0022-538X/83/070096-10$02.00/0
Copyright © 1983, American Society of Microbiology
Bacteriophage P22 In Vitro DNA Packaging Monitored by
Agarose Gel Electrophoresis: Rate of DNA Entry into Capsids
RAJALAKSHMI GOPEANDPHILIP SERWER*
Department of Biochemistry, The University of Texas Health Science Center, San Antonio, Texas 78284 Received 27 January 1983/Accepted 29 March 1983
Bacteriophage P22, like other double-stranded DNA bacteriophages, packages DNA in a preassembled, DNA-free procapsid. The P22 procapsid and P22 bacteriophage have been electrophoretically characterized; the procapsid has a
negativeaverageelectricalsurface charge density (uf) higher in magnitude than the negative uf of the mature bacteriophage. Dextrans, sucrose, and maltose were showntohave adramaticstimulatory effectonthe in vitropackaging of DNA by the P22procapsid. However, sedoheptulose, smallersugars,and smallerpolyols didnotstimulate in vitro P22 DNApackaging. These and other datasuggestthat anosmoticpressuredifferenceacross someparticle, probablyacapsid, stimulates P22 DNApackaging. After in vitro packagingwasoptimized by including dextran 40 in extracts, the entry kinetics of DNA into P22 capsids were measured. Packaged DNAwasdetectedby: (i) DNA-specific staining of intact capsids after fractionationbyagarosegel electrophoresis and (ii)agarosegel electrophoresis of DNase-resistant DNA after release of DNase-resistant DNA fromcapsids. Itwas
found that the first DNAwas packaged by 1.5 min after the startofincubation. The data furthersuggestthat either P22 capsids with DNA partially packaged in vitroare toounstable tobe detected by the above procedures or entryof DNA into thecapsid occurs inless than 0.25 min.
All
double-stranded
DNAbacteriophages
thathave been
studied package
DNAin
vivo in
apreformed, DNA-free
procapsid.
Forall
suffi-ciently
studied
bacteriophages,
theexternal
sur-face of the
procapsid
differs from
the externalsurface of
the maturebacteriophage capsid
in
having
arounder
shape and
smallerradius
(re-viewed
inreferences 8, 9, 22,
and39).
In the caseof
bacteriophage T7,
theprocapsid
wasalsoshown
to have anegative
average electricalsurface
charge
density
(cr)
higher
inmagnitude
than the
negative
crof the
maturebacteriophage
capsid (37, 38). For
analysis
of
DNApackaging,
itis desirable to havepackaging
assynchronous aspossible
and tomaximize
controlof
thecompounds
presentduring packaging.
Tohelp
accomplish
thesegoals,
DNAispackaged
in ex-tracts ofbacteriophage-infected
cells(in vitro).
DNA
packaging
invitro has been achieved withbacteriophages
4129
(2-4),
X(1, 12),
P22(23, 24),
P2 (25), T3 (11, 21), T7 (17-19, 27, 36), and T4
(5).
In allof
thesestudies, production
of aninfective
particle
wasusedastheassay forDNApackaging,
and it was found thatproduction
of an infectiveparticle required
either ATP or some otherribonucleoside
triphosphate.
In thecaseof
T7,
a10- to50-fold stimulation of in vitroinfectious
particle assembly
wasobserved in thepresence
of dextrans and
somesmaller,
relatedcompounds (36).
To
determine the
rate atwhich
DNA entersbacteriophage
capsids in vitro and
todetermine
the
effects of
addedcompounds
onthis rate, it is necessary to have an assayfor
DNAentryinto
capsids, independent of other assembly
events neededfor
aninfective
particle.
