JOURNALOFVIROLOGY,Feb. 1973, p. 157-167
Copyright i 1973 American Society for Microbiology PrintedVol.11, No. 2in U.SA.
Detection
of
Avian Tumor Virus
RNA in
Uninfected Chicken Embryo
Cells
WILLIAM S. HAYWARD ANDHIDESABURO HANAFUSA
The Public Health Research Instituteof theCity of New York, Inc., New York, NewYork 10016
Received for publication4October 1972
Uninfected chickenembryo cells were analyzed for thepresenceofviral
ribo-nucleic acid (RNA) by molecularhybridization with the single-stranded deoxy-ribonucleic acid (DNA) product of the RNA-dependent DNA polymerase
con-tained in avian sarcoma-leukosis virions. Viral RNA was detected in all cells
which contained the avian tumor virus group-specific antigen and the virus-related helper factor. The amountsof viral RNA in these cells ranged from
ap-proximately 3 to 40 copies ofviral-specific sequences per cell. In general, the
viral RNAcontentcorrelated withthelevel ofhelper activity in the cells.Cells infected with Rous-associated virus 2 contained 3,000 to 4,000 copies ofviral RNApercell. RNA from theseinfected cells hybridized with nearly 100% of the
viral 3H-DNA. By contrast, a maximum of less than 50% hybridization was
obtained with RNA from the uninfected helper-positive cells, suggesting that not all of the viral RNA sequences were present in these cells. No viral RNA
was detected in cells which lacked group-specific antigen and helper activity.
Under the conditions used in these studies, less than 0.3 viral genome
equiva-lents of RNApercell would have been detected.
Two products specific to avian
leukosis-sar-coma viruses are present in uninfected cells derived from a majority of chicken embryos
from leukosis-free flocks, although mature
virus particles are not detectableinthese cells (10, 23, 25, 43). The formation ofthese viral products-the group-specific (gs) antigen and
a component ofthe viral
envelope-is
geneti-callydetermined,
with presencebeing
domi-nant (31, 45).
Presumably
because the latter product can complement a defective function of Bryan strain Rous sarcoma virus(B-RSV),
these chicken cells provide a helper activityfor this particular virus. The level of expres-sion ofboth the
helper
function and gsantigenvaries quite widely among cells derived from
different
embryos
(25). Cells from a minorityof
embryos
contain no detectable viralprod-ucts. A new type of leukosis virus, Rous-asso-ciated virus 60
(RAV-60),
canbe isolated from the viral product-positive cells following in-fection with other known leukosis or sarcoma viruses (24). This virus contains geneticin-formation, apparently derived from the host
cell, which codes for the two viral
products
described above.Further studies, using a very sensitive
tech-nique, showed that cells derived from all of
these embryos, including those which lacked detectable viral products, were capableof
pro-ducing RAV-60 following infection with other leukoviruses(25). Thepresence ofviralgenetic
information inalltypes ofcellswasalso demon-strated
by
molecularhybridization analysis,
which showed that viraldeoxyribonucleic
acid (DNA) sequences are present in similaramounts in uninfected cells both positive and negative for the viral
products
(3, 34, 40). Thepresence ofviral DNA in cells which lack viral functions suggests that the expression of the viral genes is blocked
by
a specific control mechanism. However, it is alsopossible
that cells which lack these two viralproducts
con-tain incomplete orpartially
defective viralgenomes.
To gain additional information about the expression of viral genes, we have examined uninfected chicken
embryo
cells for the pres-ence of viral ribonucleic acid(RNA)
by
mo-lecular hybridization. Viral
product-positive
cells wouldbeexpected
to contain viralRNA,
though perhaps at very low levels. Cells whichlack detectable viral
products
might
also contain viralRNA,
since the two functions which can beassayed
may notadequately
reflect the expression of the viral genes. If
157
on November 10, 2019 by guest
http://jvi.asm.org/
the expression of viral functions is regulated by some control mechanism, the presence or
absence of viral RNA in viral product-negative cells would depend on whether control is
me-diated at the level of transcription or trans-lation.
Virus-specific RNA was detected by
mo-lecular hybridization with 3H-labeled viral DNA, which was synthesized using the
endog-enous RNA template and RNA-dependent
DNA polymerase contained in sarcoma-leuko-sis virions (2, 38). This method has been used successfully for the detection of viral RNA in infected cells of both avian and mammalian origin (9, 16, 19, 26, 30). Hybridformationwas
detected by analysis with the single-strand-specific S-1 nuclease of Aspergillus oryzae
(1, 30, 37, 41). This technique permits the
use of high concentrations of RNA, thus
in-creasing the sensitivity of the assay. The conditions used in this study are capable of
detecting less than 0.3 viral genome equiva-lents of RNA percell.
MATERIALS ANDMETHODS
Materials. Deoxyribonuclease I (ribonuclease-free) and hyaluronidase wereobtained from
Worth-ington Biochemical Corp.; crude a-amylase (A.
oryzae), trypsin, and diethylaminoethyl
(DEAE)-cellulose were from Sigma Chemical Co.; Pronase andactinomycinDwerefromCalbiochem;
hydroxyl-apatite (hypatite C) was from Clarkson Chemical
Co.; Nonidet P-40 (NP-40) was from Shell Chemi-cal Corp.; transfer RNA (tRNA) (Baker's yeast)
and salmon sperm DNA werefrom Schwarz/Mann; poly(rA) and poly(dT) from Biopolymers, Inc.; and
'H-thymidine triphosphate (TTP) was from New
EnglandNuclearCorp.
Cells and viruses. Viruses used in this study
were Rous-associated virus2 (RAV-2), RAV-60, and
the Schmidt-Ruppin strain of Rous sarcoma virus
subgroup D (SR-RSV). Conditions for growth and infection of cells and characteristics of viruses have been described previously (21, 22, 24, 42). RAV-2 andSR-RSVweregrowninchick cells which lack the
gs antigen and helper activity, and RAV-60 was
prepared in quail embryo cells. The SR-RSV used in these studies contained spontaneously appearing nontransformingvirus(equivalenttoRAV-50),which
wasdetected bytheplaqueassayof Kawaiand
Hana-fusa(28).The ratiooffocus-formingto plaque-form-ingunits in this preparationwasapproximately 3: 1.
Classification of chicken embryo cells. The titer of gs antigen in each embryo was determined
by complementfixation (15, 27, 31),usingthe viscera which were removed during the preparation of
pri-mary cultures (25). Embryos have been classified
into three categories, gs-, gsL (low), and gs+ (see Table 1). Thepresence ofhelper factor activitywas
determined by assaying for the productionof infec-tiousB-RSV(f) followinginfection withthedefective
B-RSV(-) in the presence of ultraviolet (UV)-inacti-vated Sendai virus (Method A), or with B-RSV(RAV-2) (Method B) (25). Embryos have been classified h-, hL (low), hH (high) orhE (extremely high), based on the relative titer of infectious virus in these as-says (see Table 1). Allembryos were tested by both methods.
Four relatively distinct classes of embryos have been used in these studies (Table 1). Embryos of type gs-h-, gs-hL, and gs+hH correspond to cell types
C/G',
C/O'-L, andC/O, respectively, and have been described in detail previously (25). Embryos of type gsLhE, which are relatively rare, contain low levels of gs antigen and produce infectious RSV(f) at extremely high levels. All embryos used in this study are resistant to viruses of subgroup E. Thus, they are more properly classified in a new genetic category of C/E (24, 32), rather than C/Oas reported inearlier publications.
