JOURNAL OFVIROLOGY, Aug. 2007, p. 8165–8179 Vol. 81, No. 15 0022-538X/07/$08.00⫹0 doi:10.1128/JVI.02792-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Conserved Changes in Envelope Function during Human
Immunodeficiency Virus Type 1 Coreceptor Switching
䌤
Cristina Pastore,
1Rebecca Nedellec,
1Alejandra Ramos,
1Oliver Hartley,
2John L. Miamidian,
3Jacqueline D. Reeves,
3‡ and Donald E. Mosier
1*
Department of Immunology, The Scripps Research Institute, La Jolla, California1; Department of Structural Biology and
Bioinformatics, University of Geneva, Geneva, Switzerland2; and Department of Microbiology,
University of Pennsylvania, Philadelphia, Pennsylvania3
Received 18 December 2006/Accepted 5 May 2007
We studied the evolution of human immunodeficiency virus type 1 (HIV-1) envelope function during the process of coreceptor switching from CCR5 to CXCR4. Site-directed mutagenesis was used to introduce most of the possible intermediate mutations in the envelope for four distinct coreceptor switch mutants, each with a unique pattern of CCR5 and CXCR4 utilization that extended from highly efficient use of both coreceptors to sole use of CXCR4. Mutated envelopes with some preservation of entry function on either CCR5- or CXCR4-expressing target cells were further characterized for their sensitivity to CCR5 or CXCR4 inhibitors, soluble CD4, and the neutralizing antibodies b12-IgG and 4E10. A subset of mutated envelopes was also studied in direct CD4 or CCR5 binding assays and in envelope-mediated fusion reactions. Coreceptor switch intermediates displayed increased sensitivity to CCR5 inhibitors (except for a few envelopes with mutations in V2 or C2) that correlated with a loss in CCR5 binding. As use of CXCR4 improved, infection mediated by the mutated envelopes became more resistant to soluble CD4 inhibition and direct binding to CD4 increased. These changes were accompanied by increasing resistance to the CXCR4 inhibitor AMD3100. Sensitivity to neutralizing antibody was more variable, although infection of CXCR4-expressing targets was generally more sensitive to neutralization by both b12-IgG and 4E10 than infection of CCR5-expressing target cells. These changes in envelope function were uniform in all four series of envelope mutations and thus were independent of the final use of CCR5 and CXCR4. Decreased CCR5 and increased CD4 binding appear to be common features of coreceptor switch intermediates.
The high genomic variability of the primate lentiviruses, particularly in the envelope region that determines virus inter-action with target cells, is probably responsible for the multiple cross-species transmissions of human immunodeficiency virus type 1 (HIV-1) and HIV-2 to humans (18). Envelope variabil-ity in HIV-1 is also responsible for the change in coreceptor preference from CCR5 to CXCR4 that occurs in about 50% of chronically infected humans (5, 36). In each instance, evolution of envelope protein function must occur to allow infection of a new species or a new target cell. Comparison of primate and human immunodeficiency viruses suggests that the primate viruses are more dependent on CCR5 and less dependent on CD4 than HIV-1, and use of CXCR4 by primate lentiviruses is exceedingly rare (reviewed in reference 21). While the se-quence correlates of HIV-1 coreceptor switching are known, the functional consequences for the evolution of protein func-tion are less clear. Sequential virus isolates from patients be-fore and after coreceptor switching identify the starting sub-strate and the highly selected final products, but not the intermediate steps that might pose the greatest fitness chal-lenges (37, 39). One can anticipate that increased binding to
CXCR4 is a necessary step in the evolution of envelope during coreceptor switching, but loss of CCR5 binding may or may not occur. Moreover, it is not clear that there is one evolutionary pathway from CCR5 to CXCR4 use. A combination of sto-chastic selection factors, including antibody and cytotoxic-T-lymphocyte responses directed at the envelope (14), target cell selection (56), and chemokine levels influenced by both genetic factors (17), and concurrent infections (58) may influence the probability of emergence of CXCR4-using variants.
We have reconstructed four potential pathways for HIV-1 envelope evolution from CCR5 to CXCR4 use by using site-directed mutagenesis to generate most of the possible inter-mediates separating parental R5 sequences from R5X4 or X4 variants that were generated by propagating infectious clones of R5 BaL or ADA isolates on target cells expressing only CXCR4 (37, 39). These four examples were chosen for de-tailed analysis because the coreceptor switch variants displayed different phenotypes, ranging from robust entry mediated by either CCR5 or CXCR4 to selective use of CXCR4 only. In the present study, we evaluated the impacts of these mutations in the envelope on interactions with CCR5, CD4, and CXCR4, as well as sensitivity to two broadly neutralizing antibodies, b12-IgG (6) and 4E10 (7). We present evidence that envelope evolution toward CXCR4 use is accompanied by diminished binding to CCR5, increased binding to CD4, and slowly in-creasing use of CXCR4. Sensitivity to neutralizing antibody is more variable, but each pathway of envelope evolution con-tains members that are significantly more sensitive to neutral-ization than parental R5 envelopes.
* Corresponding author. Mailing address: Department of Immunol-ogy—IMM7, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. Phone: (858) 784-9121. Fax: (858) 784-9190. E-mail: [email protected].
‡ Present address: Monogram Biosciences, 345 Oyster Point Blvd., South San Francisco, CA 94080.
䌤Published ahead of print on 16 May 2007.
8165
on November 8, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Cell lines.U87-CD4-CCR5 and U87-CD4-CXCR4 cells (4, 11) were main-tained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal
bovine serum, 1g of puromycin/ml, and 300g of G418/ml (in cell lines used
at Scripps). 293T, NP2 (59), NP2/CD4 (53), QT6, U87/CD4/CCR5 (4, 11), and T-REx/CCR5 (46) cell lines were cultured in Dulbecco’s modified Eagle’s
me-dium supplemented with 10% fetal bovine serum, 100 U of penicillin, and 100g
of streptomycin per ml. In addition, 1 mg of G418 per ml was used to maintain
CD4 expression in NP2/CD4 cells and 200g of zeocin plus 5g of blasticidin
per ml was used to maintain CCR5 and Tet repressor genes in T-REx/CCR5 cells (in cell lines used at the University of Pennsylvania). High-level CCR5 expres-sion was induced in T-REx/CCR5 cells by the addition of 10 ng of doxycycline (Sigma) per ml to the culture medium.
Cloning and mutagenesis. The ADA and BaL full-length envelope (Env) glycoproteins were amplified by PCR from the virus plasmids pNL4-3-ADA and
pR8-BaL (39), respectively, with two sets of primers spanning the 5⬘and 3⬘ends
of the ADA and BaLenvgenes and inserting a 5⬘SalI and a 3⬘XhoI restriction
site for ADA and a 5⬘SalI and a 3⬘HpaI site for BaL. The ADA SalI-XhoI and
the BaL SalI-HpaI fragments of the PCR products were cloned into the expres-sion plasmid pSVIII (54).
Each single mutation or combination of mutations was introduced into the cloned Env by site-directed mutagenesis (QuikChange Site-Directed Mutagen-esis Kit; Stratagene, La Jolla, CA) by following the manufacturer’s instructions. The sequence of each mutant Env was confirmed using four previously described primer pairs (39).
For fusion experiments, SalI-XhoI fragments of ADA or mutated ADA Envs were cloned into the XhoI site of the pSI expression vector (Promega) to generate gp160 expression constructs that contained both T7 and cytomegalovi-rus promoters. For binding experiments, stop codons were introduced at the gp120/gp41 cleavage junction of these constructs by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit) using GTGCAGAGAGAAAAA AGATAAGTGGGAACGATAGAAGCTTTGTTCCTTGG (forward) and CC AAGGAACAAAGCTTCTATCGTTCCCACTTATCTTTTTTCTCTCTGCAC (reverse) primers to generate gp120 expression constructs. These primers also introduced a HindIII restriction endonuclease site (underlined) downstream of the stop codon (underlined) for screening purposes. These constructs were verified by sequencing.