Thusfar, only
bacteriophages
T3 and4)29
have been made topackage
DNAefficiently enough
toassayphysi-cally for packaged
DNA. In these studies(T3
[21];
4)29
[3,
4]),
velocity sedimentation in
su-crosegradients,
sometimes after
DNase treat-ment, was used to detect capsids with packaged DNA. However, no attempt was made to mea-surethe rateof DNA entry. Inaddition, velocity
sedimentation
in sucrosegradients
has thefol-lowing limitations
as anassay for DNA entry.(i)
The state of the
capsid (procapsid
or itslarger,
moreangular conversion product) is not reliably
determined
(4). (ii) DNA may empty fromcap-sids
during the assay. (iii) The cost (in time andmaterials)
can become excessive during studies of entry rates.A
possible
alternative DNApackaging
assayfor
overcoming
limitations(i)
and(iii)
aboveis DNA-specific staining of DNase-resistant DNAcomigrating during
agarosegel electrophoresis
96
on November 10, 2019 by guest
http://jvi.asm.org/
P22 IN VITRO DNA 97
with bacteriophage capsids (33, 37). However, there is no reason for believing that this proce-dure would assist in overcoming limitation (ii). To help overcome this limitation, agarose gel electrophoresis of DNase-resistant DNA after release from the
capsid
(noprefractionation) is
a possible procedure. This latter procedure, un-like the former, can also be used to determinethe length of partially packaged
DNA.Bacteriophage
P22 has alinear,
double-stranded DNAgenome. P22 DNA has a nonran-dom permutation of its ends (26, 42) and a molecularweight, determined by high-resolution agarose gel electrophoresis (30) of the intact genome, of 27.9±0.3 x 106(P. Serwer and S. J.Hayes,
unpublished data). P22 has a spherical procapsid (6, 7, 10, 15, 16) with an in vitro DNApackaging activity (23, 24)
which is stable for months during storage (see Results). The P22procapsid
has aninternal
protein, p8 (P22
pro-teins are indicated by p, followed by the genenumber
of the
protein [6]),
thatleaves the
capsid
during
packaging (15). The P22 procapsid has,however,
notyet beencharacterized
electropho-retically, and in studies of
invitro
DNApackag-ing,
no assay butproduction
of an infectiousparticle
has been used for DNApackaging (23,
24).
Therefore,
in the presentstudy,
the P22procapsid
and maturebacteriophage
wereelec-trophoretically
characterized, procedures
forin-creasing
theefficiency
of in vitropackaging
weredeveloped,
and the entry of DNA intocapsids
was
observed
as afunction of time by
using the
two
procedures of
agarosegel electrophoresis
described
above.Implications
of the results forunderstanding in vitro
P22 DNApackaging
are discussed.MATERIALS ANDMETHODS
Strains. All bacterial strains usedwere derivatives
ofSalmonellatyphimurium LT2 from the collection of
D. Botstein (received from either D. Botstein or J.
King).Thepermissivehostfor P22 ambermutantswas
DB7002; the nonpermissive host for amber mutants
was DB7000. To makeDNA donor extractsfor in vitro
DNApackaging, induction of strain DB7130,
lysoge-nized withP22carryinganambermutation in gene 5
(major capsidprotein),wasused. Thisstrain has been
described previously (23). P22 carrying an amber
mutation in gene 2(referredto as2am;mutantnumber
H200) was received from J. King and has been
de-scribedpreviously (6). This bacteriophage contained,
inaddition, clear-plaque mutation CI-7 andan amber
mutation in gene 13 (lysis; mutant H101). Particles
from
lysates
ofthe nonpermissivehost infected withanambermutantarereferredtoby thenumber of the
mutant gene. Bacteriophage P22 with the CI-7 and
H101mutations (to be referredto aswild type)and
9-(tail spikeless)P22(7) werereceived inpurifiedform
fromJ.King;17-bacteriophageT7(tailfiberless) was
preparedaspreviously described (33).
Mediaandreagents.Forpreparation of capsids and
extracts,2 x LB(28) (20 gof tryptone [Difco
Labora-tories], 10 g of yeast extract, and 5 gof NaCl in 1 liter
of water) was used. Bacteriophages were stored in
Tris-Mgbuffer (0.2MNaCI,0.01 MTris-Cl [pH 7.4],
0.001 M MgCI2). Standard buffer was 0.15 M NaCl,
0.05 MTris-Cl (pH 7.4), and 0.005 M EDTA.
Electro-phoresis buffer A was 0.05 M sodiumphosphate (pH
7.4) and 0.001 MMgCI2. Electrophoresis buffer Bwas
0.05 M sodium phosphate, (pH 7.4) and 0.001 M
EDTA. Sample bufferA was 0.005 M sodium
phos-phate (pH 7.4), 0.001 M
MgC92,
4% sucrose, and 400p.g of bromophenol blue per ml. Sample buffer Bwas
the same as sample buffer A, with 0.001 M EDTA
replacing 0.001 M MgCI2. TSMB buffer used for in
vitro assembly was 0.01 M Tris-Cl (pH 7.4) 0.06 M
spermidine-hydrochloride,0.2 M
MgC92,
and 0.03 M2-mercaptoethanol. NET bufferwas0.1 MNaCI, 0.01 M
Tris-Cl (pH 7.4) and 0.001 M EDTA. Agarose was
purchased from Marine Colloids, Rockland, Maine.
