes: mRNAs and polypeptides ofwild-typevirusandputativenonsense mutants. VOL.49, 1984
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.59.298.629.708.2]378 WITTEK, HANGGI, AND HILLER
J. Virol. 45:62 72.
2. Belle Isle, H., S. Venkatesan, and B. Moss. 1981. Cell-free translation of early and late mRNA's selected by hybridization to cloned DNA fragments derived from the left 14 million to 72 milliondaltons of the vaccinia virus genome. Virology 112:306-317.
3. Berk, A. J., and P. A. Sharp. 1977. Sizingand mapping of early adenovirusmRNA's by gelelectrophoresisofS1 endonuclease-digested hybrids. Cell 12:721-732.
4. Boone, R. F., and B. Moss. 1978. Sequence complexity and relative abundance of vaccinia virus mRNA's synthesized in vivo and in vitro. J. Virol. 26:554-569.
5. Cooper, J. A., R. Wittek, and B. Moss. 1981. Hybridization selection and cell-free translation of mRNA's encoded within theinvertedterminalrepetitionofthevacciniavirus genome. J. Virol. 37:284-294.
6. DeFilippes, F. M. 1982.Restrictionenzymemapping of vaccinia virus DNA. J. Virol. 43:136-149.
7. Glisin, V., R.Crkvenjakow, and C. Byus. 1974.Ribonucleic acid isolated by cesium chloride centrifugation. Biochemistry 13:2633-2637.
8. Hiller, G., and K. Weber. 1982. Aphosphorylated basicvaccinia virion polypeptide of molecular weight 11,000 is exposed on the surface of matureparticles and interacts with actin-containing cytoskeletal elements. J. Virol. 44:647-657.
9. Jackson, R. J., and T. Hunt. 1983. Preparation and use of nuclease-treatedrabbit reticulocytelysatesfor thetranslationof eukaryotic messenger RNA. Methods Enzymol. 96:50-74. 10. Joklik, W. K. 1962. The preparation and characteristics of
highly purified radioactively labelled poxvirus. Biochim. Biophys. Acta 61:290-301.
11. Mackett, M., and L. C. Archard. 1979. Conservation and variation in orthopoxvirus genome structure. J. Gen. Virol. 45:683-701.
12. Moss,B. 1968. Inhibition ofHeLacellprotein synthesis bythe vaccinia virion. J. Virol. 2:1028-1037.
13. Moss, B. 1974. Reproduction of poxviruses, p. 405-473. In H. Fraenkel-Conrat and R. R.Wagner(ed.).Comprehensive virol-ogy, vol. 3. Plenum Publishing Corp., New York.
14. Pennington, T. H. 1974. Vacciniavirus polypeptide synthesis:
sequential appearance and stability of pre- and postreplicative polypeptides. J. Gen. Virol. 25:433-444.
15. Rigby, P. W. J., M.Dieckmann, C. Rhodes, and P. Berg. 1977. Labelling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase 1. J. Mol. Biol. 113:237-251.
16. Sanger, F., and R. Coulson. 1978. The use of thin acryl-amide gels for DNA sequencing. FEBS Lett. 87:107-110. 17. Sarov, I., and W. K. Joklik. 1972. Studies on the nature and
location of the capsid polypeptides of vaccinia virions. Virology 50:579-592.
18. Schumperli, D., A. Menna, F. Schwendimann, R. Wittek, and R. Wyler. 1980. Symmetrical arrangement of the heterologous regions of rabbit poxvirus and vaccinia virus DNA. J. Gen. Virol. 47:385-398.
19. Southern, E. M. 1975. Detection of specific sequences among DNAfragments separated by gel electrophoresis.J. Mol. Biol. 98:503-517.
20. Thayer, R. E. 1979. Animprovedmethodfordetectingforeign DNAinplasmids of Escherichia coli.Anal.Biochem. 98:60-63. 21. Venkatesan, S., B. M. Baroudy, and B. Moss. 1981. Distinctive nucleotide sequences adjacentto multipleinitiation and termi-nationsitesofan earlyvaccinia virus gene. Cell 125:805-813. 22. Venkatesan, S., A. Gershowitz, and B. Moss. 1982. Complete
nucleotidesequencesoftwoadjacent early vacciniavirusgenes locatedwithin the invertedterminalrepetition.J.Virol. 44:637-646.
23. Weaver, R. F., and C. Weissman. 1979. Mappingof RNAby a modification ofthe Berk-Sharp procedure: the 5' termini of 3-globin mRNA have identical map coordinates. Nucleic Acids Res. 7:1175-1193.
24. Weir, J. P., and B. Moss. 1983. Nucleotide sequence of the vaccinia virus thymidine kinase geneand thenatureof sponta-neousframeshift mutations. J. Virol.46:530-537.
25. Wittek, R., J. A. Cooper, E. Barbosa, and B. Moss. 1980. Expression ofthevaccinia virusgenome:analysisandmapping ofmRNA's encoded within the inverted terminal repetition. Cell21:487-493.
26. Wittek, R. 1982. Organization and expressionof the poxvirus genome. Experientia38:285-297.
J. VIROL.