Entry assay.The entry efficiencies of mutated Env proteins were measured in a single-cycle pseudovirus infection assay. Mutant Env clones inserted into the pSVIII plasmid were cotransfected with Env-negative, luciferase-positive
(NL4-3–Luc⫹E⫺R⫺[10]) reporter plasmids in 293T cells, and the resulting
pseudovi-ruses were harvested, standardized for p24 content, and used to infect either U87-CD4-CCR5 cells or U87-CD4-CXCR4 cells. The luciferase activities from triplicate wells were measured on a luminometer (EG&G Berthold LB 96V; Perkin-Elmer, Gaithersburg, MD) with the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer’s instructions. Virus infectivity was compared to that of the parental R5 virus by calculating the slope of the infec-tivity curve by plotting p24 input versus light units of luciferase acinfec-tivity after 48 to 72 h of culture. The slope was calculated using linear regression analysis (Prism 4; GraphPad Software, San Diego, CA). Determination of infectivity was repeated in three replicate experiments, and the mean infectivity was compared to that of parental ADA or BaL Env by expressing the log change from the parental Env.
Inhibitors.The CCR5 inhibitor PSC-RANTES (20) or TAK-779 (2) was used to block single-cycle infection of U87-CD4-CCR5 target cells by Env-pseudotyped viruses. AMD3100 (13) was used to block single-cycle infection of U87-CD4-CXCR4 cells. CD4-immunoglobulin G (IgG) (kindly provided by Mi-chael Franti, Progenics) was used to inhibit single-cycle infection of U87-CD4-CCR5 or -CXCR4 cells. The broadly neutralizing monoclonal antibodies (nAb) b12-IgG and 4E10 were supplied by Dennis Burton and the NIH AIDS Research and Reference Reagent Program, respectively. All inhibitors were used over a broad concentration range that spanned 0 to 100% inhibition of infection, and
50% inhibitory concentrations (IC50) were calculated for each experiment using
the sigmoidal curve-fitting program in GraphPad Prism 4 (GraphPad, San Diego,
CA). Each inhibition experiment was repeated three times, and the log mean⫾
standard error (SE) of the three IC50values was calculated. For inhibition of
infection of U87-CD4-CCR5 cells, the log change in IC50values from the
pa-rental ADA or BaL Env was calculated. These data are reported over a 4-log-unit scale to facilitate comparison of different mutant Env proteins and to
accommodate the broad variation in IC50values. For infection of
U87-CD4-CXCR4 cells, the log mean IC50values⫾SE are reported, since there is no
reference parental Env that is infectious for these target cells.
Env receptor binding assays.gp120s were produced from 293T cells that were calcium phosphate transfected with gp120 expression constructs and infected with a vaccinia virus encoding T7 polymerase (vTF1.1 [1]) to drive expression from the T7 promoter. Cell culture supernatants were harvested 24 h posttrans-fection, and gp120 concentrations were determined by enzyme-linked immu-nosorbent assay as previously described (49), with the exception that gp120 was detected with an HIV-1 Env-specific rabbit serum and a horseradish peroxidase-conjugated anti-rabbit immunoglobulin antibody (Amersham Life Science),
fol-lowed by TMB substrate (3,3⬘,5,5⬘-tetramethylbenzidine; KPL). The receptor
binding efficiencies of ADA and ADA mutant gp120s were determined with a cell surface binding assay in which bound protein was detected by immunostain-ing and flow cytometry analysis, as previously described (46, 49). CD4-negative T-REx/CCR5 cells, induced to express a high level of CCR5, were used to
determine Env-CCR5 binding efficiency in the absence and presence of 5g of
soluble CD4 per ml to induce coreceptor binding site exposure. NP2 and NP2/ CD4 cells were used to determine Env-CD4 binding efficiency. Bound gp120 was detected with an HIV Env-specific rabbit serum and a phycoerythrin-conjugated anti-rabbit immunoglobulin antibody (Pharmingen).
Cell-cell fusion assay.QT6 effector cells, transfected with Env expression plasmids and infected with vTF1.1 (1), were added to QT6 target cells cotrans-fected with a luciferase reporter construct under the control of a T7 promoter (pGEM2 T7-luc; Promega), CD4 (16), and CCR5 or CXCR4 (47) expression plasmids. Cell-cell fusion, resulting from a functional interaction between the Env-expressing effector cells and receptor-expressing target cells, was detected by assaying for T7 polymerase-driven luciferase expression within the linear range of the assay. This assay has been described in detail previously (50).
Env fusion kinetics.The fusion kinetics of ADA wild-type (wt) and mutant
Envs were determined in a-lactamase reporter cell-cell fusion assay, based on
that described by Lineberger et al. and Reeves et al. (28, 48). QT6 effector cells,
cotransfected with Env and-lactamase expression constructs and infected with
vTF1.1, were added to U87-CD4-CCR5 target cells labeled with CCF2-AM as previously described (48). Cell-cell fusion was detected by assaying for a shift
from green to blue fluorescence, indicating-lactamase cleavage of CCF2.
Flu-orescence was measured with a CytoFluor Series 4000 FluFlu-orescence multiwell plate reader.
RESULTS
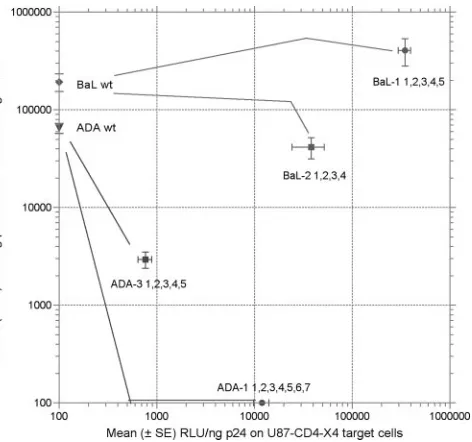
The generation of coreceptor switch mutants in vitro and an initial analysis of the impacts of different mutations in Env of the ADA- or BaL-derived variants have been described previ-ously (37, 39). Table 1 shows the sites of the mutations in HIV-1 Env for the four coreceptor switch variants examined in these studies, with each mutation site numbered sequentially from N terminal to C terminal. The mutational pathways to CXCR4 use for four independent variants were examined: ADA-1, a variant with seven mutations resulting in a pure X4 phenotype (Fig. 1); ADA-3, a variant with five mutations re-sulting in a weak R5X4 phenotype; BaL-1B, a variant with five mutations resulting in a robust R5X4 phenotype; and BaL-2A, a variant with three mutations in V2 plus V3 (Table 1) and one mutation in C5 (K490T) with a somewhat weaker R5X4 phe-notype. The assignment of entry efficiency was made by single-cycle infection of either U87-CD4-CCR5 or U87-CD4-CXCR4 target cells with molecularly cloned Env mediating entry of the reporter construct NL4-3–E⫺ Luc⫹ (10) as previously de-scribed (37). The relative entry efficiency of each final variant on both target cell lines is shown in Fig. 1. Note that the BaL Env is highly efficient at using CCR5 for virus entry, and R5X4 variants of BaL retain efficient use of CCR5 while acquiring the ability to use CXCR4. By contrast, ADA Env is somewhat less efficient at using CCR5, and acquisition of the ability to use CXCR4 is accompanied by either a major or total loss of CCR5-mediated entry. Analysis of the mutational pathways leading to these four variants thus spans the observed spectrum of R5-to-X4 transitions. We also note that these assay methods
8166 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
for assigning coreceptor use are very similar to those used in recent clinical studies (5, 22, 36) but that both target cell lines express high levels of both CD4 and CCR5 or CXCR4 and thus are able to support infection by viruses that cannot infect activated human CD4⫹T cells (39). This feature is useful for assessing the entry efficiencies of coreceptor switch intermedi-ates with very inefficient use of CCR5 or CXCR4, as well as in predicting the response to CCR5 inhibitors in current clinical trials (57), but it should be noted that no such target cells are likely to exist in human lymphoid tissue.
