0022-538X/82/120886-07$02.00/0
Copyright©1982, American Society for Microbiology
Lethal Action of Bacteriophage
X S
Gene
JINNIE M.
GARRETT
AND RY YOUNG*Departmentof Medical Biochemistry, Texas A&M University,CollegeStation, Texas 77843
Received4June1982/Accepted5 August 1982
Thefunctionsofthe bacteriophage X lysis genes S, R, and Rz were investigated.
Different combinations of wild-type and inactive alleles ofall three lysis genes
wereclonedinto the plasmid pBH20 and were expressed under the control of a lac operator-promoter. Theinvolvement ofthe Rz gene inlysiswasproposedin our
previouswork andwasconfirmedby theMg2+-dependent lysisdefect of clones in
which part of the Rz gene is deleted. Membrane vesicles preparedfrom induced S+ cells were showntohaveaseverelyreducedcapacityfor active transport of glucose; thisdefect was detectableatleast 20minbeforelysis.Cellviability was also shown to decrease verysoonafterinduction,long beforephysiologicaldeath
and lysis;this decrease inviability isabsolutely dependenton Sexpression and
independent of R and Rz. The nonviable fraction of cells at any time after
induction was demonstrated tobe equal to the fraction committed to eventual lysis. Induction ofan Sts clone showed that the S gene product is stable and
capableofinducing lysis long afterthecessation ofsynthesisof S geneproduct.A
modelfor Saction is proposed.
Theterminatingeventof the infective cycle of
many viruses is lysis of the host cell with
con-comitantrelease of the progeny virus particles.
Three genes have been directly implicated in lysis of thebacteriophage X: S, R, and Rz (16, 19). The X lysis gene region has been cloned undercontrol of the lac promoter in theplasmid
pBH20 (9). Inductionof thelysis operon by the
addition ofthegratuitouslac inducer
isopropyl-P-D-thiogalactopyranoside
(IPTG)results in celllysisin the S+R+Rz+ clone. It is mucheasierto
studythemechanismofaction ofthe individual
lysis proteins in the cloned system, since the
lysis genes are removed from themultiple con-trols ofbacteriophage development.
Two of the lysis proteins, those coded by genes R and Rz, have been implicated in the
degradation of therigid peptidoglycan cell wall
(2-4). Together these activities constitute the
bacteriolytic activity previously designated as
endolysin (18). The R gene product (pR) has
beenpurifiedandbeenshowntobea
transglyco-sylase which degrades the peptidoglycan by attacking the glycosidic linkages (3). The Rz gene product (pRz) has been tentatively identi-fied as an endopeptidase which is believed to play aminor role inpeptidoglycan degradation bydigesting oligopeptide cross-links within the peptidoglycan (19).
The role of the S gene product (pS) is un-known, but there is evidence that it isrequired
for the transport ofbacteriolytic activityto the periplasm. Ourprevious work showed that in-duction ofthe S-R+Rz+ clone results in
exten-siveaccumulationof intracellular endolysin (9);
however, nolysis or reduction in the rate of cell growth was observed, indicating that endolysin was not reaching the vicinity of the peptidogly-can.
In early work on the mechanism of A lysis,
Reader and Siminovitch(17) suggested thatpS attacks the cytoplasmic membrane to allow transport of endolysin to the periplasm. They observed that in the case of induced
S+R-lysogens the cytoplasmic membrane becomes
permeabletosucrose; thatis,thesecellscan no
longer be plasmolyzed by hypertonic sucrose. This suggests that the normal integrity of the membrane has beendisrupted. However,
elec-tronmicroscopicexamination of thin sections of
induced XS+R- lysogens beyond the normal
lysis time revealedno detectable change in the cytoplasmic membrane structure. There is no detectablephospholipase activityattributableto pS; in the absence ofSgenefunction,celllysis
by mechanical methods results in a small amount of lipid degradation indistinguishable
from that occurring in X S+-infected cells (14). Other workers have noteddeleterious effects of pS action on membrane-dependent oxidative metabolism (1, 6), whichalso suggestsaninner membrane lesion of some kind. However, no model forSaction has beenproposed.
Inthis work wehaveuseda complete setof cloned S+R+Rz+
lysis
gene allelesto study the action of theSgene.Wedocument theeffect of induction of these cloned lysis genes on cell viability, membrane permeability and active886
on November 10, 2019 by guest
http://jvi.asm.org/
LETHAL S
transport,andlysis kinetics. A general model for
pS action is proposed.
