0022-538X/88/041437-05$02.00/0
Copyright
©
1988, AmericanSociety for MicrobiologyThe Hepatitis B Virus Enhancer
Modulates Transcription of
the
Hepatitis B Virus Surface Antigen Gene from
an
Internal Location
GARY A. BULLAAND ALEEM SIDDIQUI*
Departmentof Microbiology and Immunology and Programin Molecular Biology, University of ColoradoSchool of
Medicine,
Denver,
Colorado 80262Received 5 October 1987/Accepted 20December 1987
Wehave usedatransient expressionsystem toexamine the influence of the hepatitis B virusenhanceronthe transcription of the hepatitis B virus surface antigen (HBsAg) gene. This enhancer is located3' to HBsAg
codingsequencesbut is contained withinthematureHBsAgtranscripts. Removal of the enhancer resulted in
asignificant reduction in HBsAggeneexpression in liver cells. By usinganinvitro nuclearrun-on assay,this effectwasdeterminedtobeatthe levelof transcription.We havefurthernoted that HBsAg expressioncanbe restored by thereinsertion of theenhancersequencesinthe enhancerlessvector. However, this reconstitution
ofHBsAg expression appearstodependonlocationofthe enhancer with regardtothe HBsAgpromoters.
Hepatitis B virus (HBV) infection represents a major worldwide human health problem. This viruscauseschronic liver disease and has been linked to hepatocellular
carci-noma(15). Though research has been hindered by the lack of
a suitable model for HBV infection, significant advances have been made through analysis of the cloned virus (37). Four open reading frames have been identified from HBV sequence data andaredesignated S/pre-S, C/e,
pol,
and X.Major promoter elements and RNA transcripts have been mapped for both the HBV surface antigen (HBsAg)gene(3,
25, 30, 31, 34) and the core/e gene (25, 26). Putative X
gene-specific transcripts have also been detected, and a
separate promoter element has been tentatively identified (30a, 39). In addition, an enhancerelement has been
identi-fied and partially characterized (12, 28, 38). However, there have been few studies involving the role of the enhancer element in HBVgene expression.
Enhancersareeucaryotic control elements which activate
transcription of adjacentgenes inamanner independent of
orientation and position. These control elements have been identified inanumberof viruses and mammaliangenes
(16,
27). The HBV enhancer element, likemanyotherenhancers, exhibits both host- and tissue-specific activity (12, 28). Studies inourlaboratorysuggestaninvolvement of the HBV enhancer incore/e and Xgeneexpression (26; Siddiquietal., in press). The effect of the enhancer on HBsAg gene
expression, however, has notbeendetermined.
The gene encoding HBsAg contains three conserved in-frame translational start codons whichareused to produce HBsAg polypeptides of various lengths, designated
pre-Sl,
pre-S2, and S (Fig. 1). These proteins share common car-boxyltermini. Transcription of the HBsAggeneappears tobe controlledbytwodiscrete promoter elements(23, 30, 34). Thepromoterregion upstreamofthe
pre-Sl
sequence(thepre-Sl
promoter) contains the canonical TATA sequenceand controls theexpressionof thelarge HBsAg polypeptide (p39/gp42). The secondpromoterelement(pre-S2 promoter) located within the
pre-Sl
region shares homology with the simianvirus40(SV40)
latepromoterregion (3)and controls expression of pre-S2 (p33/gp36) and major S (p24/gp27) polypeptides.Themajorityoftranscriptsfromthisgenemap tothree sites clustered around thepre-S2translational start* Correspondingauthor.
site, with barely detectable levels of transcripts originating from the pre-Sl promoter region (23, 30). The HBV en-hancer element has beenmappedtobetween nucleotides (nt) 1073 and 1235(28). This places the enhancer approximately 200base pairs (bp) 3' tothe HBsAg coding sequences but within the HBsAg transcription unit (Fig. 1). In this study,
weshowed that the HBV enhancerservestoelevate expres-sion of the HBsAggeneincells of liver origin.