Functional changes in ADA-1 Env during the transition
from R5 to X4.Figure 2A summarizes the entry efficiencies of
Env mutants representing most of the combinations of seven mutations that separate the parental ADA Env from ADA-1 (Table 1). The data in Fig. 2A extend previously published observations (37) and provide the basis for selecting Env mu-tants for further study. Env mumu-tants that had less than a⫺ 1-log-unit change in entry efficiency compared to ADA wt Env were evaluated for sensitivity to CCR5 inhibitors, nAb, and CD4 inhibition by CD4-IgG (Fig. 2B to F) on CCR5-express-ing target cells. Env mutants that had gained weak or strong use of CXCR4 were evaluated for sensitivity to AMD3100, nAb, and CD4-IgG (Fig. 3A to D) on CXCR4-expressing tar-get cells. The p24 content of each pseudovirion preparation expressing a given Env mutant was adjusted to yield⬃100,000 relative light units for all inhibition studies to correct for the differences in entry function shown in Fig. 2A. For inhibition studies with CCR5 target cells, data are expressed as the log change in IC50from ADA wt Env. Each experiment was
re-peated three times, and changes of⬎0.5 log unit were highly significant (P ⬍ 0.05) for all inhibitors studied. The same 4-log-unit scale was used for different inhibitors to facilitate
TABLE 1. Summary of envelope mutations a Mutation V1/V2 C2 V3 ADA-1 NTSTITQACPKVSFEPIPIHYCTPAGTAILK CTRPNNNTRKSIHIGPGRAFYTTGEIIGDIRQAHC D –––––––– –––––––––––––T ––––– ––––H ––––R ––––R –––––––E K –––––––– 1 2 345 6 7 197 221 301 306 313 321,322 ADA-3 CSFNITTSIRDKVKKDYALFYRLDVVPIDNDNTSYRLINC CTRPNNNTRKSIHIGPGRAFYTTGEIIGDIRQAHC –––K –––––––––––––––––– I––– N––––––––––– ––––––––––––––– R–––––– K––––––––– 12 3 4 5 160 181 185 314 322 BaL-1B CTDLRNATNGNDTNTTSSREMMGGGGEMKNCSFKITTNIRGKVQKEYALFYELD CTRPNNNTRKSIHIGPGRALYTTGEIIGDIRQAHC –––––T ––––––––––––––––––––––––––––––––––––––––K–– –––––– –––––––––– K I––– –K ––––––––– 137 178 316,7 322 BaL-2A b NCTDLRNATNGNDTNTTSSREMMGGGGEMKNCSFKITTNIRGKVQKEYALFYELD CTRPNNNTRKSIHIGPGRALYTTGEIIGDIRQAHC D–––––––––––––––––––––––––––––––––––––––––––––– K –– ––––––––––––––––––––––K ––––––––– 130 178 322 a Single-digit numbers indicate the mutation number; three-digit numbers indicate numbering according to HxB2 convention. b The BaL-2A mutated envelope had one additional mutation, K490T, in C5.
FIG. 1. Relative use of CCR5 and CXCR4 by the four coreceptor switch variants used in these studies. The data are mean (⫾ SE) relative light units (RLU) of luciferase activity in multiple single-cycle entry assays on either CCR5-expressing (yaxis) or CXCR4-expressing (xaxis) U87-CD4 cells. The lines connecting the parental BaL or ADA entry activity to the final variant activity are based on data presented in subsequent figures.
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8167
on November 8, 2019 by guest
http://jvi.asm.org/
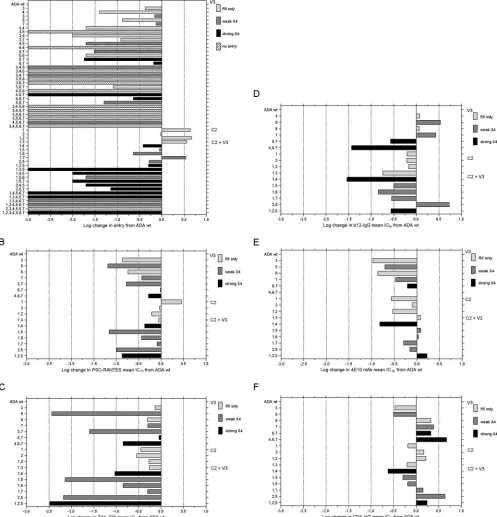
[image:3.585.48.284.67.288.2]comparison of major changes in inhibitor sensitivity. Figure 2B and C show that Env mutants that retained entry function via CCR5 were generally more sensitive to inhibition by both PSC-RANTES and TAK-779. In particular, mutation 5
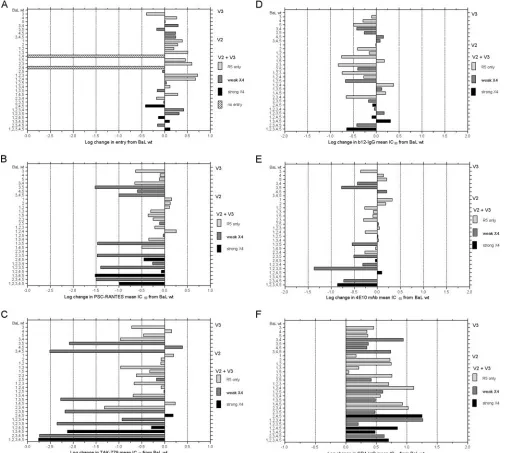
[image:4.585.44.541.83.600.2](P313R) at the crown of the V3 loop, alone or in combination with the C2 region mutations, significantly increased suscepti-bility to both CCR5 inhibitors. The asuscepti-bility of the nAb b12-IgG (directed at the CD4 binding site) or 4E10 (directed at the
FIG. 2. Entry efficiencies and sensitivities to CCR5 inhibitors, nAb, and CD4-IgG of ADA-1 mutated envelopes. Each bar depicts the log change in entry (A) or sensitivity to PSC-RANTES (B), TAK-779 (C), b12-IgG (D), 4E10 monoclonal Ab (MAb) (E), or CD4-IgG (F) of mutated envelopes relative to ADA wt envelope as assayed on U87-CD4-CCR5 cells. Entry assays were also performed on U87-CD4-CXCR4 target cells, and the efficiency of CXCR4-mediated entry is indicated by the gray scale. Envelope mutants with a⬎1-log-unit decrease in entry efficiency were too poorly infectious to use in subsequent assays. The data are mean changes in three triplicate experiments. Changes in entry efficiency or IC50
of greater than 0.5 log unit are highly significant (P⬍0.05). Mutations are numbered as in Table 1.
8168 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 3. Inhibition of ADA-1 mutated envelope-mediated infection of CXCR4-expressing target cells by AMD3100, nAb, and CD4-IgG. Log mean IC50values⫾SE in pg/ml (A) or ng/ml (B, C, and D) are calculated from three replicate experiments, and the comparable values for ADA wt envelope
infection of CCR5-expressing target cells are shown for reference in panels B, C, and D.
8169
on November 8, 2019 by guest
http://jvi.asm.org/
membrane-proximal domain of gp41) to inhibit entry mediated by the mutated Envs is shown in Fig. 2D and E. Sensitivity to b12-IgG inhibition was more variable, with the majority of mutations causing increased sensitivity. However, mutation 5 and mutations 2 plus 5 (P313R and A221T plus P313R) in-creased resistance to b12-IgG. Sensitivity to 4E10 inhibition increased for most mutated Envs and was unchanged for the remainder. Figure 2F shows the change in sensitivity of the mutated Envs to inhibition by CD4-IgG. There was less than a 0.5-log-unit change for most of the Envs, with two mutants showing increased sensitivity and two showing decreased sen-sitivity by greater than 0.5 log unit. The mutant 2 plus 5 (A221T plus P313R), which was more resistant to b12-IgG, was also
more resistant to CD4-IgG, confirming that these two muta-tions diminished access to the CD4 binding site.