MATERIALSANDMETHODS
Bacterial strains, phages, and plasmids. The host
bacterium formost experiments wastheEscherichia
coli strain WFK, which is W3101 pro phe/F-lacIq
lacZ::Tn5 (9). The methylation-deficient E. coli K-12
strain GM119 (F- dem dam-3 metBI gaIK2 gaIT22
lacY) tsx-18 supE44) (obtained from J. A. Arraj)was
usedfor the preparation of unmethylated phage and
plasmid DNA. Thebacteriophages and plasmids used
arelisted in Table1.
Chemicalsandenzymes.Ethidiumbromide, EDTA,
IPTG, phosphoenolpyruvate (PEP),
glucose-6-phos-phate dehydrogenase, lysozyme, and the restriction
enzymeEcoRI werepurchased from Sigma Chemical
Co., St. Louis, Mo. T4 polynucleotide ligase was
obtained from NewEngland Biolabs, Beverly, Mass.
Allother restrictionenzymes wereobtainedfrom
Be-thesda Research Laboratories, Gaithersburg, Md.
[14C]sucrose (477 mCi/mmol) and [14C]glucose (500
mCi/mmol)werepurchased from New England
Nucle-arCorp.
Media andgrowth conditions. Cultures for all
experi-ments except membrane vesicle preparations were growninminimal media (7) supplemented withamino
acids and thiamine. Antibiotics usedwere ampicillin
(50 ,ug/ml) andkanamycin (30 ,ug/ml).
For lysis kinetics and cell viability experiments,
culturesweregrowntoadensity of10'cellsperml and
were induced by the addition ofl0' M IPTG. Cell
viability was followed by plating dilutions of the
culture on broth plates (1% tryptone, 0.5% yeast
extract, 0.5% NaCl) atgiven times before and after induction.
Cultures for membrane vesicle preparations were
grownin rich media(3.2% tryptone, 2.0%o yeast
ex-tract, 0.5% NaCI) with vigorous aeration and were
harvestedataconcentration ofapproximately5 x 109
cellsperml.
Plasmid construction. The S+R-Rz+ (pJG1) and
S-R-Rz+ (pJG3) plasmids were constructed as
de-scribedpreviously (9).TheS+R+Rz- plasmids (pJG7,
pJG8, and pJG9) were prepared by digestion of the
unmethylatedparentplasmid DNA (prepared from the
dam- strain GM119) with BclI and BamHI and
religa-tion. TheStsR+Rz- plasmid(pJG13)wasconstructed
by digestion of unmethylated phage DNA withEcoRI
and BclI and by ligation of the isolated lysis gene
fragment into pBH20 DNA cleaved with EcoRI and BamHI.
Transformation was performed with cryogenically
stored competent cells by the method of Morrison
(13).Transformants containing the lysis plasmidswere
identifiedaspreviouslydescribed (9).
Membrane veside experiments. Membrane vesicles
werepreparedfrom bacterial cultures by a
modifica-tion of the lysozyme-EDTA methodof Kaback (11).
Cultureswereharvestedby centrifugationat16,000x
gandweresuspendedatroomtemperature in 30 mM
Tris-hydrochloride, pH 8.0, (1g[wet weight] ofcellsper
80ml) containing 20%osucrose. The cells were
con-verted to spheroplasts by adding potassium EDTA,
pH 7.0,andlysozymetofinalconcentrations of 10 mM
and 50,ug/ml, respectively, and incubating for 1 h. The
spheroplastswerecollectedby centrifugationat16,000
x g for 20 min and were suspended with a glass
homogenizerinthe smallestpossible volume of0.1M
potassium phosphate buffer, pH 6.6, containing20%
sucrose, 20 mM magnesium sulfate, and 5 ,ug of
DNaseperml. The suspension was poured into 300
volumes of 50 mM potassium phosphate buffer, pH
6.6,at37'C andwasincubated withvigorous swirling
for 45 min. After 15 min of incubation, potassium
EDTA, pH 7.0, wasaddedtoafinalconcentration of
10 mM, and after a further 15 min of incubation,
magnesium sulfatewasaddedto15 mMfinal
concen-tration.The suspensionwasthencentrifugedat45,000
x g for 1 h to collect the membrane vesicles. The
vesiclesweresuspended byhomogenization in0.1 M
potassium phosphate, pH 6.6,ataconcentration of 7
mgofproteinperml, quick-frozen in ethanol-dry ice,
and stored at -80°C. Vesicles were prepared from
inducedlysis clonesatvarious timesafter the addition
ofIPTG.