Figure 1 shows the recombinant plasmidconstructs used in this study. Plasmid pML18 contains the 2.8-kilobase (kb) BglII restriction fragment of HBV, including the entire HBsAg geneplus flanking sequences. Transientexpression
of the HBsAg gene (pML18) in a variety of cell lines
produced levels of HBsAg that were virtually undetectable (Table 1). However, in the HepG2 human hepatoma cell line (17), the levels of HBsAg expressionwereverypronounced
(Table 1). We estimate that levels of HBsAg in the HepG2 line were approximately 100-fold higher than levels in the other cell lines tested. These differentlevels of expression
arenotlikelytobe duetotheefficiency of transfection, since uptake of other plasmid vectors appears to be efficient in these cell lines(datanotshown). Although replication of the plasmidvectorin the HepG2 cells could accountfor these results, analysis of extrachromosomal DNA from
trans-fected cells indicated thatreplication of pML18 didnotoccur
(S. Jameel, unpublished results).Thispromptedusto inves-tigate the roles of HBVsequencesthatwereresponsiblefor
thisapparenttissue-specific expression of the HBsAggene.
Inaprevious study,wefound that bothHBsAgpromoter regions
(pre-Sl
andpre-S2)
of theHBsAggenelack host-or tissue-specific activity when linkedto the chloramphenicol acetyltransferasegene(30).Recent identification of the HBV enhancerelement and its tissuespecificity (12, 28) prompted us to examine the role of this enhancer in HBsAg geneexpression. It is noteworthy that this enhancer element is
locatedinthe 3'untranslatedregionof theHBsAggene(Fig. 1).
To address the question of enhancer involvement in
HBsAg expression, we transfected HepG2 cells with
en-hancer deletion plasmidspMLAE and pMLAE344 (Fig. 1). Figure 2A compares levels of HBsAg RNA produced in
HepG2 cells transfectedwith native (pML18)or
enhancer-less (pMLAE) HBsAg plasmids. A 32P-labeled antisense
riboprobe (21) wasused to detect HBsAggenetranscripts.
1437
on November 10, 2019 by guest
http://jvi.asm.org/
TATA P. Int.P.
TATA pre-S EN
pM
118
jy Bg
!
V
.
pAp Ba I )(b NpCSPHP,
I Ic
89
TATAAA,
I,
pMI.AE
pM1,AE334
pENAP
pENBS
pENAE
1
i^< ~~~~~~~~-273
--273
-.as
-2
i _.-273z
FIG. 1. Construction of recombinantplasmids. The 2.8-kb BgIII restriction fragment of HBVwasinserted into theBamHI site ofpML (19) toproduce plasmid pML18.PlasmidspMLAE and pMLAE344areenhancer deletionplasmids derived from pML18 bydeletingthe273-bp HpaI-SphI (nt 964 to 1235) or 344-bp HpaI-HpaII (nt 964 to 1310) restriction fragment, respectively. Enhancer reconstitution plasmids pENAP andpENBS contain the344-bp HpaI-HpaII enhancerregion inserted into the ApaI (nt 2603)orBstEII (nt2824) site ofpMLAE. PlasmidpENAE contains the 273-bp enhancer fragment inserted 257bp downstream of thepolyadenylation signalof theHBsAg gene. In each case, enhancerreconstitution plasmids contain the enhancer with the putative Xgene promoter directed away from the HBsAg gene transcription unit. Open boxes represent the openreading frames of the HBV genome. Allconstructsweremadebyusingstandard methods (20).Pre-S, S, C, X, andPrefer to the openreading frames ofHBV.Thelocations of the HBsAg genepre-Sl promoter (TATA P.), internal promoter(Int.P.), and polyadenylationsignal areindicated. Thearrow indicates the polarity of the enhancer fragments in relationto the HBsAg gene. Abbreviations: Ap,ApaI; Bg, BglII; Bs, BstEII; Hp, HpaI; HpII, HpaII; RI, EcoRI; Sp, SphI; Xb,XbaI; Ac, AccI; EN, enhancer.
Hybridization signals
werequantitated by measuring
peak areasfrom densitometer
tracings of
autoradiographs
andnormalized
tosignals obtained by probing
areplicate
filterwith
arabbit
P-globin
gene probe(data
not shown) which was used as aninternal
control in thetransfections.
Anapproximately
sixfold
decrease in the levels of
HI3sAg
genesteady-state
RNA wasobserved when
pMLAE
was used(Fig. 2A). This decrease of HBsAg steady-state
RNAlevels
correlates
well withHBsAg
protein
data obtainedfromthesetransfections (Fig.
2A).We found
that the extendeden-hancer
region deletion plasmid (pMLAE344) produced
HB-sAg levels
similar
tothose of
pMLAE
(data
notshown).