Mutated ADA Envs with weak or strong entry function on CXCR4-expressing target cells were evaluated for sensitivity to AMD3100, as well as nAb and CD4-IgG. Data were expressed as the mean IC50⫾SE of three replicate experiments, with the
IC50 value for the nAb and CD4-IgG for ADA wt Env on CCR5 target cells shown for reference. All Env mutants were equally or more sensitive to AMD3100 inhibition than the final ADA-1 Env with all seven mutations (Fig. 3A), but there was not a clear correlation between entry efficiency via CXCR4 and sensitivity to AMD3100 inhibition. Sensitivity to b12-IgG var-ied by almost 3 log units, with the final ADA-1 mutant among
FIG. 4. Entry efficiencies and sensitivities to CCR5 inhibitors, nAb, and CD4-IgG of ADA-3 mutated envelopes. The data were collected and are presented as in Fig. 2, with the log change in mean IC50from ADA wt Env shown. Mutations are numbered as shown in Table 1.
8170 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
the most sensitive to inhibition. Note that the Env with muta-tions 1 plus 4 (N197D plus S306R) was highly susceptible to b12-IgG inhibition on CCR5 target cells (Fig. 2D) but not so susceptible on CXCR4 target cells, implying that the choice of coreceptor impacts the neutralization sensitivity of the same R5X4 Env. The same mutated Env was the most sensitive to both 4E10 nAb and CD4-IgG inhibition of CXCR4-mediated entry (Fig. 2C and D). All Env mutations increased sensitivity to 4E10 or CD4-IgG inhibition compared to ADA wt Env on CCR5 target cells (Fig. 2C and D), but several of the mutated Envs were more resistant to both inhibitors than the final ADA-1 mutant.
Functional changes in ADA-3 Env during the transition
from R5 to R5X4.Similar inhibition studies were performed
with Env mutations involved in the R5-to-R5X4 evolution of the ADA-3 mutant (Fig. 1 and Table 1). As before, pseudovirion concentrations were adjusted to yield nearly equal levels of infection to correct for the varying levels of infectivity shown in Fig. 4A. Note that only mutant ADA-3, with all five Env mutations (Table 1), was capable of strong infection of CXCR4 target cells. The mutations in V2 tended to either decrease susceptibility to the CCR5 inhibitors or cause no change, whereas many of the combined V2 plus V3 mutations increased susceptibility to PSC-RANTES and, to a greater extent, TAK-779 (Fig. 4B and C). The sensitivities of the mutated Envs to b12-IgG were more variable, with V2 plus V3 mutations either increasing sensitivity by 1 log unit in three instances or decreasing sensitivity by 0.8 log unit in one in-stance (Fig. 4D). However, sensitivities to the nAb 4E10 were uniformly increased, with many mutated Envs showing a⬎ 0.5-log-unit decrease in IC50values (Fig. 4E). All but one of the
mutated Envs had increased resistance to CD4-IgG (Fig. 4F), with the Env with mutations 1, 2, and 5 (N160K, V181I, and E322K) showing the greatest increase in resistance to both CD4-IgG and b12-IgG (Fig. 4D and E).
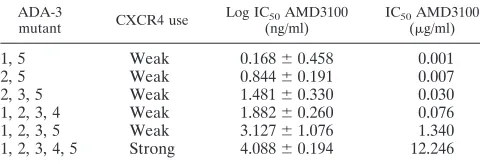
Because most of the mutated Envs were poor at mediating entry into CXCR4 target cells, we performed inhibition studies only with AMD3100. The results are presented in Table 2 and show an increase in resistance to AMD3100 inhibition as the CXCR4 entry function improved.
Impacts of ADA-3 envelope mutations on CD4 and CCR5
binding.The efficiency of Env binding to CD4 or CCR5 (in the
presence of soluble CD4) was assayed by immunostaining of appropriate target cells, as previously described (46, 48). No mutation significantly impaired the ability of Env to bind to CD4 (Fig. 5A), and several combinations of mutations signif-icantly improved CD4 binding. The combination of V3 muta-tion 5 (E322K) and either V2 mutamuta-tion 1 (N160K) or 2
(V181I) led to increased CD4 binding, which was preserved in the ADA-3 envelope with all five mutations. By contrast, sev-eral mutations on the pathway to improved CXCR4 use sig-nificantly decreased binding to CCR5 (Fig. 5B.) The V3 mu-tation 5 (E322K) alone reduced CCR5 binding substantially, but this effect was countered by the introduction of either the V2 mutation 1 (N160K) or 2 (V181I). As might have been predicted from the single-round infection results (Fig. 4A), the combination of all three V2 mutations led to increased CCR5 binding. Combining these three V2 mutations with the V3 mutation 5 (E322K) led to a significant reduction in CCR5 binding, indicating that the negative impact of the V3 mutation outweighed the positive benefits of the three V2 mutations. Binding of CCR5 was further diminished by the introduction of all five mutations. There was no consistent correlation be-tween binding of CD4 and CCR5 and sensitivity to inhibition of infection of U87-CD4-CCR5 cells by CD4-IgG (Fig. 4F). Mutations associated with the R5-to-R5X4 transition of ADA-3 thus generally improved CD4 binding and increased resistance to CD4-IgG inhibition, and reduced binding to CCR5 and increased sensitivity to CCR5 inhibitors.
Extent and kinetics of Env-mediated fusion. On average,
mutations associated with the R5-to-R5X4 evolution of ADA-3 had relatively minor impacts on the extent of Env-mediated fu-sion or the kinetics of fufu-sion (Fig. 5C and D). The combination of V2 mutation 1 (N160K) and V3 mutation 5 (E322K) accel-erated fusion kinetics by about twofold (Fig. 5C) but dimin-ished the extent of Env-mediated fusion with CCR5-expressing target cells (Fig. 5D). The introduction of all five V2 and V3 mutations slowed fusion kinetics with CCR5-expressing target cells by about 50% and promoted the most efficient fusion with CXCR4-expressing target cells (Fig. 5C and D). Compared to the major impact of mutations in the coreceptor binding region of Env on the extent of fusion and fusion kinetics (48), muta-tions in V2 and V3 largely preserved these Env-mediated func-tions. These results are also consistent with CD4 binding hav-ing a larger impact on subsequent fusion efficiency than CCR5 binding.
Functional changes in BaL-1B and BaL-2A during the
tran-sition from R5 to R5X4. The two independent coreceptor
switch mutants gained use of CXCR4 by five mutations in V2 and V3 (BaL-1B) or three mutations in V2 and V3 and one additional mutation in C5 (BaL-2A) (Table 1). By contrast with the ADA-derived coreceptor switch mutants, the two BaL-derived mutants maintained high entry function via CCR5 while gaining robust use of CXCR4. We were therefore inter-ested to determine if they showed the same pattern of dimin-ished binding to CCR5 and increased binding to CD4 as the ADA coreceptor switch intermediates discussed above.