Membrane vesiclesweresuspendedata
concentra-tion of 35 mg/ml in 40 ,ul of 0.1 M potassium
phos-phate, pH 6.6, containing4mMunlabeledsucroseand
10,uCi of [14C]sucrose andwereincubated overnight
at4°C. At timezero,thelabeledvesicleswerediluted
TABLE 1. Phagesandplasmids
Designation Genotype Sourceor Reference
Phages
X363R- XbSl9b5l5cI857nin5RTn903.979 R. Young
X363Sam7R- XbSl9bSl5cI857nin5Sam7RTn9O3.979 R. Young
ASts XcI8575ts9B D.B. Wilson
Plasmids
pBH20 AprTcr DerivedfrompBR322 (10)
pRF26 AprS+R+ Rz+ Derived frompBH20 (9)
pJH2 Apr Sam7 R+Rz+ Derived frompBH20 (9)
pJG1 Apr S+R-Rz+ Derived frompBH20 (this paper)
pJG3 AprSam7R-Rz+ Derived frompBH20(thispaper)
pJG7 Apr S+R'Rz- DerivedfrompBH20(this paper)
pJG8 AprS+R-Rz- Derived frompBH20(thispaper)
pJG9 AprSam7R-Rz- DerivedfrompBH20 (thispaper)
pJG13 AprSts9BR+Rz- DerivedfrompBH20(thispaper)
pKY2 AprTCs Derived frompBH20(thispaper)
VOL.44,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.491.46.446.517.677.2]200-fold into 0.1 Mpotassiumphosphate(pH6.6)40
mM MgCl2, and atvarious times 2-ml samples were removed, filtered through Millipore filters (pore size, 0.45 pLm),andwashed under suction with 5 x 1 ml of
0.1 MLiCl. The filters were dried and counted in a
TracorAnalytic Delta 300 liquid scintillation counter.
Glucose-6-phosphatewasassayed by the method of
Olive and Levy (15), and the protein concentration
wasdeterminedby theBradford dye-binding assay (5).
RESULTS
Construction of hybrid lysis operons. In previ-ouswork(9), the Alysisgenes S(bothS+and S-alleles) and R were cloned into the plasmid pBH20(Fig. 1). Tocomplete the set of cloned genes, the S+R- and S-R- alleliccombinations werecloned by identical methods; the resultant
plasmids carryingS+R- and S-R- hybridlysis
operons were designated pJG1 and pJG3, re-spectively. Whereas induction of the S+R+ clone with 10-3 M IPTG results in a sharply
definedlysis after40min (9;Fig.2),induction of
acultureoftheS+R- cloneresults in an eventu-al heventu-alt in mass accumulation (Fig. 2), and the cells appear tolose motility undermicroscopic
observation (data not shown). Predictably,
in-duction of the S-R- clone has no detectable
effect on culturegrowth.
Verification that no other genes
promoter-distaltoRz (19;Fig.1) areinvolved in cellkilling
orlysiswasobtainedbyconstructinganother set
of plasmids in which the region of A DNA
beyondthe BclI site within the Rz gene (Fig. 1)
wasdeleted by digestion withBclI and BamHI and by subsequent religation. This series of
clones,which alllacktheintact Rz gene,
result-ed in lysis kinetics identical to those of the
parentalclones,showingthat there are no other
X genes involved in lysis (data not shown). However, the S+R+Rz- clone did show
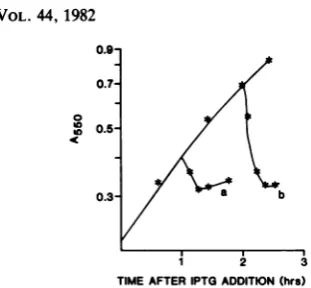
aber-rantlysis in the presenceof10 mM Mg2+ (Fig.
3).This aberrantlysispatternisassociatedwith
theformationofmechanicallyfragilespheroidal
cells and constitutes the Rz- phenotype de-scribed forbacteriophages carryingthe kanamy-cin resistance transposonTn9O3 inserted in the Rzgene (19).