Purification of viruses. Virus stocks for RNA extraction and DNA synthesis were prepared from infected secondary cultures of chicken embryo cells which lack both gs antigen and chick cell helper factoractivity. Culture fluids offully infected cells were collected at 12-hr intervals, pooled, and centri-fuged at 8,000 x g for 20 min. The supernatant fluid was centrifuged for 2 hr at 19,000 rpm in an
Internationaltype 19rotor.Thepelletwassuspended
in TEN buffer (0.01 M tris(hydroxymethyl)amino-methane [Tris], pH 7.4; 0.001 M ethylenediamine-tetracetic acid [EDTA]; 0.15 M NaCl) containing
10 Ag per ml of hyaluronidase and centrifuged at
lowspeed to remove cell debris. The pellet was re-suspended in a small volume of TEN plus hyal-uronidase, incubated for 10 min at 38 C, and again centrifuged at low speed. The supernatant fluids from these low-speed centrifugations were pooled and treated for 20min at 37 C with 20 ,ug/ml of pro-nase which hadbeen predigested for 2 hr at 37 C to
remove nucleases. The viruses were further purified by equilibrium density centrifugation, using a linear gradient of15to 50%(w/w) sucrose in TEN buffer. Virusinasingleband(p 1.16)waspooled, diluted with TEN, and concentrated by centrifugation onto
a layer of 50% sucrose overlaid with 20% sucrose. This material wasused for RNA extraction orDNA synthesis.
Extraction of RNA. Viral RNA was prepared by sodium dodecyl sulfate (SDS)-phenol extraction
at room temperature and concentrated by ethanol TABLE1. Classification of chicken embryo cells
Celltype Titer ofgs Titer ofRSV(f)
antigena atday5b
gs-h- 0 0
gs-hL 0 10-200
gs+ht, 8-16 1,000-3,000
gsLhE 2-4 10,000-30,000
aExpressedas thereciprocal ofthe terminal
anti-gen dilution which givesmorethana +3 reaction in
the complement fixation test.
°Determinedby MethodA(seereference 25).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.493.266.457.542.636.2]VIRAL RNA IN CHICKEN CELLS
precipitation. Yeast tRNA (200
gg/ml)
was used asacarrier. Viral 70sRNAwas purified by rate zonal centrifugation, usinga linear gradient of 15 to 30% glycerol in TEN buffer. For isolation of 35s RNA, phenol-extracted viral RNA was heated for 3 min at 70 C in 0.01 x SSC (SSC = 0.15 M NaCl plus 0.015 M sodium citrate) prior to centrifugation. Centrifugation was for 90 min (70s RNA) or 150 min (35s RNA) at 50,000 rpm in an SW50.1 rotor.
Fractions containing 35sor70sviral RNAwerepooled, dialyzed for 6 hr against 2 x SSC and stored at
-20 C. When necessary, RNA was concentrated by lyophilization. Whole cell RNA was prepared from secondary cultures 4 to 5 days after transfer. Cells were removedfromcultureplates by trypsiniza-tion (0.5 mg/ml, 10 min at 38 C) andwashed with Tris-saline. RNA was extracted by the hot phenol method, as described by Scherrer (36). After phenol extraction the RNA was precipitated with ethanol and resuspended in a buffer solution
con-taining 0.01 M Tris (pH 7.0), 0.01 M NaCl, and
0.001 M MnCl2. The sample was treated with 10
Mg/ml of deoxyribonuclease (ribonuclease-free) for 60 min at 4 C. Predigested Pronase (50 ug/ml) was
then added, and the sample was incubated for 30 min at 37 C. The RNAwasthen repurified by SDS-phenol extraction at room temperature and precipi-tated with ethanol. The pellet was resuspended in 2 x SSCcontaining 0.1%SDS and storedat -20C. Cellular 4 to 10s RNA was prepared by rate zonal centrifugation ofthis RNA, using a linear gradient
of15to 30%glycerolinTENbuffer. RNA concentra-tions were determined by optical density at 260 nm. The 260/280 ratio was greater than 2 for all preparations.
Synthesis and purification of Viral DNA. 3H-DNAwassynthesizedfromdetergent-treated virions
inthe presence ofactinomycinD. The reaction
mix-ture contained 20 mmTris (pH 8.1), 5 mmMgCl2,
15 mM dithiothreitol, 0.03% NP40, 0.5 mm each of deoxyadenosine triphosphate, deoxyguanosine
tri-phosphate, and deoxycytidine triphosphate; 0.0005
mM 3H-TTP (51 Ci/mmole), 200Mg/mlof actinomy-cinD, and purified virus (0.5to 1mg/mlofviral pro-tein). The mixturewasincubated for3hrat38C, fol-lowed by addition of EDTA to a concentration of 10 mM.DNA was purifiedby SDS-phenol extractionat roomtemperature, dialyzed overnightagainst0.01 x SSC,and treated with0.5 NNaOHfor 20hrat 37C. The sample was dialyzed overnight against 0.05 M (Na)PO4 buffer(pH 7.8) with twochanges. A minor fraction ofthis DNA (8to 12% in different prepara-tions)wasresistanttodigestion bythe single-strand-specific S-1 nuclease of A. oryzae, even after treat-mentwith alkali or incubation at 100 C.(Nuclease
di-gestionwasperformedin 0.3MNaCl,asdescribedfor
analysis of RNA-DNA hybrids. Under these
condi-tions the S-1 nuclease is completely active against very low concentrations of single-stranded DNA
[30].) Theseproperties suggest the presenceofsome structures containing intramolecular regions of complementarity which rapidly reanneal following
denaturation. These structures were not further
characterized, however, and their significance is not clear. The nuclease-resistant material was ef-fectively removed from the DNA preparation by fractionation on hydroxylapatite. DNA was applied to acolumn (1 by 8 cm) of hydroxylapatite equili-brated with 0.05 M (Na)PO4 (pH 7.8) at 55 C. The DNA was eluted at the same temperature, using a linear gradient of 0.05 to 0.4 M (Na)PO4 buffer. Fractions containing single-stranded 3H-DNA (eluting at less than 0.18 M PO4) were pooled, dia-lyzed against water, and concentrated by lyophyli-zation. The lyophilized DNA was resuspended in 2 x SSC-0.1% SDS, and stored at -20 C. This DNA, which constituted approximately 70% of the 3H-DNA applied to the column, was 97 to 98% hydro-lyzed by S-1 nuclease. The DNA prepared in this waycontained more than 60% of the viral sequences as indicated by hybridization experiments of the type described by Duesberg and Canaani (12), using DNA-RNA ratios as high as 250: 1. (The specific activity of the 32P-RNA was 107counts per min per jg,based on thespecific activityof the 32P used in the growth medium during virus production.) How-ever, the majority (70-80%) of this 3H-DNA repre-sentedonlyasmall fraction(ca. 15-20%) ofthe viral genome,asindicatedbythekinetics of theannealing reaction in this study(unpublished results). Similar results have been reported for viral DNA products synthesized in the absence of actinomycin D (17, 39). Isolation of S-1 nuclease. S-1 nuclease was purified by ammonium sulfate precipitation and DEAE-cellulose column chromotography, as de-scribed by Ando et al. (1). The starting material
was crude a-amylase from A. oryzae. Enzyme prep-arations obtained from the DEAE-cellulose column werestored on ice, or at -20 C in 50% glycerol. The activity against double-stranded DNA with these preparations was less than 0.1% of the single-strand activity, under the conditions used in the hybridiza-tionanalysis (see below).
Hybridization. The annealing mixture contained
0.3 M NaCl, 0.03 M sodium citrate, 0.1% SDS, 37% formamide, 0.005
Ag
per ml of 3H-labeled DNA (except where indicated), and RNA as indicated in legends. The specific activity of the 3H-DNA was 1.8 x 107 counts per min per;g.