Unless otherwise indicated. ME-grade agarose was
used. Dextrans werepurchased from Pharmacia Fine
Chemicals. The osmotic pressure (P) of sugar
solu-tions was obtained by linearextrapolation of data (14).
Productionandpartialpurification of procapsids.To
prepare 2- procapsids, a1-liter, log-phase culture of
S.typhimurium DB7000 wasinfected at 2 X 108/cm3at
37°C with 2am P22(multiplicityofinfection,5).
Aera-tion of the culturewascontinuedat37°C for 2 h. The
infected cellswerepelletedat10,000 rpm and4°Cfor
10 min in a Beckman JA-14 rotor. The pellet was
suspended in2 mlofstandard buffer,and 67 1.l of
1-mg/mllysozyme in standard bufferwas added. After
incubationat4°C for 20 min, 67 pd of10%oBrij 58was
added, and incubation was continued at4°C for 20
min, followed by incubation for 60 min at room
temperature (25 ± 3°C). Atthistime, atleast99%oof
the cells hadlysed. After the addition of 10 ±1 of1 M
MgCl2, thelysate wasdigestedwith 15,ul of1-mg/ml
DNase I(Millipore Corp.)at30°C for1h,followed by
67,ul of1-mg/ml boiled pancreatic RNase in 0.05 M
Tris-Cl (pH 7.4)at roomtemperaturefor15min.
Capsidsin thelysatewerepartiallyfractionatedby
being layered on a discontinuous cesium chloride
gradient previously described (29) and centrifuged for
180minat40,000 rpm and 18°C inaBeckman SW41
rotor.Thecapsids(density =1.3g/ml)wereremoved
from the gradient through a punctured tube bottom,
werediluted 1:1withTris-Mgbuffer, and were
clari-fiedbycentrifugationat5,000 rpm and4°C for 5 min in
aBeckman J-21 rotor. Thesupernatantwas brought to
avolume of5.2 cm3 andadensity of 1.28
g/cm3
withcesium chloride andwassubjected to buoyant density
sedimentation at40,000 rpm and 10°Cfor 20 h in a
Beckman SW50.1 rotor. The capsid band was
re-moved by pipetting from the top and was dialyzed
against Tris-Mg buffer.
Production and purification ofwild-typeP22
bacte-riophage.Lysates ofwild-type P22-infected cells were
prepared as described above. Bacteriophages were
purified fromtheselysatesaspreviously described for
bacteriophageT7(29).
Electrophoresis in metrizamide density gradients.
Electrophoresis in metrizamide density gradients was performedbydilutingone part ofsample inTris-Mg
buffer with 1.2 parts of sample buffer A and by
subjecting this mixturetoelectrophoresiswith
electro-phoresis bufferA at25°Caspreviouslydescribed (38).
on November 10, 2019 by guest
http://jvi.asm.org/
Forthe largerpreparations, a 1.0-cm (innerdiameter)
gel tube was used instead of the 0.6-cm tube used
previously (38). As much as 20 mg of procapsid had
been fractionated with a single 1-cm tube without
obvious distortion caused by high concentrations.
Dimerizationof a T7 procapsid does not alter its solid
support-free electrophoreticmobility(p.)(31),
indicat-ing that P22 procapsidaggregation would not alter its
profile during electrophoresis in metrizamide density gradients.
Fractionation ofcapsidsby agarose gel
electrophore-sis.To 35
RI
ofasamplewasadded 10 ,u1ofsamplebuffer A. Of thismixture, 30,u1waslayered insample
wellsof a horizontal agarose gelcastin andsubmerged
beneathelectrophoresisbufferA.Electrophoresiswas
performed at0.%V/cm andatroomtemperaturefor
16 h as previously described (33). Gelswere stained
withethidium bromide (DNAspecific) after the
pack-aged DNAwasemptied (33). Subsequently,gelswere
stained with Coomassie brilliant blue(protein specific)
(37).
Fractionation of DNase-resistant(packaged)DNAby
agarose gelelectrophoresis after release fromcapsids.