0.8-
0.4-
0.2-c
b
a
1 2
[image:3.491.277.446.61.209.2]THMEAFTERFTGADDITKON (a)
FIG. 2. Induction of lysis gene clones. Cultures
were grown inglycerol-minimalmedia (7) at 37°C to a
concentrationof108cells per ml. Thelysisgenes were
induced by adding 1 mM IPTG and culture mass
accumulation followed. (a) S+R+Rz+; (b) S+R-Rz+;
(c)Sam7R+Rz+; A550, absorbance at 550 nm.
The StsR+Rz- clone was constructed by
di-gestion of
XSts
DNA with identical restrictiondigestions andligation;the clonescontaining the
lysis genes were screenedfor inducible loss of
viability at the permissive temperature (30°C).
Inductionofthetemperature-sensitive Sgeneat
therestrictive temperature (42°C)hadno effect
on culture growth (Fig. 4). Subsequent shift of
the culture to 30°C resulted in immediate
lysis
(Fig. 4).
The functional stability of the
temperature-sensitive S protein was investigated. Cells that
had been induced for 1 h at the restrictive temperature were diluted 25-fold into
nonin-ducer medium(Fig. 5).Thisdilution(5 x
10-4
Mto 2 x 10-5 M) was shown to decrease
1B-galactosidasesynthesis50-foldfromtheinduced
level and to suppress gene S killing activity
(Table 2) and should therefore turnoffpS
syn-thesisatthe timeof dilution. Temperatureshift
ofthe cultureto30°Cafterincubationindiluted
mediumfor1 hresulted inimmediatelysis(Fig.
5). This indicates that pSts is stable within the
cellattherestrictivetemperature.
0.4-"' 0.2-H/ndE
Tn905 PR' TR
. S R z
4.5. l 46 465 47 T 47.5 48 485(KIo-bp)
EwRI Bc/I
FIG. 1. Genetic mapof thelysisregion of phageX.
Thepositions of the lysis genes and relevant
restric-tion enzyme sites are shown. The stem and loop
structurerepresents the transposonTn9O3 inserted at
48.5kilobasepairs.
1 2
TIME AFTERPTGADDMTON(w)
FIG. 3. The effect of magnesium on lysis in the
S+R+Rz- clone. Cultures of strain WFK(pJG7) were
grown and induced as described in thelegend to Fig. 2
inglycerol-minimalmedium without (a) and with (b) 10
mMmagnesium chloride.
b
a
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.491.282.426.524.633.2] [image:3.491.53.247.568.675.2]t
0.5-oc
0.3 b
1 2 3
TIME AFTERIPTG ADDITION (hrs)
FIG. 4. Induction of the temperature-sensitive S
protein. A cultureof strain WFK(pJG13)(StsR+Rz-)
was grown at42°C in glycerol-minimal medium and
wasinduced by adding 1 mM IPTG. Portions of the
culturewereshiftedto30°Cafter incubation in
induc-ing conditions for1 h (a)or2 h(b).
Effect of gene S on membrane permeability.
Since gene S is implicated in the transport of bacteriolytic activity totheperiplasmand in the permeability of the inner membrane to sucrose
(9, 17), it would be reasonable to expect that membranes isolated from induced S+ lysis cloneswould have altered permeability
proper-ties. To characterize the action of pS on the membrane, we prepared membrane vesiclesby
the method of Kaback (11) from the induced S+R- and S-R- lysis clones as well as from
cells containing no cloned lysis genes. Mem-brane vesicles prepared by this method have
0.10-
08-
0.6-
0.4-
02-
0.1-06.
0.04-
0.02-I
PI
3TC
1 2 3 4 5
TME Ow.)
FIG. 5. Stability of the temperature-sensitive S
protein. A culture ofWFK(pJG13) (StsR+Rz-) was
grownandinducedasdescribed in the legendtoFig. 4. After1 hofincubation, aportion of theculture was
[image:4.491.51.207.55.199.2]diluted25-fold intononinducermedium; 2hlater, the culturewasshiftedto30°C.