The reaction was performed at 45 C, using either 5 or 10Aliters
of annealing mixture in sealed 50-uliter capillary tubes. ForCrt analysis of cellular or viral RNA samples, a series of annealing reactions was performed, usingasingle RNA concentration and varying the incuba-tion time. In mostcases, samples were frozen follow-ing incubation and stored at -80 C untilincubation was completed for all samples in the series. The
extent of hybridization was analyzed by digestion with thesingle-strand-specific S-1 nuclease. Anneal-ing mixtures were diluted into 0.1 ml of a buffer solution containing 0.025 M potassium acetate (pH 4.4), 0.005 M ZnSO4, 0.3 M NaCl, 60
jig/ml
of double-stranded salmon sperm DNA, and 10 units/ml of S-1 nuclease. (One unit of nuclease is equal to thatamount which hydrolyzes 1
Mlmole
ofnucleo-tide per min under the conditions described here, 159 VOL. 11, 1973
on November 10, 2019 by guest
http://jvi.asm.org/
using250yg/ml of denatured salmonspermDNAas
substrate.) The native salmon sperm DNA was
in-cluded during the nucleasetreatmentof thehybrids
to saturate any double-strand-specific nuclease
which mightbe present in the S-1 nuclease
prepara-tion.Thesampleswereincubated for 60minat38C, and precipitated with cold 10% trichloroacetic acid, using tRNA (150 Ag/ml) as a carrier. The
hybrid-ized 3H-DNA was collected on nitrocellulose filters
as acid-insoluble material, and theradioactivitywas
determined by scintillation spectrometry. In
dupli-cateexperiments,hybridization valueswerein
agree-mentwithina rangeof ±5%.
A low level of self-annealing (up to 2-3% above
background) was observed when 3H-DNA was
incu-bated for long periods of time in the absence of
RNA. To correct for this time-dependent
self-annealing,and forvarying backgroundlevelswith
dif-ferentDNApreparations,controlmixturescontaining only 3H-DNAwere incubated inparallel with
DNA-RNA hybridization mixtures. All data have been
corrected forthebackground levels obtained in
con-trol samples incubated for comparable periods of
time. This control levelwaslessthan 5%ofthe total
3H-DNA. A positive control mixture, containing both cellular RNA(20 mg/ml)and viral RNA(1 gg/ ml), was also incubated underannealing conditions to test each RNA sample for the presence of
ribo-nuclease or other inhibitors. In every case, greater than 90% hybridization was obtained with these positive controls. Hybridization between viral 3H-DNA and RNA from helper-positivecells was
com-pletely abolished by pretreatingthe RNAsamplewith
ribonuclease. Thus, the virus-specific sequences
which annealed to the 3H-DNA probe were
con-tained inRNA, notin DNA.
Thehalf-Crt (see below) for hybridization between
RAV-2 3H-DNA and RAV-2 RNA was 8 x 10-2 mole sec/liter, usingthe conditionsdescribedabove. In 2 x SSC(without formamide), valuesof10- (60
C)and 2 x 102(68 C)wereobtained.
RESULTS
Specificity of the hybridization assay.
RAV-2 3H-DNA was synthesized in the pres-enceofactinomycin D, using detergent-treated RAV-2virions as a sourceofenzyme and RNA
template (see above). This DNA hybridized almost completely with 70s or 35s RNA from
RAV-2 (98 and 93%, respectively) (see Table
2). Thus, essentially all of the 3H-DNA
pre-pared inthiswayiscomplementarytothe viral
plus (+)-strand RNA. The RAV-2 3H-DNA
also hybridized extensively with 70s RNA from two other leukoviruses, RAV-60 and
SR-RSV (96 and 88% hybridization, respectively). This demonstrates thata considerable amount
of cross-homology is present in the RNAs of these three related viruses.
RAV-2 3H-DNAwas tested forhybridization withheterologous RNA from 3T3 and hamster embryo cells to determine the degree of
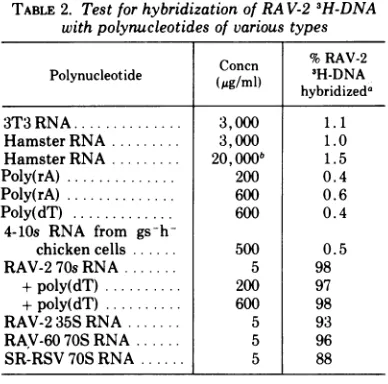
spec-TABLE 2. TestforhybridizationofRA V-23H-DNA withpolynucleotides of various types
Cnn %RAV-2 Polynucleotide Concn 3H-DNA hybridizeda
3T3 RNA... 3,000 1.1
HamsterRNA... 3,000 1.0
Hamster RNA..20,OOOb 1.5
Poly(rA) ..200 0.4
Poly(rA) ..600 0.6
Poly(dT) ..600 0.4
4-lOs RNA from
gs-h-chicken cells 500 0.5
RAV-2 70s RNA 5 98
+poly(dT) .. 200 97
+poly(dT) .. 600 98
RAV-235S RNA 5 93
RAV-60 70S RNA ... 5 96
SR-RSV 70S RNA ... 5 88
aReaction mixtureswereincubatedfor 150hr.
"Thekinetics ofhybrid formation are not affected
by this high RNA concentration. The presence of 20 mg/ml of heterologous RNA did not affect either the rate or extent of the annealing reaction between RAV-2 RNA andRAV-2 3H-DNA (see Fig. 2).
ificity in the annealing reaction. The level of
hybridization with these RNAs was less than 2% above background (Table 2). (The
back-ground level in the experiments was approxi-mately 3%.) Low levels of hybridization were also found with poly(rA) (0.6%) and with 4 to lOs RNA (0.5%) isolated from chicken
embryo cells of the type used for the virion preparations. These latter controls were in-cluded to test specifically for the possibility
that a portion of the 3H-DNA might have been transcribed from the adenine-rich se-quences of the viral RNA (18, 20, 29, 35), or from low-molecular-weight cellular RNA which
might be present in the virion (5, 13, 14, 33). Such transcripts wouldhybridize with the
ade-nine-rich sequences of cell messenger RNA
(mRNA), and with the 4 to lOs RNA in the
cell, thus interfering with ouranalysisof
viral-specific RNA in these cells. The results de-scribed above demonstrate, however, that transcripts of this type do not constitute a significant fraction of the 3H-DNA. The ab-sence of sequences in the 3H-DNA which were
complementary to adenine-rich regions of the viral RNA was further confirmed by the fact that excess poly(dT) did not compete in the annealing reaction between RAV-2 3H-DNA and RAV-2 RNA (see Table 2). Approximately 98% hybridization was obtained in the pres-ence of poly(dT) at a concentration 120-fold in excess of viral RNA and 120,000-fold in excessofRAV-2 3H-DNA.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.493.265.459.72.264.2]VIRAL RNA IN CHICKEN CELLS
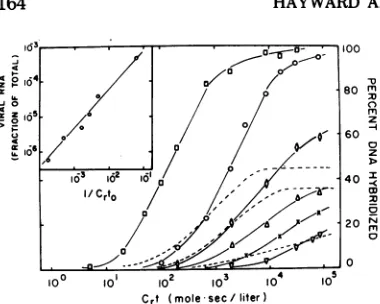
Characteristics
of the annealingreac-tion. Hybridization data can be conveniently represented on a semilogarithmic graph, plot-ting the percent DNA hybridized against the product of initial RNA concentration x time
(Crt)
(8, 30). The parameter Crt, suggested byBirnstiel et al. (4), is analogous to
Cot
(DNAconcentration x time) introduced by Britten
and Kohne (8) to describe DNA-DNA inter-actions. With this representation, the curves
obtained for a given polynucleotide species
will beidentical over a wide range of RNA
con-centrations since the incubation time (t)
re-quired to reach a given level of hybridization
is inversely proportional to the initial RNA
concentration
(Cr)
when RNA is in sufficient excess. The Crt value at which 50% maximalhybridization is attained
(half.Crt)
ischarac-teristic ofthe hybridizing RNA species and is
roughly proportional to its genetic complexity
(4,
6).