Before releasing DNA fromcapsidsfor
electrophore-sis,itwasnecessarytoinhibit DNases. Thiswasdone
by addinga0.06volumeof 0.2 M EDTA (pH7.4) and,
after 10 min at room temperature, adding a 0.09
volumeof10% Sarkosyl NL97(Ciba-Geigy Corp.) in
water. To releaseDNAfromcapsids, this mixturewas
incubated at 75°C for 15 min. Some samples were
subsequently diluted in NET buffer. To 39 p.1 of the
final mixturewasadded 10 .1lofsamplebufferB,and
30p.lof this mixturewaslayeredinsamplewellsofa
horizontal agarosegelcastinandsubmergedbeneath
electrophoresis bufferB,asdescribed above. Forhigh
resolution ofmatureP22-sizedDNAs,electrophoresis
wasconducted at 0.34V/cm andat roomtemperature
ina0.25%gel for 3 to4days, as previouslydescribed
(30). For all other DNAfractionations, electrophoresis
wasconducted at 1V/cm and at room temperature for
16h.
Measurement of solid support-free ,u and related
parameters. To determine the p. ofaparticle in the
absence ofasolid support
(pO),
p. values measuredbyagarosegelelectrophoresisat25.0°Cwere
extrapolat-edto anagaroseconcentration ofzeroandcorrected for electroosmosis, as previously described (34), by
using either MEor
HGT[P]agarose.
From p0and theparticle radius (see Table1),aand total surfacecharge
number(z; ifRis theparticleradius,z=
4[1rR2)
werecalculated as previously described (37).
Preparationof DNA donorextractsfor in vitro
assem-bly. S.typhimurium DB7130wasgrownto2x 108/cm3
in 2x LB with aeration at 28°C. This culture was
shiftedto38°Candincubatedforanadditional90min.
The cellswere pelletedat 10,000 rpmand4°Cfor10
min in a J-21 rotorand were suspended in a1/200
volume of0.05 M Tris-Cl (pH 7.4)containinga
suffi-cientconcentration ofthe indicateddextran,sugar,or
polyol to give the indicated final concentration. The
cellswereslowlyfrozenina-20°Cfreezerovernight
andwerethawed at35°C. A1/10volume of
3-mg/ml
lysozymein 0.25 MTris-CI(pH 7.4) wasadded, and
the mixturewas incubated for30min onice.
Subse-quently,a1/10 volume of TSMB buffer and of0.3 M
ATPin0.25 MTris-CI(pH 7.4)wereadded.
In vitroassembly. Unless40%odextranwasused,in
vitro assembly was conducted by adding 15 p.l of
procapsid preparation to 15 p.l of the DNA donor
extractand by incubating at the indicated temperature
for the indicated time. Toterminate packaging,
sam-ples weretransferred to an ice bath, and 2,ul of 1-mg/
mlDNase I in water was added, followed by
incuba-tion at30°C for 30 min.Termination of packaging with
EDTA was also used in some experiments, but it did
notaffect the results. For packaging in40% solutions
ofdextrans,10p.l of procapsid preparation and 20p.l of
extract wereused as described above.
Sodium dodecyl sulfate-polyacrylamide gel
electro-phoresis. Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis was performed as previously
de-scribed (41). A10o gel was used, and proteins were
detectedbystaining with Coomassie blue (29).
Quantitation of DNA packaged. To determine the
amountofDNApackaged in capsids after agarose gel
electrophoresis, the integrated intensity of ethidium
bromide-stained bands was determined by scanning
fluorimetry of the orange fluorescence of the bands.
Fluorescence wasexcited by a short-wave UV light
bulb in an Helena R and D scanning
densitometer-fluorimeter. A Wratten 23A (orange) filter was placed
overthe entranceslits for the photomultiplier tube. To
convertintegrated intensities into amounts of DNA,
theintegrated intensities of bands formed by serially
diluting a known concentration of 17- T7 were ob-tained in the same gel. From these data, a calibration
curve wasconstructedfor determining the amount of
DNAfromintegrated intensities of the DNA packaged
in vitro by P22. The amount of protein forming
Coo-massie blue-stained bands was determined by
densi-tometry, performed with the above-mentioned
densi-tometer.