We
nextcharacterized
theHBsAg
genetranscripts from
HepG2 cells transfected with either pML18
or pMLAE. Northern(RNA) blot
analysis of poly(A)+ purified
RNA(Fig. 3A) revealed
a2.1-kb HBsAg transcript from
pML18-transfected cells
and asmaller transcript
from
pMLAE-transfected
cells,
the latterbeing consistent with
the extentof
the enhancerdeletion.
The0.8-kb band (Fig.
3A, lane 1) isprobably
an Xgene-related transcript
(30). To determine whether enhancer removal altered the specificity oftran-script
initiation,
Si
nuclease mapping
was carried out(Fig. 3B). Thetranscriptional
initiation sites for both native (30)TABLE 1. Transient expression of HBsAg gene in variouscelllinesa
Cell line Source HBsAgS/Nb
APB Ratfibroblast 1.3
3T3 Mouse fibroblast 1.1
HeLa Human cervical carcinoma 1.2
T47D Human breast carcinoma 1.2
MCF-7 Human breast carcinoma 1.1
HepG2 Human hepatoma 33.3
a Cellsweretransfected with20 ,ug ofplasmidpML18byusing theCaPO4 procedure (9), and culturesupernatantswereassayed forHBsAg48 h laterby theAusriaIIradioimmunoassay(AbbottLaboratories).
bS/N, Ratio ofcounts per minute of sample to counts per minute of negativecontrol serum, asdeterminedbyradioimmunoassay.
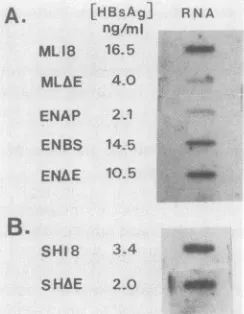
A.
[HBsAg]
ng/mi MLIB 16.5
RN A
em,
MLAE 4.0 ENAP 2.1
ENBS 145 V
ENhE 105 _
B.
SHIS
3-44U_
[image:2.612.64.551.68.225.2]SHAE 2.0
am
FIG. 2. HBsAg gene expression from recombinant plasmids in HepG2 and COS cells. Unconcentrated cell supernatants were
assayed by acommercially available radioimmunoassay (Abbott), and levels were determined from a standard curve. For mRNA quantitation, 10 F±g of total cytoplasmic RNA isolated from
trans-fected cellsby the method of Chirgwin etal. (4)wasdenaturedat
65°Cfor 10min in 6.15 M formaldehyde-lOx SSC (1x SSC is1.5 M
NaClplus 0.15 M sodium citrate) and immobilized onto nitrocellu-lose membranes (14) by usingaslot blotapparatus (Schleicher& Schuell, Inc.). BoundRNAwasprobed with a32P-labeled antisense RNA generated from a 744-bp HincIl (nt 222 to 966) restriction fragment. This probe includes HBsAg gene coding sequences but
excludes the enhancer region. As an internal transfection control, a duplicate filterwasusedto hybridize with a
P-galactosidase
nick-translated probe. Filters were exposed to XAR5 film (EastmanKodakCo.). Relativelevels of HBsAg expression were determined by densitometer scanning (Quickscan R & D) of the autoradio-graphs, and signals were normalized to
P-galactosidase
signals. HBsAgproteinandRNA levelsfrom transfected HepG2 cells (A)andfrom COS cells transfected with pSH18 and pSHAE (B) were
used.
I
2.A
I
I
I I
I
I I
I Ii
i
I
I
on November 10, 2019 by guest
http://jvi.asm.org/
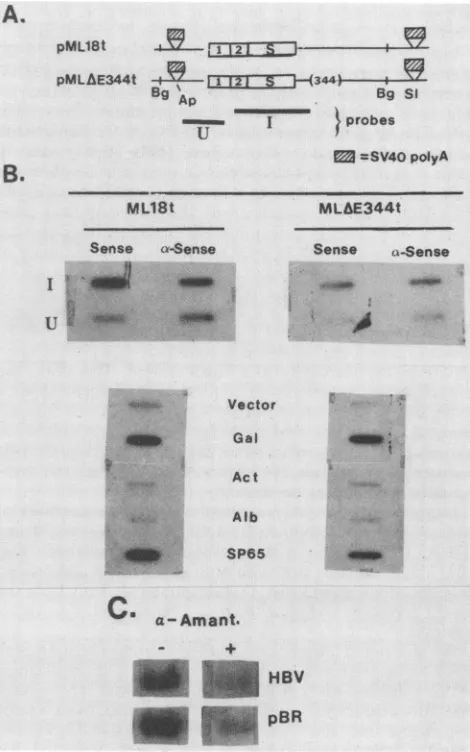
[image:2.612.370.492.373.530.2]A.