When theRNA sequenceshomologous tothe radiolabeled DNA are present in a mixture of RNA species (e.g., cellular RNA), a
Crt
curve can be constructedby
calculatingCrt
values based on total RNA concentration. The curveobtained inthiswaywill be
displaced
tohigher
Crt
values, in proportiontothe dilution ofthe homologous RNA species with heterologous RNA. The concentration of thehybridizing
RNA can be calculatedby
comparing thehalf-Crt
obtained for the RNA mixture with thatobtained
usingonly
RNAhomologous
tothe DNA
probe.
The kinetics of
hybrid
formation between RAV-2 3H-DNAand RAV-2RNAwerestudiedover a
10,000-fold
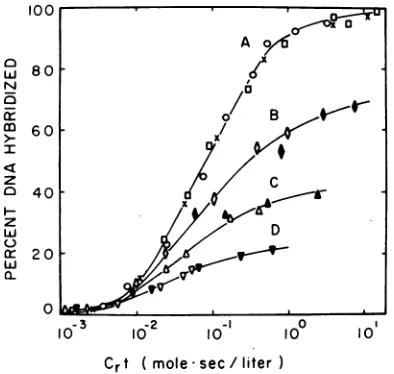
range ofviral RNA concen-trations to determine the limits ofsensitivity of ourassay (seeFig.
1). At concentrations of0.75 (curve A, open circles) to 100
Ag/ml
(X)
the points fall on the same curve,reaching
a maximum ofnearly
100%hybridization.
The linear portion of this curve,extrapolated
to 0and 100%, covers
approximately
2logs
ofCrt,
asexpected
for an ideal second-order reaction (8). Viral RNAwasreadily
detectable atlevelsas low as0.012
,ug/ml
(curveD). However,
thecurve
obtained
with this low RNA concentra-tion differedconsiderably
from the idealrep-resented by curve A, and less than 25% of the
3H-DNA was
hybridized
at thelongest
incu-bation time tested. These non-ideal kinetics are a result of the
low-input
ratio of RNA toDNA, which is
only
2.4: 1 in thesesamples
(curve D). The RNA-DNA ratio appearsto beextremely critical in these
experiments,
pre-sumably because of the
heterogeneity
of the DNA probe. Although the 3H-DNA used inthese experiments contains sequences homol-ogous to at least 60% of the viral RNA, the
majority of the DNA is homologous to only a
limited region (15-20%) of the viral genome (see above). At low RNA-DNA ratios many of
the DNA species would be in considerable
excess over the homologous RNA sequences, whereas other DNA species would not. Thus,
the kinetics of the annealing reaction would be heterogeneous because of the different re-action rates ofthe various DNA species. The heterogeneity of the DNA is reflected in the
decreased slope of curve D, and, to a lesser
extent, in the slopes of curves C and B, in
which RNA concentrations of 0.10 and 0.23
,ug/ml
were used. Apparently, evenat a ratio of45:1 (curve B), some ofthe RNAsequencesarenot insufficient excess.
Itseemed possible that the hybridization val-ues obtained with RNA-DNA ratios of 45:1 or
less might be artificiallylow due to thelong
in-cubation times(up to 500 hr) required atthese
RNA concentrations. To test this possibility,
100
--,,
A
,80 7
N o
5 /
cr ~~~~o/B <
40 4 z
ui ~~~~~~~D
20
0
-3
-°
II
,g,
10-3
1 2 10O 10° 10Cr
t ( mole*sec/literFIG. 1. Hybridization of RA V-2 3H-DNA with RAV-2 RNA. Hybridization was performed as de-scribed in Materials andMethods, using RNA-DNA ratios of 150 to 20,000 (curve A), 45 (curve B), 20
(curveC),and2.4(curveD).
3H-DNA
concentrationswere 0.060
;ig/ml
(V), 0.038,g/ml
(A, *), or0.005 ug/ml (all others). Viral RNA -was used atconcen-trations of 100 jug/ml (X), 9.0
jig/ml
(0), 0.75;tg/
ml (0), 0.225
jig/ml
(0),
1.70jig/ml
(*),
0.10jg/ml
(A), 0.75jg/ml
(A), 0.012 iAg/ml (V), and0.144
;tg/ml
(v). Annealing reactions were per-formed in the presence of heterologous RNA fromhamsterembryo cells(0) oryeast tRNA(others) at a concentration of20 mg/ml. Crt values and RNA-DNA ratios were calculated from viral RNA
con-centrations and thus do not reflect the presence of heterologous RNA.
VOL. 11, 1973 161
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.493.244.441.321.508.2]80
C)80
~~~A
Bco
1 60 C
C) D
40-CE 20 a----0
LU +
I ~~~~~~~~~~G
0 4- ---T
10-2 I 10 210P 3 4 15
10- 10 10° 10 102 103 10 10
C
rt
(mole
sec/liter)
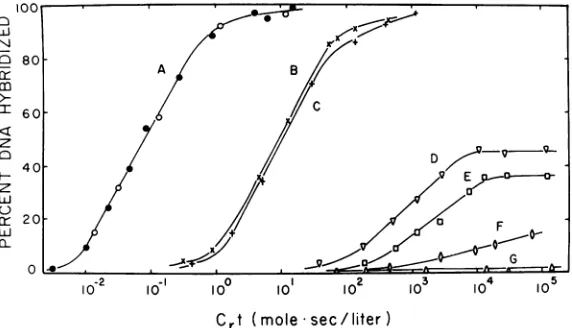
FIG. 2. Hybridization of RA V-2 3H-DNA with RNA isolatedfrominfected anduninfectedchickenembryo cells. 3H-DNAwashybridizedwith RNA isolatedfrom thefollowingsources:RA V-2 virus(curveA);RA V-2-infectedcells oftypegs-h- (curveB); andgs+hH (curve C); uninfectedcellsoftypegsLhE (curve D), gs+hH (curveE), gs-hL (curveF), andgs-h- (curveG). RNAconcentrations were9.0,ug/mlofviral RNA (curve A) in the presence(0)orabsence(0) ofwhole cell RNA(20mg/ml)isolatedfromhamsterembryo cells;3mg/ml of cellular RNA (curves B and C); and20mg/ml ofcellular RNA (curvesD-G).Approximately 1,000counts
per minof 3H-DNAwereused in eachhybridreaction. Crt valuesforcurveA were based on viral RNA concen-tration; all otherswerecalculatedfromtotal cellular RNA concentration.
samples were prepared containing
higher
con-centrations of'both RNA andDNA, butmain-tainingthesameinputratiosused in the
experi-mentsabove. The incubation times
required
toreachagivenCrt valuewillbe decreasedin pro-portion to the increase in RNA concentration. (The increase in DNA concentration will not affect the Crt curves. The data are normalized to DNA concentration
by
expressing thehy-bridization level in terms ofpercent DNA hy-bridized.) The values obtained at these higher nucleic acidconcentrations (Fig. 1,closed
sym-bols)fell onth;- same curvesobtained with sim-ilar RNA-DNA ratios at lower concentrations
and longer incubation times (open symbols). Thus,it seemsunlikelythat
hybridization
levels were significantly affectedby
nucleic acid deg-radation or other artifacts resulting frompro-longed incubation.