RESULTS
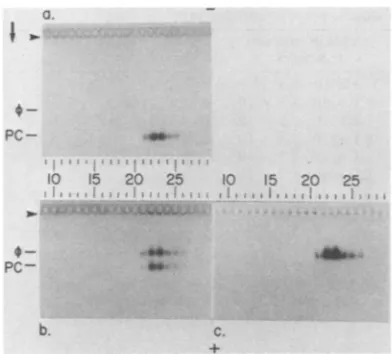
Electrophoresis of the P22 procapsid. To
iso-late
the
P22procapsid,
2-procapsids,
prefrac-tionated
asdescribed
above,
weresubjected
toelectrophoresis in
ametrizamide
density
gradi-ent. Toassay
for
procapsids after fractionation
of this
gradient, portions of each fraction
weresubjected
toagarosegel
electrophoresis. A band
further from
the origin
of
electrophoresis
than
the
band formed
by
maturebacteriophage
P22
was
observed in fractions
21through 25 of the
metrizamide
gradient (Fig. la). Equal portions
of
these andadjacent
fractions
werediluted into
capsid-defective
extractsfor the
assembly of
infective P22. The titer
of
infective particles
obtained
wasroughly
proportional
to theinte-grated intensities of the band
inFig.
lafor
allfractions
(data
notshown;
seealso
below).
Thisobservation
suggests that the bandof
Fig.
lais
formed
by
the P22procapsid,
aconclusion
con-firmed
by
electronmicroscopy
and sodiumdo-decyl
sulfate-polyacrylamide gel electrophoresis
(7,
10, 16)of fractions
in thegradient of Fig.
1(data
notshown).
In the
preparation of
Fig.
1, thedistance
migrated by
procapsids
in the agarosegel
wasindependent of
themetrizamide
gradient
on November 10, 2019 by guest
http://jvi.asm.org/
a. I
PC-10 15 20 25 10 15 2
Pc-FIG. 1. Electrophoresis of theP22 proi
metrizamide density gradient. A sample
procapsids, preparedandprefractionateda
in the text, was subjected to electroph4
metrizamide density gradientfor 7 h. Aport
fractionwassubjectedtoagarosegelelect
followed by staining with Coomassie bl
quently,anequalportionof eachfraction M
invitro assemblyat35°Cfor 1 h in theprese
dextran40,asdescribed in thetext.After
thereaction mixtures were subjected to;
electrophoresis, followed by staining wit
bromideand Coomassie blue.(a)Noincuba
ing withCoomassieblue.(b)Incubation,st
Coomassie blue.(c)Incubation, stainingwi bromide(contrast reversed).Theoriginsar byarrowheads.Procapsids (PC)and bacter are indicated. The direction ofelectrophoi
agarosegel is indicatedbythearrow.The(
electrophoresis in the metrizamidegradiei
right. The voltage gradient for (b) and (I
during electrophoresis becauseofa defec
supply. It is for thisreason that theproca
tion distance in (b)is less than it is in(a).
tion from which theprocapsidwastak However, the distance migrated in ag by 2- procapsids from other prepar creased significantly with the distance in the metrizamidegradient,upto7%.
c.
heterogeneity
of the T7 procapsid has been observed (19), but thecauseof theheterogeneity is not known in either case. P22 procapsids, purified by electrophoresis in metrizamide den-sity gradients, usually do not lose more than 25% of theirDNA-packaging
activityduring
storage for timesupto 3 monthsat4°C without
dialysis of metrizamide (longer storage times
o 25 have not been tried).
The p. of the P22 procapsid in metrizamide gradients is higher in magnitude than the IL of
most negatively charged, contaminating host
46b*
proteins. By sodium dodecyl sulfate-polyacryl-amidegelelectrophoresis, itwasfound that the electrophoresis shown in Fig. 1 separated the P22 procapsid from more than95% of the con-taminating host proteins. No host proteins (<2% of the amount of p5, the major P22 capsid tcapsid in a protein[7])weredetected in thecapsid-contain-of 2 .P22
ing regions
of the gradient of Fig. 1 (data notLsdescribed shown).
tionofeach The p.o, a, and z of the P22 procapsid, phage
rophoresis,
P22,
and 9- P22have
been determined(Ta-ue. Subse- ble 1). As previously found for bacteriophage
vasused for T7 (37, 38), the P22 procapsid hasanegative p.O
nceof20% higher in magnitude than the negative ofthe
incubation, maturebacteriophage. Themagnitude of the
9-agarose gel P22 p. was slightly, but significantly, higher h ethidium than the magnitude of the P22 p..Thisindicates
ation,
staih
anetpositive
external
charge
ofp9
(tail
spikes
th ethidium
[7]).