pML18t
I1
.2.IS pMLAE344t t 5l7--T---I---[( 344)-- -- ------BgAp ~~~BgSi
lgI tprobes
= =SV40polyA
ML18t
Sense a-Sense
I v
,1
U:
'....MLfE344t
Sense a-Sense
FIG. 3. RNA analyses of pML18- and pMLAE-directed
tran-scriptsinHepG2cells. Poly(A)+ RNAwaspurified by chromatog-raphy through an oligo(dT)-cellulose column (1). For Northern
analysis(36), 5 ,ug ofpoly(A)' RNAwasfractionatedon a
formal-dehyde-agarose gel. RNA was transferred to nitrocellulose and
probed with 32P-labeled HBV DNA. ForS1nuclease analysis (41), 20,ugof totalcytoplasmicRNAwasincubated witha5'-end-labeled BglII-XbaI restriction fragment(nt2432to250)(30). Hybridswere
digestedwith Si nuclease and analyzed on an 8%
urea-polyacryl-amidegel. (A) Northern (RNA) blot analysis.Lane 1, pML18 RNA; lane2, pMLA&ERNA.Molecular sizesareindicated in kilobases.(B) Si nuclease analysis.A20-,ug sample of cytoplasmicRNAwasused
for each lane. Lane 1, pML18 RNA; lane 2, pMLAE RNA.
Molecular size of protected fragments is indicated inbasepairs. and enhancerless HBsAg genes werefoundto be identical. These results indicate that thedeletion of the enhancer does
not influence the specificity of initiation of HBsAg gene transcription.
Theobserved decrease in HBsAg expressionas aresult of enhancer deletion could be due to either a decrease in transcription rate of the HBsAg gene or a post transcrip-tional event (e.g., altered mRNA transport or stability).
Because analysis of steady-state mRNA cannotdistinguish between theseprocesses,weusedtwoindependent methods
to account for the enhancer deletion effect. Our first
ap-proachwas to useacell line in which measurable levels of HBsAg synthesis are produced but in which the HBV
enhancer has been foundto be relatively inactive. For this
purpose, we used plasmids pSH18 (29) and enhancerless pSHAEin which the native orenhancerless HBsAggene is clonedinto theplasmidvectorpSVO10. pSVO10contains the SV40origin ofreplication and willreplicate toa high copy
number whenintroduced into COS cells(18).Transfection of COS cells with pSH18 has been previously shown to
pro-duce detectable levels ofHBsAg (29).Levels ofHBsAggene
mRNA and HBsAg proteinfrom both pSH18- and pSHAE-transfected COS cells wereapproximately equal (Fig. 2B). Althoughdifferential RNAprocessingandstabilitybetween the COS andHepG2cellscannotbe ruledout,these results
suggest that enhancerdeletion does notsubstantially affect
HBsAg expressionin the nonliver cell line.
Wenextcarriedoutnucleartranscriptionrun-onassaysto
determine if the observedeffect of enhancer deletionwasat
the level oftranscription. For these experiments, the
liver-derived Huh-7 (22) andHepG2 cell lines were used. While
both cell linesgavesimilarresults, the results obtainedwith
the Huh-7 cell line arepresentedhere. Nucleiwereisolated
Vector
*
Gal
Act
.,z. .Alb
;;)i: SP65
C
a-Amant.HBV pBR
FIG. 4. Effect ofenhanceron nuclear transcription ofHBsAg
gene. Nuclear run-on assays weredone by amodification of the
method ofClaytonetal.(5).(A) Huh-7cellsweretransfectedwith
pML18torpMLAE344t,which contain theHBsAggeneconstructs
flankedby SV40 polyadenylationsequences. PlasmidpCH110 (11),
encoding the 0-galactosidase gene, was used as a transfection
control. At 48 hposttransfection, nucleiwereisolated and used in
the nuclearrun-onprocedure (5). (B)Effectof enhancer deletionon
HBsAggenetranscription. Run-on-generatedRNA from transfected cellswashybridizedtoeithersingle-stranded HBsAg gene-specific
DNA fragments (fragments I and U indicated in panel A) or
double-stranded restrictionfragmentsof controlgenesandexposed
tofilm. Abbreviations: Act, Actin; Bg, BgIII; Gal,
0-galactosidase;
S1,Sall; Alb, albumin; SP65, plasmidDNAcontrol; Vector,
single-strandedpTZ18RDNA(Pharmacia, Inc.). (C) Transcriptionrun-on
ofpML18-transfected cells in the presenceorabsence of 3 ,ugof
a-amanitinperml. Each laneorslot contains 3to5 ,ugof DNA.