Very high RNA concentrations can be used in the annealing mixture without affecting the kinetics of the reaction. TheCrtcurveobtained
with RAV-2 RNA in the presence of 20 mg of heterologous RNA/ml fromhamster cells (Fig.
2, curve A, closed circles) was identical to that obtained in the absence of heterologous RNA (open circles). Neither whole cell RNA, as mentioned above, nor low-molecular tRNA
(data not shown) affected the rate or extent of the reaction at a concentration of20mg/ml,and both have been used as heterologous RNA in experiments described below.
Hybridization analysis of RNA isolated
from infectedanduninfected chickenembryo cells. When RNAisolatedfromRAV-2-infected cells was tested forthe presence ofviral RNA
by
hybridization
with RAV-2 3H-DNA(Fig.
2,curves B andC),nearly100%hybridizationwas
obtained, demonstrating that essentially all of
the nucleotide sequences which were
repre-sentedin the viral DNA were present inthe in-fected cells. The half-Crt values obtained with these infected cell RNA sampleswere 0.9 and 1.1 x 101 mole sec/liter, as compared with 8 x 10-2 mole
sec/liter
forpurified RAV-2 RNA(curve A). Based on these half-Crt values, we
have calculated that approximately 0.7 to0.9% of the infected cell RNA is viral specific, in
good agreement with previously published re-ports (9, 16, 19). This would correspond to
approximately 3,000 to4,000viral RNAcopies
percell, based on a value of1.7 x 10- I,ugof
RNA per viral genome (11) and an estimated 8 x 10-6 ,ug of total RNA per cell. The slight
differences in the
Crt
curves obtained with the infected helper-positive and helper-negative cells are notsignificant with this assay.Noviral-specific RNA wasdetected in unin-fected chicken embryo cells which lacked gs
antigenorhelperactivity (Fig.2, curve G), even at Crtvalues ofgreater than 105. However,
hy-bridformation wasdetected with RNA from all cells which contained these viral functions
(curves D-F). The levels ofhybridization with RNAs from cells with high levels of helper
ac-tivity (gs+hH and gsLh E) reached maxima of
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.493.117.403.84.248.2]VIRAL RNA IN CHICKEN CELLS
approximately 36 and 45%, respectively. The fact that these curves reached plateau levels
of lessthan100%suggeststhatmanyofthe viral
sequences represented in the 3H-DNA are not
present in the RNA from these cells. Low but significantlevelsofhybridizationwereobtained
with RNA from cells with low helper activity (gs-hL cells) (curve F). However, no plateau
value was reached at the level of sensitivity used in these experiments.
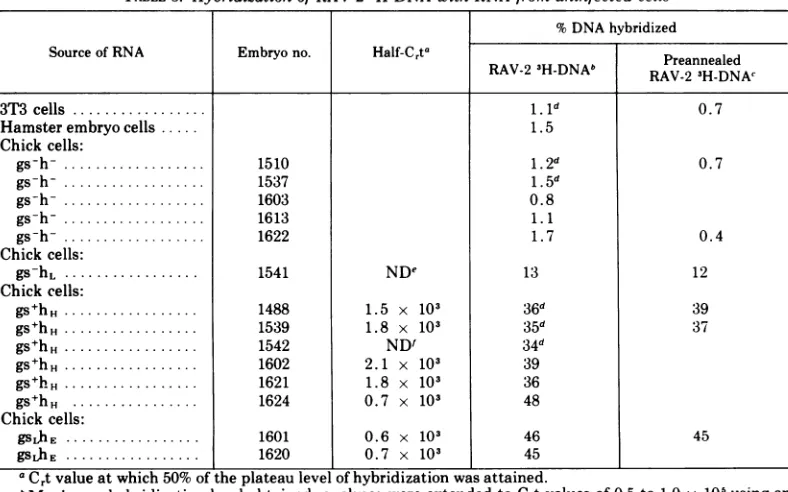
Atotal of14 embryos of differenttypeswere
examined for the presence of viral RNA (see
Table 3). No hybrid formation (<2%) was
de-tected with RNA from any of five embryos
which lackedgsantigen and helper activity. Of
nine helper-positive embryos, all contained detectable levels of viral RNA. The maximum levels ofhybridization and the half-Crt values for embryos of each type weresimilartothose shownin Fig. 2.
To exclude the possibility that some ofthe
hybridization observed with cellular RNAwas
duetothepresenceofhost cellsequencesin the
3H-DNA preparation, asampleof3H-DNAwas
pre-annealedtoviral 35sRNA and then treated with S-1 nuclease. Thistreatmentwould
elimi-nate any DNA sequences which would not
hy-bridize with the viral RNA. The DNA-RNA complexes which remainedafternuclease
treat-ment were extracted with SDS-phenol and
treated with NaOHtohydrolyze the RNA.This preannealed 3H-DNA was then used in
hybrid-izationexperiments with RNA from each of the celltypes testedabove. Levels ofhybridization obtained with the preannealed DNA (Table 3)
were essentially the same as those obtained
with DNA which had not been preannealed. Slightly lower values (0.4 and 0.7% versus 1.2
and 1.7%) were obtained with the RNA from gs-h- cells. However, values of less than 2% above
backgroun'd
arenotsignificant under the conditions used;in these experiments. Wecon-clude that the hybridization obtained using RAV-2 3H-DNA represents virus RNA-specific
sequences.
Estimation ofviral RNA content in unin-fected cells. Although the amount of RAV-2 RNA in infected cells was calculated by
com-TABLE3. Hybridization of RA V-23H-DNA with RNA from uninfected cells
%DNAhybridized
Source of RNA Embryo no. Half-Crt |
Preannealed
RAV-2 3H-DNAb RV23H-N
3T3cells 1l.d 0.7
Hamsterembryocells 1.5
Chick cells:
gs-h-.1510 1.2d 0.7
gs-h-.1537 1.5d
gs-h-.1603 0.8
gs-h-.1613 1.1
gs-h-.1622 1.7 0.4
Chickcells:
gs-hL.1541 NDe 13 12
Chick cells:
gshH *1488 1.5 x 103 36d 39
gs hH1539 1.8 x 103 35d 37
gs+hH.1542 ND' 34d
gs+hH.. 1602 2.1 x 103 39
gs+hH. 1621 1.8 x 103 36
gs+hH .1624 0.7 x 103 48
Chick cells:
gSLhE 1601 0.6 x 103 46 45
gsLhE 1620 0.7 x 103 45
aCrtvalueatwhich50% oftheplateaulevel ofhybridizationwasattained.
bMaximumhybridizationlevel obtained; analyseswereextendedtoCrtvalues of 0.5to1.0 x 105usingan
RNAconcentrationof 20mg/ml,except whereindicated.
cDetermined at a
Crt
value of 0.5 x 105. 3H-RAV-2 DNA was preannealed to viral 35s RNA and thentreated withS-1 nuclease. The DNA-RNAcomplexes whichremainedafternuclease treatmentwere phenol extracted andthentreated withNaOHtohydrolyzetheRNA.
dAnalyses extendedtoCrtvalues of 0.2to0.4 x 105using4 to5mg/mlofRNA.
eNotdetermined; analysiswas notextendedtohigh enoughCrtvaluestoreachaplateau level.
fNotdetermined; hybridization levels weredeterminedonlyathigh Crtvalues.