TABLE 2. Effect of IPTG concentrationonlossof
cellviabilitya
% Survivalattime after
IPTG concn (M) induction:
20min 60min
1 x lo-, 27 3.6
5 x 10-4 35.6 4.4
1 x 10-4 75.7 7.6
2 x10-5 128.0 285.0
a Cultures of strain WFK(pRF26) (S+R+Rz+)were
induced with various concentrations of IPTG. Cell
viability was followed by plating dilutions of the
cultureon noninducer rich medium atvarious times
afterIPTG addition. Cell viability is expressedasthe
percentageofsurvivingcellsatthattime,with cellsat
timezero as100o.
been shown tobe whole cell vesicles, withthe samepermeabilitybarriersasthe intact cell(11). These vesicles were used in passive sucrose diffusion experiments. Vesicles from the S+ clonewereapproximatelyfourfold more perme-able to sucrose thanwerevesiclespreparedfrom control bacteria(Table3).Surprisingly,the
vesi-clesaffected by Sam7 (S-) were alsofound to
have increased permeability to sucrose (Table
3).Webelievethis is an indication that the Sam7 nonsense fragment retains some function; in
fact, induction of the Sam7 clone results in
accumulation oflargequantitiesof thenonsense fragmentpolypeptide (55 amino acidresidues;8; D. L.Daniels, Ph.D. thesis,Universityof Wis-consin, Madison, 1981) in the membrane (E. A. Altman, J.Garrett,R.Grimaila,R.Schultz, and R. Young, manuscriptinpreparation).
Effect ofgene S on PTS-mediated active trans-port.In E.coli membranevesicles,theuptakeof severalsugars (e.g.,glucose,fructose)occursby
TABLE 3. Passive diffusion of ["4C]sucrose from
membrane vesiclesa
Initial Strainsupplyingvesicles t1/2(min)b rateof
effluxc
WFK(pBH20) 2 7.5
WFK(pJG)(S+R-) 0.46 32.6
WFK(pJG3) (Sam7R-) 0.43 34.9
a Membrane vesicles were passively loaded with
[14C]sucrose overnight, washedquickly, diluted into
label-free medium, and assayed for the retention of
[14C]sucroseatvarious times(14). Maximumcapacity
of the vesicle samples was estimated as 30 pmol of
[14C]sucrose per mgofprotein.
btj/2, Time taken for one half of the maximum
capacity of vesicle samples to diffuse out of the
vesicles.
cCalculated from [14C]sucroselost in the first30 s
ofincubation;expressed aspicomolesofsucroseper
minute permilligramofprotein.
VOL.44,1982
II I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.491.86.210.430.624.2]phosphorylation via the
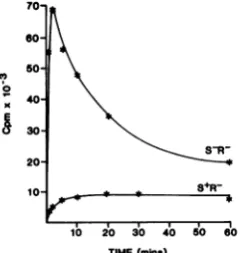
PEP-phosphotransfer-ase system (PTS). We used the S+-induced vesicles to determine the effect of pS PEP-driven [14C]glucose uptake in these membrane vesicles. S+-affected vesicles were completely
deficient in PTS activity, in contrast to
S--affected and control vesicles (Fig. 6). This result was verified by assaying the
glucose-6-phosphate produced by this system. Each
ex-periment was allowed to continue for 2 h to
allow all theglucose-6-phosphate todiffuseout
of the vesicles before starting the assay. In vesicles madeafter only 15 min of S+ induction, the membraneloses the ability to support PTS-mediated phosphorylation of glucose (Table 4). No effect is observed on glucose-6-phosphate
production in the control and S--affected vesi-cles. Thus,theloss of theabilitytosupport PTS-mediated activetransportof glucoseoccurswell
before normal lysis time in S+-affected
mem-branes. Furthermore, the Sam7-affected
mem-braneswereessentially normal in their abilityto
support PTS-mediated active transport, despite
theirincreased permeabilityto sucrosein vitro.