and incubated in thepresenceof
[IP]rUTP
to allow elonga-tion of RNAtranscripts by previouslybound RNA polymer-aseIImolecules. Extracted32P-labeled RNAwashybridizedtosingle-strandedHBVfragmentsand double-stranded
con-trol genes immobilized on nitrocellulose filters. Relative
HBsAg gene transcription was normalized to signals
ob-tained with both 3-galactosidaseand actin DNAprobes.The
,B-galactosidase signal serves as a control for transfection
efficiency. This method allows foranestimationof the RNA
A.
NORTHERNB.
1 2
Si
1 2
4probe
B.
.4c
235-270
29.1 i
-0-m
4
4m
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.65.305.75.258.2] [image:3.612.328.563.78.454.2]polymerase loading on
a gene (10) andtherefore indicates
whether an enhancer affects the initiationof
transcription.
Initial experiments
indicated
that plasmid vector pBR322sequences
were beingefficiently
transcribed,
making HBV
transcription
results difficult to interpret. Tolimit
transcrip-tion from possible crypticpBR322
promotersinterfering
with HBsAg genetranscriptional
analyses, weflanked
thenative (pML18)
and enhancerless (pMLAE344)HBsAg
genes with theSV40 polyadenylation
sequencescontained in
aBamHI-BclI fragment
(Fig.4A). Run-on analysesof nuclei
from Huh-7 cells transfected with these HBsAg gene plas-mids showed readilydetectable
HBV-and
pBR322-specific
RNA being produced. In addition,levels of
transcription of
both HBV and plasmid sequences appear tobe increased
by
the presence of the linkedHBV
enhancer.Transcription of
vectorsequences
does not appear to influencethe signal
obtained
from theinternal S
gene sequences(designated I),
however,
since the upstream S gene sequences(designated
U) are
transcribed
only to asmall
degree.
Therefore,
thetranscriptional
activitydetected for the HBsAg
gene appears to be initiated within the HBV sequences. As aninternal
control,
signalsdetected
byusing
theP-galactosidase-co-transfected
control gene wereapproximately equal. This
indicates
that the observed increase in HBsAg genetran-scription
is enhancerdependent.
Densitometry of
autoradiographs
and directcounting of
filters show anapproximate
fivefold increase in sense-strandHBsAg transcription
in the presence of the enhancer(Fig.
4B).This
correlates wellwith both steady-state
andprotein
data describedpreviously. Transcription
occurring from the
pre-Sl promoter
was near the background level(Fig.
4B, Ufragment).
We also noted a significant level
of antisense
HBsAg
gene RNA being produced by the HBsAg gene. Thesignificance
of this
antisense
RNAproduction
isnot
known.
Itis
unlikely
that this representstranscription
originating from
vectorsequences,
since the SV40polyadenylation
signal is located
3' to the HBsAg genefragment.
It has been suggested that the antisense message from thisregion is produced in
murine
cells containingmultiple
HBVintegrates
(42). Inaddition,
an RNApolymerase
IIItranscript
derived from the opposite strand has been previously reported from the 3' region of the HBsAggene
region (33).As afinal control, the run-on analysis was carried out in the presence of 3
p,g
ofcx-amanitin
per ml. Results indicate thattranscription
of both HBsAg gene and plasmid se-quences was reduced byapproximately
80%, indicating that the majority oftranscription
isdirected by RNApolymerase
II molecules (Fig.4C).
We therefore conclude that the majority ofenhancement
ofHBsAg geneexpression
by the HBV enhancer is at the level oftranscription.