163 VOL. 11, 1973
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.493.47.441.354.600.2] [image:7.493.48.440.355.598.2]_o/_g,a- / O80
-jo 0
9 m
60
3
-1~~~~~~-10° 10 10 10 10
Crt ( mole sec /liter )
FIG. 3. Analysis ofhybridization levels in recon-struction experiments using heterologous RNA and various amounts of viral RNA. Reaction mixtures contained 0.005,ag/ml of RA V-2 3H-DNA, 20 mg/mI of heterologous RNA from hamster embryo cells(0), gs-h- chicken embryo cells (X), or yeast tRNA (0, , A,IV);and RA V-2 RNA at concentrations of 9.0
ygIml (A), 0.05 yg/ml (X), and 0.012 Mg/mi (C'). These amounts of viral andheterologous RNA would be equivalent to approximately 210 (0), 18 (0), 5
(O'), 2.4 (A), 1.2 (X), and 0.29 (V) copies of viral RNA per cell (see text).C,tvalues in the experiments were calculated from total RNA concentrations, rather thanfrom viral RNA concentration as in Fig. 1. Crt curves for uninfected cell RNA, shown in Fig. 2 (curves D, E, F), have been superimposed (broken lines) to facilitate comparison with the curves ob-tained in these reconstruction experiments. The presence of 20 mg/mI of heterologous RNA, whether from hamster cells, gs-h- chicken cells, or yeast tRNA, does not affect the kinetics of the annealing reaction. (Inset) Relationship between relative viral RNA concentration and Cdto values obtained in re-construction experiments.Cuti values were
deter-minedfromthe C,t curves shown above, by
extrapo-latingthe linear portionofeach curve to the baseline
(i.e., to 0% hybridization). The linearportions used for this analysis were takenfrom points 3-6on the
firsttwo curves (O, O)andpoints2-4on the remain-ing curves.
paring the half-Crt values with that of RAV-2
RNA, this method might notbevalid for
unin-fected cells containing much lower
concentra-tionsofviral RNA. To evaluateresultsobtained
with uninfected cell RNA, we have plotted a
series of Crt curves based on reconstruction
experiments in which RAV-2 3H-DNA was
hy-bridized with mixtures containing various low
amounts of viral RNA, and 20 mg/ml of
het-erologous RNA (see Fig. 3). At low RNA-DNA
ratios the
half-Cft
does not accurately reflectthe initial concentration of viral RNA, since
the kinetics of hybrid formation are affected by
the change in viral RNA concentration as the
reaction progresses. However, a Crt value
which more nearly reflects the initial reaction
rate can be obtained by extrapolating the Crt
curve to the base line. This
extrapolated value,
which we shall call Crto, appears to be a morevalidparameter for
comparison
ofhybridization
dataatlow RNA-DNAratios.When1/Crto
wasplotted against the relative concentration of
viral RNA inthe reconstruction experiments,a
nearlylinearrelationshipwasobtained,evenat
the lowest RNAconcentrationsused(seeinset,
Fig. 3). Thus, instead of half-Crt values, we
have used these Crto values for estimating the
amount ofviral RNA in uninfectedcells. A second problem inestimating the amount ofviral RNAinuninfected cellsarises fromthe
fact that only a fraction of the viral RNA se-quences were detected in these cells. Both the plateaus and the slopes ofthe Crt curves for
uninfected cell RNA (Fig. 3,broken
lines)
weresignificantly lowerthan thoseobtainedat
com-parable Crt values with RAV-2 RNA (solid lines) in the reconstruction experiments. The precisefraction ofviralRNA sequencespresent in the uninfected cells cannot be determined because of the heterogeneity of the 3H-DNA
(see Discussion). For this reason, the
concen-tration of viral RNA in these cells cannot be calculated. However, the average number of
copies of virus-specific sequences can be
esti-mated from these data. The rate of
hybridiza-tion is directly proportional to the number of
the copies of unique RNA sequences in the
reaction mixture which hybridize with the
DNA, irrespective of the molecular weight of
the RNA (4, 6). The numberofcopies of viral
RNA sequences in uninfected cells was
esti-mated from
Crto
values, using the relationship betweenCrt0
and relative viral RNA concen-tration shown in Fig. 3 (inset) as a standard. One genome equivalent ofviral RNA per cellcorresponds
to approximately 2.1 x 10-6Mg
ofviral RNA per ,ug oftotal RNA, based on a
value of 1.7 x 10- " ,g of RNA per viral
ge-nome (11) and an estimated 8 x 10-6
Ag
of total RNAper cell. Based on thesecalculations we have obtainedvalues of 36, 15, and 3copies ofviral-specific RNA percell forgs,hE,
gs+hH,
and gs-h, cells, respectively. These numbers represent average values for those sequences which hybridize specifically with the RAV-23H-DNA. The range of values for the four
em-bryos of gs+hH type was 14 to 20 copies per
cell; values of 36 and 40 copies wereobtained for the two embryos of type
gs,hE
. Oneem-bryo oftype
gs+h,,
(embryo 1624) gave a value similar to those obtained for embryos of typegsl,hE
(38 copies percell).on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.493.61.251.56.208.2]VIRAL RNA IN CHICKEN CELLS
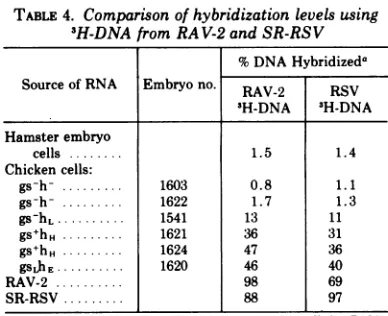
TABLE 4. Comparison ofhybridizationlevelsusing 3H-DNAfrom RAV-2 andSR-RSV
%DNAHybridizeda
SourceofRNA Embryono. RAV-2 RSV 3H-DNA 3H-DNA Hamsterembryo
cells 1.5 1.4
Chickencells:
gs-h- 1603 0.8 1.1
gs-h-.1622 1.7 1.3
gs-hL.1541 13 11
gs+hH. 1621 36 31
gs+hH ... . 1624 47 36
gsLhE. 1620 46 40
RAV-2 98 69
SR-RSV 88 97
aDetermined at aCrtvalueof 0.5 x
10'
forcellularRNA; hybridization levels for viral RNA are maximum plateau values.Hybridization
of cell RNA with 3H-DNA from SR-RSV virions. Approxi-mately 98% of the 3H-DNA synthesized fromSR-RSVvirions
hybridizes
withSR-RSVRNA, but a maximumofonly69% hybridizationwasobtained with RAV-2 RNA(see Table
4).
Thus,
asignificant fraction oftheRSV 3H-DNA rep-resents sequences which are not present inRAV-2 RNA. These additional sequences may
include viral genes involved inthe transforma-tionprocess.
It seemed
possible
that some ofthese addi-tionalsequencesmight
bepresent incells which do notcontain RNAhomologous
to the RAV-2 3H-DNA.Forthisreason,samplesofRNAfromcellsofeach typewere testedfor
hybridization
with RSV 3H-DNA. As in theprevious
experi-ments, no viral-specific RNA was detected inthegs-h- cells (see Table 4).
Significant
levelsofhybridization were observed with RNA
iso-lated from cells whichcontained gsantigen or
helper activity. The values obtained with the RSV 3H-DNAweresomewhat lower than those obtainedwith RAV-2 3H-DNA. This difference
would be expected ifthe additional sequences intheRSV DNAwerenotpresent inuninfected cells.
DISCUSSION
Previous investigators have detected
virus-specific RNA in infected chicken
cells,
butnot inuninfected cells (9, 16). The failuretodetectviralRNA inthe uninfectedcells may have been due to limits insensitivityunder theconditions employed. However, cells used in the
previous
studies were not examined for gsantigen
orhelper activity, sothe resultscannotbe
directly
compared with thosereported
here.Recently,
Leong et al.(30)havebriefly describedprelimi-nary observations suggesting the presence of
viral RNA insomenormal chicken cells.