[image:7.585.43.283.91.173.2]Figure 6A summarizes the entry activities of the BaL-1B series of mutated Envs. Note that few mutations increased or decreased entry function⬎0.5 log unit, with the exception of two lethal combinations of mutations, 1 plus 4 and 2 plus 4 (A137T plus L317I and E178K plus L317I). Nonetheless, many of the Env mutations showed highly significant increased sen-sitivity to CCR5 inhibitors, with the final BaL-1B sequence with all five mutations being 2.75 log units (562-fold) more sensitive to TAK-779 than BaL wt Env (Fig. 6B and C). Many of the BaL-1B Env mutants were highly sensitive to entry inhibition by both b12-IgG and 4E10 nAb (Fig. 6D and E), and
TABLE 2. AMD3100 inhibition of entry into U87-CD4-CXCR4 cells
ADA-3
mutant CXCR4 use
Log IC50AMD3100
(ng/ml)
IC50AMD3100
(g/ml)
1, 5 Weak 0.168⫾0.458 0.001
2, 5 Weak 0.844⫾0.191 0.007
2, 3, 5 Weak 1.481⫾0.330 0.030
1, 2, 3, 4 Weak 1.882⫾0.260 0.076
1, 2, 3, 5 Weak 3.127⫾1.076 1.340
1, 2, 3, 4, 5 Strong 4.088⫾0.194 12.246
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8171
on November 8, 2019 by guest
http://jvi.asm.org/
8172 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
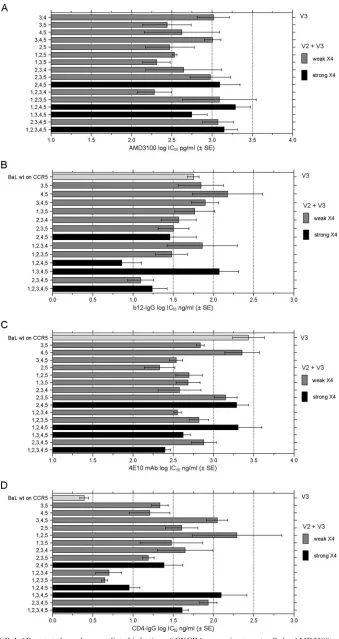
all Env mutants were more resistant to CD4-IgG inhibition than the parental BaL Env (Fig. 6F). When the functions of all the mutated Envs capable of CXCR4 use were tested on CXCR4-expressing target cells, similar results were seen. The
sensitivity of nAb inhibition generally increased, and suscepti-bility to CD4-IgG inhibition decreased (Fig. 7B, C, and D). However, only a few of the BaL1-B mutated Envs were signif-icantly more sensitive to AMD3100 inhibition than the final
[image:9.585.42.547.70.523.2]FIG. 5. CD4 and CCR5 binding and fusion properties of ADA-3 mutated Env. Details of these assays are presented in Materials and Methods. The CD4 (A) and CCR5 (B) binding efficiencies of ADA and ADA mutated Envs were determined by immunostaining and flow cytometry analysis of gp120 binding to the surfaces of NP2/CD4 cells (for NP2, only background was subtracted) for CD4 binding and TREx/CCR5 cells in the presence of soluble CD4 for CCR5 binding. The results are the mean plus SE of two independent experiments. The data are normalized to the percentage of ADA wt Env binding (100%). The geometric mean fluorescence intensity (⫾standard deviation) for the ADA wt Env CD4 binding to NP2/CD4 cells was 42.98⫾13.13, and for binding to TREx/CCR5 cells it was 38.82⫾3.35. (C) Kinetics of cell-cell fusion on each mutated Env with CCR5-expressing target cells. (D) The extent of cell-cell fusion was measured with both CCR5-expressing targets (top bar for each Env) and CXCR4-expressing targets (bottom bar for each Env). The level of entry via CXCR4 is indicated by the gray scale.
FIG. 6. Entry efficiencies and sensitivities to CCR5 inhibitors, nAb, and CD4-IgG of BaL-1B mutated envelopes. Data collection and display were as for Fig. 2. Mutations are numbered as indicated in Table 1.
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8173
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 7. Inhibition of BaL-1B mutated envelope-mediated infection of CXCR4-expressing target cells by AMD3100, nAb, and CD4-IgG. The data are expressed as in Fig. 3.
8174 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 8. Entry efficiencies and sensitivities to CCR5 inhibitors, nAb, and CD4-IgG of BaL-2A mutated envelopes. Data collection and display are as in Fig. 2. Mutations are numbered as indicated in Table 1.
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8175
on November 8, 2019 by guest
http://jvi.asm.org/
mutant with all five mutations. This may reflect the substan-tially better entry function of these Envs via CXCR4 than either ADA-1 or ADA-3 Envs (Fig. 1).
Figure 8 summarizes the impacts of the Env mutations re-sulting in the BaL-2A coreceptor switch mutant. Mutation 4 in C2 had little impact on Env function, so it was not examined in every possible combination. The remaining mutations in V2 plus V3 resulted in less dramatic changes in sensitivity to in-hibitors than those seen with ADA-1, ADA-3, or BaL-1B, but similar trends were observed. There were modest increases in susceptibility to CCR5 inhibitors, particularly PSC-RANTES (Fig. 8B), and some mutated Envs were more sensitive to b12-IgG. Only the final BaL-2A mutant with all four mutations was significantly more sensitive to 4E10 nAb, suggesting that the C5 mutation has some impact on Env function (Fig. 8D and E). Finally, almost all mutated Envs showed increased resistance to CD4-IgG inhibition (Fig. 8F), although the in-crease was more than⫹0.5 log unit for only two mutants in or adjacent to the V2 region (2 and 1 plus 2; E178K and N130D plus E178K).
DISCUSSION
HIV-1 evolved from primate lentiviruses that used CCR5 in a CD4-independent manner (3, 8, 21), so CCR5 may be viewed as the primordial receptor, and adaptation to humans involved acquisition of CD4 binding. The second step in adaptation to human hosts was the ability to use CXCR4 as an additional or alternative coreceptor, although this adaptation may be more evident in subtype B and subtype D viruses than in some other subtypes (9, 34; W. Huang, S. Eshleman, J. Toma, S. Fransen, E. Stawiski, B. Jackson, J. Whitcomb, N. Parkin, and C. J. Petropoulos, presented at the 2nd International Workshop Targeting HIV Entry, Boston, MA, October 20–21, 2006). The acquisition of CD4 binding can be reasonably explained by
selective pressure to hide the coreceptor binding site from neutralizing antibody, since CD4-independent isolates are gen-erally more sensitive to neutralization (15, 25, 32, 55). The switch in coreceptor preference from CCR5 to CXCR4 is more difficult to explain, with the primary advantage to the virus being the expansion of the target cell pool to include naı¨ve CD4⫹T cells that express very low levels of CCR5 but higher levels of CXCR4 (27, 56). However, the long delay from in-fection until emergence of CXCR4-using variants, their rela-tively low incidence in non-subtype B infections, and their poor transmissibility (29) all suggest that evolution to CXCR4 use may be a dead-end pathway for the virus.
Prior studies from this laboratory have emphasized that the envelope mutations that drive coreceptor switching from CCR5 to CXCR4 reduce the fitness of switch intermediates (37, 39). It is possible that many poorly fit switch intermediates are generated in patients and are never detected because they fail to compete with larger and more robust R5 virus popula-tions, even with the potential advantage of establishing infec-tion of naı¨ve CD4⫹T cells. This would be consistent with the finding that coreceptor switch mutants selected in vitro often have few mutations (19, 39) while sequential isolates before and after coreceptor switching in patients have more dramatic sequence changes (31, 52, 56). The primary goal of the present study was to determine if potential coreceptor switch interme-diates gained any functional properties that would aid their survival during the evolution from CCR5 to CXCR4 use. For example, there are prior studies indicating that the level of CD4 expression impacts the efficiency of CCR5 use (44), and it is possible that reduced binding to CCR5 is compensated for by increased binding to CD4 (42).