If the enhancer region is
functionally
able toincrease
transcription
of the HBsAg gene,placement
of these se-quences outside of theenhancer-deletion
HBsAg transcrip-tion unit should recover expression. To test this, we used plasmids with the enhancer region inserted 5' or 3' to the HBsAg gene inpMLAE
(Fig. 1). Insertion of theenhancer
element(HpaI-HpaII)
5' to thepre-Sl
promoterelement
(plasmid pENAP) resulted in no increase in HBsAg geneexpression
(Fig. 2).However,
insertion of theenhancer
between thepre-Sl
promoter and thepre-S2
promoter at nt 2824 resulted in levels of HBsAg expression similar to levelsproduced
with the native HBsAg gene (Fig. 2).Enhancer
insertion 3' to the HBsAg gene (pENAE) led to a similar increase in HBsAgexpression.
These results indicate that
enhancer-mediated activation
of HBsAg gene expression
can berestored by insertion
of theenhancer
element
but ispartially
location
specific.
Theinability of the enhancer insertion
5' tothepre-Sl
promoterto reconstitute
expression
is notentirely unexpected
be-causeof
the apparentnegative regulatory
constraintsplaced
onthe
pre-Sl
promoterduring
normalHBsAg
expression(3,
23, 30).These constraints
may beinterfering
with directactivation of the transcription
control elements. Preferential actionof enhancers on proximal rather than distal promoters (6, 13, 40) mayalso
contributeto this effect.In
this study,
wehave
addressed the question of the involvementof the
HBVenhancer
element in expression of the HBsAg gene. Using a transient expression system, we demonstrated that(i) the
HBVenhancer functions to elevate HBsAg geneexpression
atthe levelof transcription, (ii) thedeletion of
enhancer sequences neither alters the specificityof
initiation of stable HBsAg transcripts
norappreciably
affects mRNA stability, and (iii) enhancer reconstitution restores theoriginal level of HBsAg expression.The HBV enhancer is strategically located within the 3' untranslated
region of the
HBsAg gene and 5' to the X andcore/e
genes as well as within the coding region of thepolymerase
gene. It istheonly
enhancer, to ourknowledge,
whose sequences are partof a mature transcript, in this case the HBsAg geneandpolymerase
genetranscripts.
Theonly otherexample of
aninternal
enhanceris theimmunoglobulin
enhancer. However, with theimmunoglobulin enhancer,
the enhancersequences aresubsequently spliced
out anddo not appear in the stabletranscript (2,
8, 24).The HBV enhancer has been suggested as dramatically increasing expression from the HBV core promoter as well as from a heterologous promoter gene in the human hepa-toma cell line
PLC/PRF/5
(28).However,
we have consis-tently found the HBV enhancer toincrease
expression
ata more modest level (8- to 10-fold) both in the context ofhomologous
(core and HBsAg) andheterologous (SV40)
promoters fused to a reporterchloramphenicol acetyltrans-ferase gene (12; unpublished results). That the enhancer alone cannot account for the large difference between HB-sAg gene expression in liver versus nonliver cell lines suggeststhe involvement of multifactorial mechanisms in theregulation
of this gene.However,
theenhancer may contrib-ute to the abundant synthesis of HBsAgduring
HBV infec-tion. Similarly, theenhancerprobably
alsoplays
asignificant role in the generation of pregenomic RNA, thus regulating replication of the viral genome.If the major role of the HBV enhancer is to potentiate
transcription
of HBVtranscription
units located 3' to the enhancer (core, X, andpot), then the activation of HBsAg gene expression from this strategiclocation
of the HBV enhancer 3' totheHBsAg open reading frame is perhaps due to the constraints of the small size (3.2 kb) of the HBV genome. Also, the spatialarrangement
of this enhancer between HBsAg andX/core promoters
may ensure properproportions
ofpre-Sl,
pre-S2, and major S polypeptides. The apparent livercell-specific
regulation of HBV gene expression by this enhancer further suggests a pivotal role of the enhancer in overall viral generegulation
during HBV infection. Such a role for an enhancer in viral pathogenesis has been described with other viruses (7).Thisinvestigationwassupported by Public Health Service grants A121182 and CA33135 from the National Institutes of Health and by agrant from the MilheimFoundation forCancer Research to A.S. We thank Scott Stockert for technical help and J.E. Mapoles for
criticalreading of this manuscript.
on November 10, 2019 by guest
http://jvi.asm.org/
LITERATURE CITED
1. Aviv, H., and P. Leder. 1972. Purification of biologically active globinmessenger RNA by chromatography on oligothymidylic acidcellulose. Proc. Natl. Acad. Sci. USA 69:1408-1413. 2. Banerji, J., L. Olson, and W. Schaffner. 1983. A
lymphocyte-specific cellular enhancer is located downstream of the joining region in the immunoglobulin heavy chain genes. Cell 33:729-740.