The sensitivity of the hybridization analysis is primarily determined by three factors:
incu-bation time, RNA concentration, and specific
activity oftheradiolabeled DNA.We have tried tooptimizethe conditions forthisassay by per-forming the reaction at a relatively low tempera-ture (45 C) in thepresence of formamide, thus permitting prolonged incubation without serious thermaldegradation of RNA (4, 7). The use of S-1 nuclease to analyze hybrid
forma-tion provides several advantages over other
methods (30, 37, 41). The assay is relatively
simple, permitting the handling of large
num-bersofsamples; low and consistent background
levels areobtained; and high concentrations of
RNAcanbe used, thus increasing the sensitiv-ity ofthe assay. RNA concentrations of up to 20
mg/ml
were used in our experiments. Thishigh concentration did not affect the kinetics ofthe annealingreaction. The conditions used in these studies permit the detection of less
than 0.3viral genomeequivalents/cell, as
dem-onstrated in reconstruction experiments (Fig.
3).
In our studies we have used only
single-stranded DNA complementary to the viral RNA. Thus, the results presented here do not
apply to minus-strand RNA. Reports from
other laboratories indicate that only the plus strand of viral RNA is synthesized in virus-infected cells (9, 30). One seriouslimitation to
the use ofvirion-synthesized DNA is the het-erogeneity of the DNA product. Essentially
all ofthe viral sequences are transcribed into
DNA (12, 17). However, the transcription is
nonsymmetrical, with the majority of DNA representing a small fraction (5-25%) of the viral genome (17, 39). Thus, viral sequences
which are represented at very lowlevels inthe 3H-DNA may not be detected with this assay.
Likewise, the plateau levels of hybridization obtained with helper-positive cell RNAdo not
necessarily reflect the fraction of viral RNA
present in these cells.
No viral RNA (<<0.3 copies per cell) was
detected in uninfected cells which lacked gs
antigenor
helper
activity.However,
significantlevels of viral RNA were found in all cells which contained either of these viral functions. The amountof RNA inhelper-positive cells,ranging from 3 to 40copies per cell, correlated
reason-ablywell with the level ofhelperactivity. Only
afraction of the viral RNAsequencesappeared
tobe present in thesecells, however. This was indicated bothbythe lowplateaulevels andby
the decreasedslope ofthe
Crt
curves forcellu-VOL. 11, 1973 165
on November 10, 2019 by guest
http://jvi.asm.org/
lar RNA as compared with those for RAV-2 RNA at similar C,t values (see Fig. 3). The
analysis of RNA from cells with low helper activity was not carried to a high enough Crt
value to determine a plateau level. However,
the slope obtained with this sample was lower than that forcomparablereconstructioncurves,
suggestingthat a similarplateau would be ob-tained if the assay were extended to higher
levels of sensitivity. Several
possibilities
can be offered to explain these results. (i) A sig-nificant portion of the viral genes in these cells may be repressed or expressed at levels toolow to be detected in this assay. (ii) The viral genes in these cells may contain significant
regions which are not homologous with the RAV-2 3H-DNA. However, similar plateau levels were obtained with 3H-DNA from
SR-RSV. Furthermore, a high degree of
cross-homology was observed between RAV-2
3H-DNA and RNA from two other leukoviruses, SR-RSVand RAV-60. The RAV-60preparation
was derivedfromcellsofthetypestudiedhere
and presumablycontains viral
genetic
material fromthe host cell.(iii)Thecellsmay containin-complete viral genomes. This could explain why spontaneous
production
ofvirus particlesis never observed in these cells. RAV-60 is
recoveredonly after infectionwith other leuko-sis or sarcoma viruses.
Furthermore,
the induc-tion ofvirus production has notbeen observed inthese cellsby
treatmentwithvariouschemi-cal or physical agents (T.
Hanafusa,
personal communication). Cells used in these studiesappear to differ in this respect from those
studied by Weiss et al. (44), which
produced
low levels of virusfollowing
treatment withchemicaland physicalagents.
The average numberofviral RNA copies in
uninfected cellsorwhether it is inducedbythe
mately 1% ofthat in infected cells. Thiscould
mean either that 1% ofthe cells contain viral
RNA at high
levels, equal
to that in infectedcells, orthatall cellscontain low levelsofviral RNA. No virus-specific RNA sequences were
detected in uninfected helper-negative cells.
However,the presence of one cell per 105 which contained viral RNA in amounts equal to that in the infected cells would not be detected at the levels ofsensitivityused in thisassay.
The presence of helper activity in chicken cells is determined by abiological assay which
involves infection withB-RSV. It is notknown
whether this function is normally expressed in uninfected cells or whether it isinduced by the
infectingvirus. Thehybridizationanalysis used in these studies does not permit identification
of the individual RNA species which code for this function.However,thecorrelationobserved
between helper activity and viral RNA levels inuninfected cells suggests that the viral genes are expressed even in the absence of'viral in-fection.
Previous experiments have demonstrated
that helper-negative cells from these chicken
flocks contain viral DNA which can be de-tected both by molecular hybridization (34)
and byrescue ofRAV-60 following
superint'ec-tion (25). The absence of detectable levels of viral RNA in these cells suggests that viral gene expression is controlled at the
transcrip-tion level. However, a mechanism which in-volves rapid breakdown of viral RNA cannot be excluded.
ACKNOWLEDGMENTS
We thank T. Hanafusa for helpful discussions, and
Grazina Boeckforexcellent technical assistance. Wewould
also liketothank H. FanandD.Baltimore forproviding
in-formation concerning the use of single-strand-specific
nu-clease forhybridization analyses. This work was supported
by U.S. Public Health Serviceresearch grants CA08747 and
CA12177 from theNationalCancer Institute.
LITERATURE CITED
1. Ando, T. 1966. A nuclease specific forheat-denatured
DNA isolated from a product ofAspergillus oryzae.
Biochim. Biophys. Acta 114:158-168.
2. Baltimore, D. 1970. RNA-dependent DNA polymerase in virions of RNA tumor viruses. Nature (London)
226:1209-1211.
3. Baluda, M. A. 1972. Widespread presence in chickens
ofDNA complementary to the RNA genome of avian leukosisvirus.Proc. Nat. Acad.Sci. U.S.A. 69:576-580.
4. Birnsteil, M. L., B. H. Sells, and I. F. Purdom. 1972. Kinetic complexity ofRNA molecules. J. Mol. Biol.
63:21-39.
5. Bishop, J. M., W. E. Levinson, N. Quintrell, L. Fan-shier, and J. Jackson. 1970.Thelowmolecularweight RNAs of Roussarcoma virus. I. The 4S RNA. Virology 42:182-195.
6. Bishop, J. O. 1969. The effect of genetic complexityon
thetime-courseofRNA-DNAhybridization. Biochem.
J. 113:805-811.
7. Bonner,J., G. Kung, and I. Bekhar. 1967. A method of hybridization of nucleic acid molecules at low
temper-ature.Biochemistry 6:3650-3653.
8. Britten, R. J., and D. E. Kohne. 1968. Repeated
se-quences inDNA. Science 161:529-540.
9. Coffin, J. M., and H. M. Temin. 1972. Hybridization of Rous sarcoma virus DNA polymerase product and
RNA fromchicken and rat cells infected with Rous sarcomavirus. J. Virol. 9:766-775.
10. Dougherty, R. M., and H. S. DiStefano. 1966. Lack of relationship between infection with avian leukosis virus and the presence of COFAL antigen in chick embryos. Virology 29:586-595.