[image:12.585.42.543.81.306.2]The changes in Env properties for the four coreceptor switch pathways analyzed are summarized in Table 3. We infer that increased sensitivity to CCR5 inhibitors reflects reduced bind-ing to CCR5, an assumption validated for ADA-3 mutants
TABLE 3. Common and distinct features of R5-to-(R5)X4 transition
Feature
Relative level of sensitivity for indicated virusa:
ADA-1 ADA-3 BaL-1B BaL-2A
CCR5 entry Reduced Reduced Varies Increased
⫺1.5 to⬎⫺3.0 log ⫹0.5 to⫺2.4 log ⫹0.7 to⫺0.4 log ⫹0.5 to⫹0.7 log CCR5 inhibitors More sensitive More sensitive More sensitive Some more sensitive
⫺0.5 to⫺2.5 log ⫹0.5 to⫺2.3 log ⫺0.5 to⫺2.7 log ⫺0.5 log
MAb (b12) Most more sensitive Varies Most more sensitive Most more sensitive
⫺0.5 to⫺1.5 log ⫹0.8 to⫺1.0 log ⫺0.5 to⫺0.7 log ⫺0.5 log
MAb (4E10) More sensitive More sensitive More sensitive Final more sensitive
⫺0.5 to⫺1.0 log ⫺0.5 to⫺1.4 log ⫺0.5 to⫺1.4 log ⫺0.7 log CD4 inhibition Varies Most less sensitive All less sensitive All less sensitive
⫺0.7 to⫹0.7 log ⫺0.7 to⫹1.0 log ⫹0.5 to⫹1.3 log ⫹0.4 to⫹0.7 log
CXCR4 entry Increased Increased Increased Increased
CXCR4 inhibitors More sensitive More sensitive Varies More sensitive
⫺0.5 to⫺2.0 log ⫺0.9 to⫺4.8 log ⫺0.8 to⫹0.3 log ⫺0.3 to⫺0.7 log
MAb (b12) Varies ND More sensitive More sensitive
⫺1.2 to⫹1.7 log ⫺0.5 to⫺1.0 log ⫺0.5 to⫺1.2 log
MAb (4E10) More sensitive ND More sensitive Varies
⫺0.5 to⫺2.5 log ⫺0.5 to⫺1.0 log ⫺0.5 to⫹0.6 log
CD4 inhibition Varies ND Less sensitive Less sensitive
⫺0.9 to⫹0.5 log ⫹0.3 to⫹1.9 log ⫹0.4 to⫹0.9 log
CD4 binding ND Increased ND ND
aND, not determined.
8176 PASTORE ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
(Fig. 5). Almost all mutated Envs with V3 substitutions had increased sensitivity to both PSC-RANTES and TAK-779, CCR5 inhibitors with two distinct modes of action (20, 26, 38). This was true even for the BaL-1B pathway that led to im-proved use of CCR5 compared to the parental BaL Env. It may not be surprising that Env mutations that allow some level of CXCR4 use come at the expense of CCR5 use, but the entry activity on CCR5-expressing target cells may not be impacted until the CCR5 binding affinity falls below a threshold required for productive infection (24, 40). Our data suggest that the threefold-less-efficient entry mediated by the ADA Env com-pared to the BaL Env may put the ADA Env mutants closer to this threshold. The evolutionary pathway of the ADA-1 mutant from pure R5 to pure X4 phenotype is littered with nonfunc-tional Envs (Fig. 2A) (37), suggesting that abandoning CCR5 use entirely is perilous. This is consistent with the small frac-tion of patient isolates that type as pure X4 viruses using similar entry assays (5, 36). Env mutants representing interme-diate stages of gaining CXCR4 use were generally highly sen-sitive to AMD3100 inhibition. This implies that efficient use of CXCR4 is generally dependent upon several Env mutations, as is most obvious for the ADA-3 series of mutations (Table 2). Most pathways of Env evolution led to apparent increased binding to CD4, as directly measured for ADA-3 Env mutants (Fig. 5A) and inferred by increasing resistance to inhibition of entry by CD4-IgG for most other Env mutants (Table 3). Only the ADA-1 series of Env mutants did not show consistent increases in apparent CD4 binding affinity, but this may reflect the smaller selection of functional Env clones remaining after elimination of the nonfunctional mutants. Increased binding to CD4 has been observed during adaptation of HIV-1 IIIB to replication in infected laboratory workers (33), as well as lab-oratory adaptation of patient isolates to growth in T-cell lines (23). All four coreceptor switch mutants studied here were initially selected for growth on U87-CD4-CXCR4 cells that have abundant CD4, but they were subsequently passaged on MT-2 cells with lower levels of CD4 (39). However, the nearly uniform increase in apparent CD4 binding of all mutant Envs representing intermediate stages of coreceptor switching im-plies that improved CD4 binding is a necessary prerequisite for subsequent improvements in CXCR4 binding. This would be compatible with a model in which the loss of CCR5 binding is compensated for by a gain in CD4 binding prior to or during improved CXCR4 binding.
We have previously shown that the Env mutations studied here lead to different patterns of chimeric coreceptor use, with binding to the second extracellular domain (EC2) of CXCR4 being the most common event during coreceptor switching (37). The hypothesis that mutations in V3 and at other sites in Env can cause differential engagement of CCR5 extracellular domains has received support from several studies (41, 43, 45). One can envision a multistep coreceptor-switching process in which Env binding to CCR5 EC2 is initially disrupted but binding to the CCR5 N-terminal domain is preserved, followed by mutations that allow improved engagement of CXCR4 EC2, and finally binding to multiple CXCR4 extracellular do-mains. A similar, if somewhat simpler, model has been pro-posed by Lu et al. (30) based on studies with CXCR4-CXCR2 chimeric coreceptors. It is also possible that other cell surface attachment receptors, e.g., proteoglycans, can contribute to
Env binding and support the survival of coreceptor switch intermediates with poor affinity for either CCR5 or CXCR4 (12, 35, 51), although these impacts may be limited to infection of selected cell types. Improved CD4 binding of these postu-lated coreceptor switch intermediates could serve two func-tions: (i) to enhance the entry function of intermediates with poor binding to both CCR5 and CXCR4 and (ii) to protect more exposed coreceptor binding sites from immune recogni-tion. The finding that many of the coreceptor switch interme-diates were more sensitive to neutralizing antibodies suggests that autologous neutralizing antibodies could select against early R5X4 viruses and be one factor in the long delay pre-ceding the emergence of X4 variants in patients.
ACKNOWLEDGMENTS
We thank Phillip Arca and Fang-Hua Lee for assistance with cloning and sequence analysis.
This work was supported by NIH grant AI058701 and amfAR fel-lowship 106437-34-RFGN (J.D.R.), NIH grant AI051649 and Swiss National Science Foundation grant 3100A0-110042 (O.H.), and NIH grants AI052778 and AI051649 and The James B. Pendleton Charita-ble Trust (D.E.M.).
This is publication number 17989-IMM from The Scripps Research Institute.
REFERENCES
1.Alexander, W. A., B. Moss, and T. R. Fuerst.1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA
polymerase and theEscherichia coli lacrepressor. J. Virol.66:2934–2942.
2.Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, K. Meguro, and M. Fujino.
1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and
selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA96:5698–5703.
3.Bhattacharya, J., P. J. Peters, and P. R. Clapham.2003. CD4-independent infection of HIV and SIV: implications for envelope conformation and cell
tropism in vivo. Aids17(Suppl. 4):S35–S43.
4.Bjorndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyo.1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to
biological phenotype. J. Virol.71:7478–7487.
5.Brumme, Z. L., J. Goodrich, H. B. Mayer, C. J. Brumme, B. M. Henrick, B. Wynhoven, J. J. Asselin, P. K. Cheung, R. S. Hogg, J. S. Montaner, and P. R. Harrigan. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis.
192:466–474.
6.Burton, D. R., J. Pyati, R. Koduri, S. J. Sharp, G. B. Thornton, P. W. Parren, L. S. Sawyer, R. M. Hendry, N. Dunlop, P. L. Nara, M. Lamacchia, E. Garratty, E. R. Stiehm, Y. J. Bryson, Y. Cao, J. P. Moore, D. Ho, and C. F. Barbas.1994. Efficient neutralization of primary isolates of HIV-1 by a
recombinant human monoclonal antibody. Science266:1024–1027.