Kinetics of the loss of cell viabilityoninduction
of the Sprotein. The observed effect of thegene
Sprotein on the PTS before normal lysis time
led us toinvestigate the kinetics of the loss of cellviability in bacteria exposedtotheaction of the gene S protein. The sharply defined lysis curveobservedat40minfortheS+R+ clone is accompaniedby the loss ofcolony-forming units beginning much earlier, about 8minafter induc-tion(Fig. 7). Identical resultsareobtained with
x
70- 60-50
40
30
20
10
lb io 3'0 io 5o 60
TIME(mine)
FIG. 6. The effect ofpSonPTS-active transport in membrane vesicles. Membrane vesiclespreparedfrom
the S+R- and S-R- clones were suspended at a
concentration of 20mgofprotein perml in 50 mM
potassium phosphate buffer(pH 6.6)-10mM
magne-siumsulfate-0.3 MLiCI-0.1 M PEP(adjusted topH
6.5 to7.0 with sodiumcarbonate). Thesamples were
incubatedat40°C. Atvarious timepoints,40-,ul
sam-pleswereremoved, dilutedinto 2 ml of0.5 MLiCl,
rapidly filtered through a Millipore filter (pore size,
0.45 Lm)undersuction,and washedwithanother 2 ml of 0.5 MLiCl. The filtersweredried andcounted.
TABLE 4. Effect of gene S expression on
PTS-mediated active transporta
Time Glucose PTS
Relevant ~~of transport
Relevant Strain
incuba-genotype tion Amtb %of
(min) control
Host WFK 16.2 81
Control WFK(pKY2) 20.9 100
S-R- WFK(pJG3) 0 14.9 74
15 14.9 74
30 14.3 71
45 16.7 83
S+R- WFK(pJG1) 0 10.4 52
15 0.12 0.6
30 0.09 0.4
45 0.08 0.4
a Transportof glucose by the
PEP-phosphotransfer-asesystem was measuredbyassayingthe
accumula-tionofglucose 6-phosphate. Purified membrane
vesi-cles were preincubated with PEP and then were
suspended in a reaction buffer containingglucose by
the methodof Kaback (12). Glucose 6-phosphate was
assayed in a coupled system by using
glucose-6-phosphate dehydrogenase and NADP+. Strain WFK is the lacIq host; plasmid pKY2 is a
tetracycline-sensitive derivative of pBH20, and pJG1 andpJG3are
lysis gene clones (9).
bExpressed as nanomoles of glucose 6-phosphate
permilligram of protein.
the S+R- clone (Fig. 7), so actual lysis is not
required for loss of viability. The loss of viability isabsolutely dependent on a functional S allele (Fig. 7); in fact,S-R+ clones can be subcultured indefinitely in the presence ofinducer without detectablekilling (9).It isclear fromcomparison of the results in Fig. 7 that the loss of viability function of the gene Sprotein takes effect much
sooner than the observable effect on the
accu-mulation of culture mass.
Loss ofviability and commitment to lysis. To pursue the relationship between loss of cell viability and lysis, induced cells were diluted into noninducer medium at various times after induction(Fig. 8). The dilutionwas asdescribed for the Sts dilution experiment (Fig. 5). Com-parison ofFig.7 and 8shows that thepercentage of cells no longer viable at a given time after induction will lyse at the normal lysis time. These results suggest that the function of the
gene S protein consists oftwodistinct phases: initial attack on the cytoplasmic membrane, which is required for the destruction of PTS-mediated activetransport andcausesloss of cell viability, and subsequentlysisingeneR+ condi-tionsorphysiological death inR- conditions.
DISCUSSION
Wehave shownhereandinpreviouswork(9)
that theexpressionof thebacteriophage A S and
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.491.90.211.440.567.2]X 891
U,
< 0.1- -100
0.06- 50
0.04--20
-10 AB
-5
-2
1 2 34
TIME(hrs)
FIG. 7. The effect of pSoncellviability
ofS+R+Rz+ (a,*), S+R-Rz+ (b,A), and
(c,(D) were grown and induced as descrit
legendtoFig. 2. Portions of the culturewere
diluted, andplatedonrichmediatoassayce
at times before and after dilution. Cell v
plotted as percentage of survival with th
induction as 100% for S+R+Rz+ (A,),
(B,O), and S-R-Rz+ (C,).
R genes will result in a scheduled ly:
normalgrowth conditions. The expres
thirdgene,Rz,isrequiredinX-infected orininducedlysisgeneclones(Fig. 3)
containingahigh
Mg2'
concentration.phage proteins are requiredforlysis c
timing of lysis;theexpression ofthely is necessary and sufficient for the phenomenon.