3. Cattaneo, R., H. Will, N. Hernandez, and H. Schaller. 1983. Signals regulating hepatitis B surface antigen transcription. Nature(London) 395:336-338.
4. Chirgwin, J. M., A.Przybyla, R. MacDonald, and W. J. Rutter. 1978. Isolation of biologically active ribonucleic acid from sources rich in ribonuclease. Biochemistry 18:5296-5297. 5. Clayton, D. F., A. L. Harrelson, and J. E. Darneli, Jr. 1985.
Dependence of liver-specific transcription on tissue organiza-tion. Mol. Cell. Biol. 5:2623-2632.
6. de Villiers, J., L. Olson, J. Banerji, and W. Schaffner. 1982. Analysis of the transcriptional enhancer effect. Cold Spring Harbor Symp. Quant. Biol. 47:911-919.
7. Feigenbaum, L., and G. Khoury. 1985. The role of enhancer elements in viral host range and pathogenicity. In B. Fields et al. (ed.), Genetically altered viruses and the environment, p. 181-194. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
8. Gillies, S. D., S. L.Morrison, V. T.Oi,and S. Tonegawa. 1983. Atissue specifictranscription enhancer element is located in the majorintron of arearranged immunoglobulin heavy chain gene. Cell 33:717-728.
9. Graham, F. L., and J.van der Eb. 1973. A new technique for the assay ofinfectivity of human adenovirus 5 DNA. Virology 52:456-457.
10. Groudine, M., M. Peretz, and H. Weintraub. 1981. Transcrip-tionalregulation of hemoglobin switching in chicken embryos. Mol.Cell. Biol. 1:281-288.
11. Hall, C., T. Jacob, G.Ringold,and F. Lee. 1983. Expression and regulation of Escherichia coli lac-Z gene fusion in mammalian cells. J. Mol. Appl. Genet.2:101-109.
12. Jameel, S., and A.Siddiqui. 1986. The human hepatitis B virus requires trans-acting cellular factor(s) for activity. Mol. Cell. Biol.6:710-715.
13. Kadesch, T., and P. Berg. 1986. Effects of the position of the simian virus 40 enhancer on expression of multipletranscription units in asingleplasmid. Mol. Cell. Biol.6:2593-2601.
14. Kafatos, F. D., C. W. Jones, and A. Efstradiadis. 1979. Deter-mination ofnucleic acidhomologies and relative concentrations by a dot blotprocedure. Nucleic Acids Res. 7:1541-1560. 15. Kew, M. C. 1981. The hepatitis B virus and hepatocellular
carcinoma. Semin. Liver Dis. 1:59-67.
16. Khoury, G., and P. Gruss. 1983. Enhancer elements. Cell 33:313-314.
17. Knowles, B., C. C. Howe, and D. P. Aden. 1980. Human hepatocellular carcinoma cell lines secrete the major plasma proteinsandhepatitis B surface antigen. Science 209:497-499. 18. Learned,R.M., R. M.Myers, and R. Tjian. 1978.Replication in
monkey cells of plasmid DNA containing the minimal SV40 origin. ICN-UCLA Symp. Mol. Cell. Biol. 22:555-566. 19. Lusky, M., and M.Botchan. 1981.Inhibition of SV40 replication
in simian cells by specific pBR322 DNA sequences. Nature (London) 293:79-81.
20. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory,
ColdSpring Harbor, N.Y.
21. Melton, D. A., P. A.Kreig, M. R. Rebagliati, T. Maniatis, K. Zinn, and M. R. Green. 1984. Efficient in vitro synthesis of biologically active RNA and RNAhybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 12:7035-7056.
22. Nakahayashi, H., K. Taketa, K. Miyano, T. Yamane, and J.
Sato. 1982. Growth of human hepatoma cell lines with differen-tiated functions in chemically defined medium. Cancer Res. 42:3858-3863.