7.Cardoso, R. M., M. B. Zwick, R. L. Stanfield, R. Kunert, J. M. Binley, H. Katinger, D. R. Burton, and I. A. Wilson.2005. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved
fusion-associated motif in gp41. Immunity22:163–173.
8.Chen, Z., A. Gettie, D. D. Ho, and P. A. Marx.1998. Primary SIVsm isolates use the CCR5 coreceptor from sooty mangabeys naturally infected in West Africa: a comparison of coreceptor usage of primary SIVsm, HIV-2, and
SIVmac. Virology246:113–124.
9.Cilliers, T., J. Nhlapo, M. Coetzer, D. Orlovic, T. Ketas, W. C. Olson, J. P. Moore, A. Trkola, and L. Morris.2003. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from
subtype C. J. Virol.77:4449–4456.
10.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau.1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in
mono-nuclear phagocytes. Virology206:935–944.
11.Deng, H. K., D. Unutmaz, V. N. KewalRamani, and D. R. Littman.1997. Expression cloning of new receptors used by simian and human
immunode-ficiency viruses. Nature388:296–300.
12.de Parseval, A., M. D. Bobardt, A. Chatterji, U. Chatterji, J. H. Elder, G. David, S. Zolla-Pazner, M. Farzan, T. H. Lee, and P. A. Gallay.2005. A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans
and CCR5 via sulfated motifs. J. Biol. Chem.280:39493–39504.
13.Donzella, G. A., D. Schols, S. W. Lin, J. A. Este, K. A. Nagashima, P. J. Maddon, G. P. Allaway, T. P. Sakmar, G. Henson, E. De Clercq, and J. P.
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8177
on November 8, 2019 by guest
http://jvi.asm.org/
Moore.1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the
CXCR4 co-receptor. Nat. Med.4:72–77.
14.Draenert, R., T. M. Allen, Y. Liu, T. Wrin, C. Chappey, C. L. Verrill, G. Sirera, R. L. Eldridge, M. P. Lahaie, L. Ruiz, B. Clotet, C. J. Petropoulos, B. D. Walker, and J. Martinez-Picado.2006. Constraints on HIV-1 evolution and immunodominance revealed in monozygotic adult twins infected with
the same virus. J. Exp. Med.203:529–539.
15.Edwards, T. G., T. L. Hoffman, F. Baribaud, S. Wyss, C. C. LaBranche, J. Romano, J. Adkinson, M. Sharron, J. A. Hoxie, and R. W. Doms.2001. Relationships between CD4 independence, neutralization sensitivity, and exposure of a CD4-induced epitope in a human immunodeficiency virus type
1 envelope protein. J. Virol.75:5230–5239.
16.Edwards, T. G., S. Wyss, J. D. Reeves, S. Zolla-Pazner, J. A. Hoxie, R. W. Doms, and F. Baribaud.2002. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human
immunodefi-ciency virus type 1 envelope protein. J. Virol.76:2683–2691.
17.Gonzalez, E., H. Kulkarni, H. Bolivar, A. Mangano, R. Sanchez, G. Catano, R. J. Nibbs, B. I. Freedman, M. P. Quinones, M. J. Bamshad, K. K. Murthy, B. H. Rovin, W. Bradley, R. A. Clark, S. A. Anderson, J. R. O’Connell, B. K. Agan, S. S. Ahuja, R. Bologna, L. Sen, M. J. Dolan, and S. K. Ahuja.2005. The influence of CCL3L1 gene-containing segmental duplications on
HIV-1/AIDS susceptibility. Science307:1434–1440.
18.Hahn, B. H., G. M. Shaw, K. M. De Cock, and P. M. Sharp.2000. AIDS as
a zoonosis: scientific and public health implications. Science287:607–614.
19.Harrowe, G., and C. Cheng-Mayer.1995. Amino acid substitutions in the V3 loop are responsible for adaptation to growth in transformed T-cell lines of
a primary human immunodeficiency virus type 1. Virology210:490–494.
20.Hartley, O., H. Gaertner, J. Wilken, D. Thompson, R. Fish, A. Ramos, C. Pastore, B. Dufour, F. Cerini, A. Melotti, N. Heveker, L. Picard, M. Alizon, D. Mosier, S. Kent, and R. Offord.2004. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc.
Natl. Acad. Sci. USA101:16460–16465.
21.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore.2005. V3: HIV’s
switch-hitter. AIDS Res. Hum. Retrovir.21:171–189.
22.Hunt, P. W., P. R. Harrigan, W. Huang, M. Bates, D. W. Williamson, J. M. McCune, R. W. Price, S. S. Spudich, H. Lampiris, R. Hoh, T. Leigler, J. N. Martin, and S. G. Deeks.2006. Prevalence of CXCR4 tropism among anti-retroviral-treated HIV-1-infected patients with detectable viremia. J. Infect.
Dis.194:926–930.
23.Kozak, S. L., E. J. Platt, N. Madani, F. E. Ferro, Jr., K. Peden, and D. Kabat.
1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus
type 1. J. Virol.71:873–882.
24.Kuhmann, S. E., E. J. Platt, S. L. Kozak, and D. Kabat.2000. Cooperation of multiple CCR5 coreceptors is required for infections by human
immuno-deficiency virus type 1. J. Virol.74:7005–7015.
25.Labrijn, A. F., P. Poignard, A. Raja, M. B. Zwick, K. Delgado, M. Franti, J. Binley, V. Vivona, C. Grundner, C. C. Huang, M. Venturi, C. J. Petropoulos, T. Wrin, D. S. Dimitrov, J. Robinson, P. D. Kwong, R. T. Wyatt, J. Sodroski, and D. R. Burton.2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on
primary human immunodeficiency virus type 1. J. Virol.77:10557–10565.
26.Lagane, B., S. Ballet, T. Planchenault, K. Balabanian, E. Le Poul, C. Blan-pain, Y. Percherancier, I. Staropoli, G. Vassart, M. Oppermann, M. Par-mentier, and F. Bachelerie.2005. Mutation of the DRY motif reveals dif-ferent structural requirements for the CC chemokine receptor 5-mediated
signaling and receptor endocytosis. Mol. Pharmacol.67:1966–1976.
27.Lee, B., M. Sharron, L. J. Montaner, D. Weissman, and R. W. Doms.1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived
macro-phages. Proc. Natl. Acad. Sci. USA96:5215–5220.
28.Lineberger, J. E., R. Danzeisen, D. J. Hazuda, A. J. Simon, and M. D. Miller.
2002. Altering expression levels of human immunodeficiency virus type 1 gp120-gp41 affects efficiency but not kinetics of cell-cell fusion. J. Virol.
76:3522–3533.
29.Long, E. M., S. M. Rainwater, L. Lavreys, K. Mandaliya, and J. Overbaugh.
2002. HIV type 1 variants transmitted to women in Kenya require the CCR5 coreceptor for entry, regardless of the genetic complexity of the infecting
virus. AIDS Res. Hum. Retrovir.18:567–576.
30.Lu, Z., J. F. Berson, Y. Chen, J. D. Turner, T. Zhang, M. Sharron, M. H. Jenks, Z. Wang, J. Kim, J. Rucker, J. A. Hoxie, S. C. Peiper, and R. W. Doms.
1997. Evolution of HIV-1 coreceptor usage through interactions with distinct
CCR5 and CXCR4 domains. Proc. Natl. Acad. Sci. USA94:6426–6431.
31.Lukashov, V. V., and J. Goudsmit.1997. Evolution of the human immuno-deficiency virus type 1 subtype-specific V3 domain is confined to a sequence
space with a fixed distance to the subtype consensus. J. Virol.71:6332–6338.