Neither the mechanism oflysis nor
is understood. The results presented I
gest that there is damage to the cyl
membrane mediatedby thegeneSpro
lesion is manifested as an increased
permeabilitytosucroseandadestructi
PTS-mediated active transport of gluc Sam7 nonsense fragment polypeptid
has a length of 55 amino acid resi(
accumulates in the membrane as does man et al., manuscript in preparati increases the passive sucrose permeal
does not destroy the PTS active trai
glucoseand hasno effectoncell viabif
Surprisingly,themembranelesionsa ent invesicles preparedvery soon aft
tionofthebacteriophage lysisgenes ra
just in vesicles prepared from the normal lysis
timeorlater. Thisresult ledus toinvestigatethe
kinetics of cell viability after induction of the
lysis genes since a membrane lesionleadingtoa
collapse ofactivetransportmightbeexpectedto
be alethal event. The results (Fig.7) show that
cell viability, measured as colony-forming
po-tential, begins to decrease dramatically very
soon after induction of the lysis genes, much
_1% earlierthan the onsetlysisinS+R+organismsor
cessation of mass accumulation in S+R-. The
>_ simplestinterpretation ofthese resultsis that the early membrane lesion detected in the vesicle
6 transport
experiments
and theearly
loss invia-bility are manifestations ofanearly membrane
ul action ofpS.Once asufficientquantityofpS has
w
o beensynthesizedorhas exerted its effectonthe
membrane, the cell is committed to lysis at a
latertime(Fig. 8), eventhoughprotein synthesis
(and thus respiration) continue atexponentially
increasing rates for at least 30 min after the
lethal event.
What is theeventuallysistrigger, then, ifthe cell iscommittedtolysisatsuchanearlier time?
.Cultures Ourresults with the Sts9B allele suggest that pS
S-R-Rz+ is stable;furthermore, once acell has
accumu-bed
in the latedenough pS nofurthersynthesis isrequired,,,°removed,
for the eventual lysis to occur (Fig. 8). In1viabiityiYprevious studies, we have shown that the
phe-e
time
of nomenonofpremature
lysis
isduplicated
in theS+R-Rz+
cloned
lysis
gene system(9).
Prematurelysis
was first observed in cells infected with the
bacteriophageT4,which haslysisgenestand e,
apparently analogoustothephageXgenes S and
sis under 0.3
sion of a cells(19) inmedia
Noother b
rforthe
0.2-sis genes
complete I
0.1-itstiming
here sug-
<0008
toplasmic
tein. The 0.06
I passive a
ionofthe
ose. The 0.04
e, which
dues and ; 2 3
pS
(Alt-on), also TIME(hra)
biity but FIG. 8. Loss of cell viability results in commitment
bsYty
bof
tolysis.
Acultureof strainWFK(pRF26)(S+R+Rz+)
nsport wasgrown and induced asdescribed in the legend tolity. Fig. 2 (a). After 10 min (b) and 20 min (c) ofinduction,
ireappar- portions of the culture were removed and diluted into
Lerinduc- noninducermedium. TheA550was normalized to the
tther than undiluted culture in (b) and (c).
VOL.44, 1982
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.491.59.219.60.316.2] [image:6.491.282.421.425.614.2]R;severallaboratorieshavedocumented prema-ture lysis in X infections. The phenomenon is simple and dramatic; ifcyanide orany
respira-torypoison is addedtocells in which the Sgene hasbeen sufficientlyexpressed (but long before thenormallysis time), lysis occursimmediately (6). No lysis is observed ifcyanide is addedto
S--affected cells(6). Thus, poisoning respiration
apparently acts to trigger lysis prematurely by
subvertingthe normalscheduling mechanism.
Takentogether with the results in this work,
theprematurelysis phenomenonsuggestsa gen-eralmodelforS-mediated scheduled lysis. Once
acertainquantityofpShas accumulated in the
membrane, themembrane is thensusceptibleto
alysis-triggering event. Thistriggering eventis
likelytobecollapse of the chemiosmotic
gradi-ent across the cytoplasmic membrane since it can be emulated by respiratory poisons. Since nofurtherpS accumulationisnecessaryfor the
triggerto occur, one possibilityis that the
trig-geringeventoccursduringtheinception ofcell
divisionor septumformation,which is theonly
knownsingularityinmembranebiogenesis.That is, the onset of septation in the presence of sufficient pScausesa catastrophiclesion in the
membrane, resulting in the immediate collapse
of the membrane potential. Alternatively, the accumulation ofpSmayresult inadeterioration of theabilityof the celltomaintain the chemios-moticgradient.Thecellcanresistcollapseof the
gradient bycompensatoryhydrolysisof ATPin
the cellularpools or by increased utilization of the oxidative metabolic pathways. The resist-ancewould beovercomebyfurtherpS
synthesis
orbyany substance whichpoisonsrespiration.This modelpredictseitherdecreasingATPpool
levels as the time oflysis approaches or more
rapidoxygenconsumption.