11.Duesberg, P. H. 1970. On the structure of RNAtumor
viruses.Curr. Top. Microbiol.Immunol. 51:79-104.
12. Duesberg, P. H., and E. Canaani. 1970. Complementar-ity between Rous sarcoma virus (RSV) RNA and the
invitro-synthesized DNA of the virus-associated DNA polymerase. Virology 42:783-788.
on November 10, 2019 by guest
http://jvi.asm.org/
VIRAL RNA IN CHICKEN CELLS
13. Erikson, E., and R. L.Erikson. 1971.Association of 4S ribonucleic acid with oncornavirus ribonucleic acid.
J.Virol. 8:254-256.
14. Erikson, R. L. 1969. Studies on the RNA from avian
myeloblastosisvirus. Virology 37:124-131.
15. Fleissner, E. 1971. Chromotographic separation and antigenic analysis of proteins ofthe oncornaviruses. I. Avianleukemia-sarcomaviruses. J. Virol. 8:778-785. 16. Garapin, A. C., J. Leong, L. Fanshier, W. E. Levinson,
and J. M.Bishop. 1971.Identificationof virus-specific
RNA in cells infected with Roussarcomavirus.
Bio-chem.Biophys. Res. Commun. 42:919-925.
17. Gelb, L. D., S. A. Aaronson, and M. A. Martin. 1971.
Heterogeneityof murineleukemiavirusin vitroDNA;
detection of viralDNA in mammalian cells. Science 172:1353-1355.
18. Gillespie, D., S. Marshall, and R. C. Gallo. 1972. RNA
ofRNAtumorviruses contains polyA.Nature N.Biol.
236:227-231.
19. Green, M., H. Rokutanda, and M. Rokutanda. 1971.
Virus specific RNAincellstransformed by RNAtumor
viruses.Nature N. Biol. 230:229-232.
20. Green, M., and M. Cartas. 1972. ThegenomeofRNA
tumor viruses contains polyadenylic acid sequences.
Proc. Nat. Acad. Sci. U.S.A. 69:791-794.
21. Hanafusa, H. 1965.Analysis of the defectivenessofRous
sarcoma virus. III. Determining influence of a new
helper virus on the host range and susceptibility to
interferenceof RSV.Virology25:248-255.
22. Hanafusa, H., and T. Hanafusa. 1966. Determining
fac-tor in the capacityofRous sarcoma virusto induce
tumorsin mammals. Proc. Nat.Acad. Sci. U.S.A.55:
532-5.38.
23. Hanafusa. H., T.Miyamoto, and T. Hanafusa. 1970. A cell-associated factor essential for formation of an
infectious form of Rous sarcoma virus. Proc. Nat.
Acad. Sci.U.S.A. 66:314-321.
24. Hanafusa, T., H. Hanafusa, and T. Miyamoto. 1970.
Recoveryofa newvirus fromapparentlynormal chick
cellsbyinfection with aviantumorviruses. Proc. Nat.
Acad.Sci. U.S.A. 67:1797-1803.
25. Hanafusa, T., H. Hanafusa, T. Miyamoto, and E. Fleissner. 1972. Existence and expression of tumor virusgenesin chickembryocells.Virology47: 475-482.
26. Hehlman,R.D.,D.Kufe,and S.Spiegelman.1972. RNA inhumanleukemic cells relatedtothe RNA ofamouse
leukemia virus. Proc. Nat. Acad. Sci. U.S.A. 69:435-439.
27. Huebner, R.J.,D.Armstrong,M. Okuyan,P.S. Sarma,
and H. C. Turner. 1964. Specific complement fixing viralantigens in hamster and guinea pigtumors
in-ducedbytheSchmidt-Ruppinstrainof aviansarcoma
virus. Proc. Nat. Acad. Sci. U.S.A.51:742-750. 28. Kawai, S., and H. Hanafusa. 1972. Plaque assay for
somestrains of avian leukosis virus.Virology 48:126-135.
29. Lai,M. M.C.,andP. H.Duesberg. 1972.Adenylic
acid-rich sequences in RNAs of Rous sarcoma virus and Rauschermouseleukemia virus. Nature(London)235: 383-386.
30.Leong, J., A. Garapin, N. Jackson, L. Fanshier, W.
Levinson, andJ.M.Bishop. 1972.Virus-specific ribo-nucleic acid in cells producing Rous sarcoma virus: detection andcharacterization. J.Virol.9:891-902.
31. Payne, L. N., and R. C. Chubb. 1968. Studies on the
nature and genetic control of an antigen in normal
chick embryos which reacts in the COFALtest. J. Gen.Virol. 3:379-391.
32. Payne, L. N., P. K. Pani, and R. A. Weiss. 1971. A
dominant epistatic gene which inhibits cellular
sus-ceptibilitytoRSV(RAV-O). J. Gen. Virol. 13:455-462. 33. Robinson, W. S., A. Pitkanen, and H. Rubin. 1965.The nucleicacid of the Bryanstrain ofRoussarcomavirus:
purification of thevirus and isolation ofthe nucleic acid. Proc. Nat.Acad. Sci. U.S.A.54:137-144.
34. Rosenthal, P. N., H. L. Robinson, W. S. Robinson, T. Hanafusa, and H. Hanafusa.1971. RNAin uninfected
andvirusinfectedcellscomplementarytoaviantumor virus RNA. Proc. Nat. Acad. Sci. U.S.A. 68:2336-2340.
35. Ross, J., S. R. Tronick, and E. M. Scolnick. 1972.
Polyadenylate rich RNA in the70S RNA of murine leukemia-sarcoma virus. Virology49:230-235. 36. Scherrer, K. 1969.Isolation andsucrosegradient
analy-sis of RNA. pp.413-432. InK. Habel and N. P.
Salz-man (ed.),Fundamental techniques in virology.
Aca-demic PressInc., New York.
37. Sutton, W. D. 1971. A crude nucleasepreparation suit-able forusein DNAreassociation experiments. Bio-chim. Biophys. Acta 240:522-531.
38.Temin, H. M.,and S. Mizutani. RNA-dependent DNA polymerase invirions of Rous sarcomavirus. Nature
(London)226:1211-1213.
39. Varmus, H. E., W. E. Levinson, andJ. M.Bishop. 1971.
Extentoftranscription by the RNA dependent DNA polymerase of Rous sarcoma virus. Nature N. Biol. 233: 19-21.
40. Varmus, H. E., R. A. Weiss, R. R. Friis, W. Levinson, and J. M. Bishop. 1972. Detection of avian tumor
virus-specificnucleotidesequencesin avian cell DNA. Proc. Nat. Acad. Sci. U.S.A. 69:20-24.
41. Verma, I. M.,G. F. Temple, H. Fan, and D. Baltimore.
1972. In vitro synthesis of DNA complementary to
rabbit reticulocyte 10S RNA. Nature N. Biol. 235: 163-167.
42. Vogt,P.K. 1965. Aheterogeneity of Roussarcomavirus revealed by selectivity resistant chick embryo cells. Virology 25:237-247.
43. Weiss, R. A. 1969 The hostrangeofBryanstrain Rous
sarcoma virus synthesized in the absence of helper virus. J. Gen.Virol. 5:511-528.
44. Weiss, R.A.,R. R.Friis,E.Katz,and P. K.Vogt.1971. Induction of avian tumor viruses in normalcells by physical and chemical carcinogens. Virology 46:920-938.
45. Weiss, R. A., and L. N. Payne. 1971. The heritable
na-tureandgeneticcontrol of the factor in chicken cells
which acts asahelpervirus forRous sarcomavirus.
Virology 45:508-515.
VOL. 11, 1973 167