32.Lusso, P., P. L. Earl, F. Sironi, F. Santoro, C. Ripamonti, G. Scarlatti, R. Longhi, E. A. Berger, and S. E. Burastero.2005. Cryptic nature of a con-served, CD4-inducible V3 loop neutralization epitope in the native envelope glycoprotein oligomer of CCR5-restricted, but not CXCR4-using, primary
human immunodeficiency virus type 1 strains. J. Virol.79:6957–6968.
33.Miller, E. D., K. M. Duus, M. Townsend, Y. Yi, R. Collman, M. Reitz, and L. Su.2001. Human immunodeficiency virus type 1 IIIB selected for repli-cation in vivo exhibits increased envelope glycoproteins in virions without
alteration in coreceptor usage: separation of in vivo replication from
mac-rophage tropism. J. Virol.75:8498–8506.
34.Morris, L., T. Cilliers, H. Bredell, M. Phoswa, and D. J. Martin.2001. CCR5 is the major coreceptor used by HIV-1 subtype C isolates from patients with
active tuberculosis. AIDS Res. Hum. Retrovir.17:697–701.
35.Moulard, M., H. Lortat-Jacob, I. Mondor, G. Roca, R. Wyatt, J. Sodroski, L. Zhao, W. Olson, P. D. Kwong, and Q. J. Sattentau.2000. Selective interac-tions of polyanions with basic surfaces on human immunodeficiency virus
type 1 gp120. J. Virol.74:1948–1960.
36.Moyle, G. J., A. Wildfire, S. Mandalia, H. Mayer, J. Goodrich, J. Whitcomb, and B. G. Gazzard.2005. Epidemiology and predictive factors for chemokine
receptor use in HIV-1 infection. J. Infect. Dis.191:866–872.
37.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier.
2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J.
Vi-rol.80:750–758.
38.Pastore, C., G. R. Picchio, F. Galimi, R. Fish, O. Hartley, R. E. Offord, and D. E. Mosier.2003. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob.
Agents Chemother.47:509–517.
39.Pastore, C., A. Ramos, and D. E. Mosier.2004. Intrinsic obstacles to human
immunodeficiency virus type 1 coreceptor switching. J. Virol.78:7565–7574.
40.Platt, E. J., J. P. Durnin, and D. Kabat.2005. Kinetic factors control effi-ciencies of cell entry, efficacies of entry inhibitors, and mechanisms of
ad-aptation of human immunodeficiency virus. J. Virol.79:4347–4356.
41.Platt, E. J., S. E. Kuhmann, P. P. Rose, and D. Kabat.2001. Adaptive mutations in the V3 loop of gp120 enhance fusogenicity of human immu-nodeficiency virus type 1 and enable use of a CCR5 coreceptor that lacks the
amino-terminal sulfated region. J. Virol.75:12266–12278.
42.Platt, E. J., N. Madani, S. L. Kozak, and D. Kabat.1997. Infectious prop-erties of human immunodeficiency virus type 1 mutants with distinct affinities
for the CD4 receptor. J. Virol.71:883–890.
43.Platt, E. J., D. M. Shea, P. P. Rose, and D. Kabat.2005. Variants of human immunodeficiency virus type 1 that efficiently use CCR5 lacking the tyrosine-sulfated amino terminus have adaptive mutations in gp120, including loss of
a functional N-glycan. J. Virol.79:4357–4368.
44.Platt, E. J., K. Wehrly, S. E. Kuhmann, B. Chesebro, and D. Kabat.1998. Effects of CCR5 and CD4 cell surface concentrations on infections by mac-rophage-tropic isolates of human immunodeficiency virus type 1. J. Virol.
72:2855–2864.
45.Pontow, S., and L. Ratner.2001. Evidence for common structural determi-nants of human immunodeficiency virus type 1 coreceptor activity provided through functional analysis of CCR5/CXCR4 chimeric coreceptors. J. Virol.
75:11503–11514.
46.Reeves, J. D., S. A. Gallo, N. Ahmad, J. L. Miamidian, P. E. Harvey, M. Sharron, S. Pohlmann, J. N. Sfakianos, C. A. Derdeyn, R. Blumenthal, E. Hunter, and R. W. Doms.2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion
kinetics. Proc. Natl. Acad. Sci. USA99:16249–16254.
47.Reeves, J. D., A. McKnight, S. Potempa, G. Simmons, P. W. Gray, C. A. Power, T. Wells, R. A. Weiss, and S. J. Talbot.1997. CD4-independent infection by HIV-2 (ROD/B): use of the 7-transmembrane receptors
CXCR-4, CCR-3, and V28 for entry. Virology231:130–134.
48.Reeves, J. D., J. L. Miamidian, M. J. Biscone, F. H. Lee, N. Ahmad, T. C. Pierson, and R. W. Doms.2004. Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and
entry inhibitor sensitivity. J. Virol.78:5476–5485.
49.Reeves, J. D., and T. F. Schulz.1997. The CD4-independent tropism of human immunodeficiency virus type 2 involves several regions of the enve-lope protein and correlates with a reduced activation threshold for enveenve-lope-
envelope-mediated fusion. J. Virol.71:1453–1465.
50.Rucker, J., B. J. Doranz, A. L. Edinger, D. Long, J. F. Berson, and R. W. Doms.1997. Cell-cell fusion assay to study role of chemokine receptors in
human immunodeficiency virus type 1 entry. Methods Enzymol.288:118–
133.
51.Saphire, A. C., M. D. Bobardt, Z. Zhang, G. David, and P. A. Gallay.2001. Syndecans serve as attachment receptors for human immunodeficiency virus
type 1 on macrophages. J. Virol.75:9187–9200.
52.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins.1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol.
73:10489–10502.
53.Soda, Y., N. Shimizu, A. Jinno, H. Y. Liu, K. Kanbe, T. Kitamura, and H. Hoshino.1999. Establishment of a new system for determination of core-ceptor usages of HIV based on the human glioma NP-2 cell line. Biochem.
Biophys. Res. Commun.258:313–321.
54.Sullivan, N., Y. Sun, J. Li, W. Hofmann, and J. Sodroski.1995. Replicative function and neutralization sensitivity of envelope glycoproteins from pri-mary and T-cell line-passaged human immunodeficiency virus type 1 isolates.
J. Virol.69:4413–4422.
55.Sullivan, N., Y. Sun, Q. Sattentau, M. Thali, D. Wu, G. Denisova, J. Gershoni, J. Robinson, J. Moore, and J. Sodroski.1998. CD4-induced con-formational changes in the human immunodeficiency virus type 1 gp120
8178
on November 8, 2019 by guest
http://jvi.asm.org/
glycoprotein: consequences for virus entry and neutralization. J. Virol.72:
4694–4703.
56.van Rij, R. P., H. Blaak, J. A. Visser, M. Brouwer, R. Rientsma, S. Broersen, A. M. de Roda Husman, and H. Schuitemaker.2000. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and
syncytium-inducing HIV-1. J. Clin. Investig.106:1039–1052.
57.Westby, M., M. Lewis, J. Whitcomb, M. Youle, A. L. Pozniak, I. T. James, T. M. Jenkins, M. Perros, and E. van der Ryst.2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (1) variants in a minority of
HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is
from a pretreatment CXCR4-using virus reservoir. J. Virol.80:4909–4920.
58.Williams, C. F., D. Klinzman, T. E. Yamashita, J. Xiang, P. M. Polgreen, C. Rinaldo, C. Liu, J. Phair, J. B. Margolick, D. Zdunek, G. Hess, and J. T. Stapleton.2004. Persistent GB virus C infection and survival in HIV-infected
men. N. Engl. J. Med.350:981–990.
59.Yamanaka, R., R. Tanaka, and S. Yoshida.1993. Effects of irradiation
on cytokine production in glioma cell lines. Neurol. Med. Chir.33:744–
748.
VOL. 81, 2007 ENVELOPE FUNCTION DURING CORECEPTOR SWITCHING 8179