Specific
predictionsof theseptationtriggerarelessclear,exceptthat
some septation delay mutants should show
se-verely retarded lysis scheduling. These
predic-tions are currently being tested in this labora-tory.
ACKNOWLEDGMENTS
Thisstudywassupported byPublic Health Service grant 1-R01-GM27099-03 from the National Institutes of Healthand by aBiomedical ResearchSupportgrant from Texas A&M UniversityCollegeof Medicine.
J.VIROL.
LITERATURECITED
1. Adhya, S., A. Sen, and S. Mitra. 1971. On the role of gene S, p. 743-746. In A. D.Hershey (ed.), The bacteriophage lambda. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
2.Bienkowska-Szewczyk, K., B. Lipinski, and A. Taylor. 1981. The Rgene product of bacteriophage lambda is the mureintransglycosylase. Mol. Gen. Genet. 184:111-114. 3. Bienkowska-Szewczyk, K., and A. Taylor. 1980. Murein
transglycosylase from phage lambdalysate; purification andproperties. Biochim. Biophys. Acta615:489-4%. 4. Black, L. W., and D. S. Hogness. 1969. Thelysozymeof
bacteriophage X.I.Purification and molecular weight. J. Biol. Chem. 244:1968-1975.
5. Bradford, M. 1976. Arapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-252.
6. Campbell, J. H., and B. G. Rolfe. 1975. Evidence for a dual control of the initiation of host-celllysiscaused by phage lambda. Mol. Gen. Genet. 139:1-8.
7. Clark, A. J., and0.Maaloe.1967.DNA replication and the divisioncycle in Escherichia coli. J. Mol. Biol. 23:99-112.
8. Daniels,D.L., and F. R. Blattner. 1982. Nucleotide se-quenceof the Q gene and the Q to S intergenic region of bacteriophage lambda. Virology 117:81-92.
9. Garrett, J. M., R. Fusselman, J. Hise, L. Chiou, D. Smith-Grillo, J. Schultz, and R. Young. 1981. Cell lysis by induction of cloned lambdalysis genes. Mol. Gen. Genet. 182:326-331.
10. Itakura, K., T. Hirose, R.Crea, A.Riggs, H. L. Heyneker, F.Bolivar, and H. W.Boyer. 1979. Expression in Esche-richia coli of a chemically synthesized gene for the hormonesomatostatin. Science 198:1056-1063. 11. Kaback, H. R. 1971. Bacterial membranes. Methods
En-zymol. 22:99-120.
12. Kaback, H. R. 1974. Transport in isolated bacterial mem-brane vesicles. Methods Enzymol.31:698-709. 13. Morrison, D. A. 1979.Transformation and preservation of
competent bacterial cells by freezing.Methods Enzymol. 68:326-331.
14. Mukherjee, P. K., and R. K. Mandal.1976. Role of Sgene ofbacteriophagelambda in hostlysis.Biochem. Biophys. Res.Commun. 70:302-309.
15. Olive, C., and H. R. Levy. 1967. The preparation and someproperties of crystalline glucose-6-phosphate dehy-drogenase from Leuconostoc meserteroides. Biochemis-try6:730-736.
16. Reader, R. W., and L.Siminovitch.1971. Lysisdefective mutantsofbacteriophage lambda: genetics and physiolo-gyof Scistronmutants.Virology43:607-622.
17. Reader, R. W.,and L.Siminovitch.1971. Lysisdefective mutants ofbacteriophage lambda: on the role ofthe S functioninlysis.Virology43:623-637.
18. Taylor,A.1971.Endopeptidaseactivity of phage-endoly-sin. Nature(London) New Biol.233:144-145.
19. Young, R., J. Way, J. Yin, and M. Syvanen. 1979. Transposition mutagenesis of phage lambda: a new gene affectingcelllysis.J. Mol.Biol. 132:307-322.
on November 10, 2019 by guest
http://jvi.asm.org/