23. Ou, J., and W. J. Rutter. 1985. Hybrid hepatitis B virus-host transcripts in a human hepatoma cell. Proc. Natl. Acad. Sci. USA 82:83-87.
24. Queen, C., and D. Baltimore. 1983. Immunoglobulin gene tran-scription is activated by downstream sequence elements. Cell 33:729-740.
25. RaIl, L. B., D. N. Standring, 0. Laub, and W. J. Rutter. 1983. Transcription of hepatitis B virus by RNA polymeraseII. Mol. Cell. Biol. 3:1766-1773.
26. Roossinck, M., S.Jameel, S. H. Loukin, and A. Siddiqui. 1986. Expression of hepatitis B viral core region in mammalian cells. Mol. Cell. Biol. 6:1393-1400.
27. Serfling, E., M. Jasin, and W. Schaffner. 1985. Enhancers and eukaryotic gene transcription. Trends Genet. 1:224-230.
28. Shaul, Y., W. J.Rutter, and0.Laub. 1985. A human hepatitis
B viral enhancer element. EMBO J. 4:427-430.
29. Siddiqui, A. 1983. Expression of hepatitis B virus surface antigen gene in cultured cells by using recombinant plasmid vectors. Mol. Cell. Biol. 3:143-146.
30. Siddiqui, A., S. Jameel, and J. E. Mapoles. 1986. Transcriptional control elements of hepatitisBsurface antigen gene. Proc. Natl. Acad. Sci. USA 83:566-570.
30a.Siddiqui, A., S. Jameel, and J. E. Mapoles. 1987. Expression of hepatitis BvirusX gene in mammalian cells. Proc. Natl. Acad.
Sci. USA84:2513-2517.
31. Simonsen, C. C., and A. D. Levinson. 1983. Analysis of
pro-cessing and polyadenylation signals of the hepatitis B virus surfaceantigen gene byusing simian virus 40-hepatitis B virus chimeric plasmids. Mol. Cell. Biol. 3:2250-2258.
32. Sninsky,J.,A.Siddiqui, W. S. Robinson, and S. N. Cohen. 1979.
Cloning and endonuclease mapping of the hepatitis B viral genome.Nature(London)279:346-348.
33. Standring,D. N., L. B.Rall, 0.Laub, and W. J. Rutter. 1983.
Hepatitis B virus encodes an RNA polymerase III transcript.
Mol. Cell. Biol. 3:1774-1782.
34. Standring, D. N., W. J.Rutter, H. E. Varmus, and D. Ganem.
1984. Transcriptionofthe hepatitis B surface antigen gene in cultured murine cells initiates within the presurface region. J.
Virol.50:563-571.
35. Summers,J., and W. S.Mason. 1982.Replication of the genome
of ahepatitis B-like virus by reverse transcription of an RNA intermediate. Cell29:403-415.
36. Thomas, P. S. 1980.Hybridization of denatured RNA and small fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci.
USA77:5201-5205.
37. Tiollais, P., C. Pourcel, and A. Dejean. 1985. The hepatitis B
virus. Nature(London) 317:489-495.
38. Tognoni, A., R.Catteneo,E.Serfling, andW. Schaffner. 1985. A
novelexpression selection approach allows precise mapping of
the hepatitis B virus enhancer. Nucleic Acids Res.
13:7457-7472.
39. Treinin, M., and 0. Laub. 1987. Identification ofa promoter
element located upstream from the hepatitis B virus X gene.
Mol. Cell. Biol. 7:545-548.
40. Wasylyk,B.,C.Wasylyk,P. Augereau, and P. Chambon. 1983.
The SV40 72 bprepeat preferentially potentiates transcription
from proximal natural or substitute promoter elements. Cell 32:503-514.
41. Weaver, R., and C. Weissman. 1979. Mapping of RNA by a modificationof theBerk-Sharp procedure: the 5' termini of 15S beta-globin mRNA and mature 10S beta-globin mRNA have
identical mapcoordinates. Nucleic Acids Res. 7:1175-1182.
42. Zelent, A. Z., M. A.Sells,P. M.Price, A.Mohamad, G. Acs, and
J. K.Christman.1987.Murinecells carryingintegrated tandem
genomes ofhepatitis B virus transcribe RNAs fromendogenous promoterson bothviral strands andexpress middle and major
viralenvelope proteins. J. Virol.61:1108-1115.