Thedifference in the and sizes ofprocap-re indicated
sid
andbacteriophage
(Table 1)
results in
aiophage
niae difference inbanding position
during
agarosegel
resis in the electrophoresis. Agarose gelelectrophoresiscan
directionof be used to distinguish these twoparticles (Fig.
nt isleftto 1). The of the mature P22 capsid is
indepen-c) dropped dent of the presence of packaged DNA (see ,tive power below), aspreviously shownforT7 (37).
Lpsid migra- Detection ofpackaged DNA after in vitro
as-sembly.Toassayphysically for theentryof P22 DNA into capsids, DNA-specific staining of capsidsfractionated by agarose gel
electropho-cen, ±2%. resis might be used (see
above).
To test this ;arose gels procedure, in vitro assembly, optimized asde-*ations in- scribed below, was performed with portions of migrated fractions of the metrizamide gradient shown in Asimilar Fig. 1. Assembly was followed by agarose gel
TABLE 1.
p.o,
radius, a, and zStructure Radius(nm) -O(cm2/V*sx 10-4)a -( (ESU/cm2 x 1O3)a -z(x 102)a
P22 31.4±
0.2b
1.47± 0.08 4.86 12.59 P22 31.4± 0.2' 1.52 ±0.08d 5.03 13.0
Procapsid 29.6 ±
1.0b
1.72± 0.08 5.69 13.0a pO,a, and zweredeterminedasdescribed inthe text.
bRadii were previously determined by low-angle, X-rayscattering (10). ESU, Electrostatic charge units.
'Theradius isassumed tobe thesame asthe radius ofP22.
dThat the
p.
of 9- P22 is significantly higher in magnitude than theP.0
of P22 has been shown bycoelectrophoresis in thesame slabgels. Differences in
pO's
aredetermined withanaccuracy higherthan theaccuracyofpodetermination.
b.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.491.48.244.73.250.2]TABLE 2. Effects on assembly of sugars, polyols, and dextransa
Compound %Present Yield ofparticlesinfectious P(atm)
None added 3 x 10'-5 x 10'
Ethylene glycol 5, 10, 20, 40 1.1 x 102-1.3 x 102
Glycerol 5, 10, 20, 40 1.0 x 102-1.2 x 102
Ribitol 5, 10, 20, 40 1.8 x 102-2.1 x 102
Sorbitol 5, 10, 20, 40 1.1 x 102-1.2 x 102
Inositol 5, 10,20,40 1.5 x 102_1.9 X 102
Methylglucopyranoside 5, 10,20, 40 1.4 x 102-1.7 x 102
Mannitol 5, 10,20,40 1.1 x 102-1.2 x 102 6.5, 13.1
Glucose 5, 10, 20,40 2.1 x 102-2.2 x 102 6.6,13.2, 29.2,74.8
Sedoheptulose 5, 10, 20, 40 1.3 x 102-1.5 x 102
Maltose 5 5.3 x 106
10 8.4 x 106
20 1.16 x 107
40 1.20 x
107
Sucrose 5 6.7 x 106 3.9
10 1.08 x 107 7.7
20 1.28 x 107 16.1
40 1.27 x 107 41.1
Dextran 10 5 1.5 x 107
10 2.4 x 107
20 2.8x 107
40 2.5 x 107
Dextran 40 5 1.6x 107
10 2.0 x 107
20 2.8 x 107
40 2.6 x 107
a In vitro assembly was conducted for 60 min, as
concentration of the indicatedcompound.
electrophoresis and staining for DNA (Fig. lc)
and
protein (Fig. lb). The procapsid-containing
fractions of Fig.
lareacted
toform
bacerio-phage-migrating particles which stained with
ethidium bromide, specific for DNA (Fig.
lc).Conversion
of
40
to50%
of the procapsids
tobacteriophage-migrating capsids occurred (Fig.
lb). Thus,
asexpected, the procedure used in
Figs. lb and
csuccessfully monitored
thepack-aging
of DNA.
Effects of dextrans, sugars, and polyols. Dex-trans, some sugars,
and
somepolyolsstimulate
in vitro
T7 DNApackaging
(36). Toincrease
invitro
DNA packagingefficiencies
and to helpdetermine mechanisms of
DNApackaging,
theassembly-promoting capabilities of
dextrans and several sugars andpolyols
weredetermined.
Sedoheptulose, glucose, sorbitol, mannitol,
andsmaller polyols
wereallineffective in
stimulating
the
formation of infective
particles (Table 2). Thesecompounds
werealsoineffective
instimu-lating either production of particles with
pack-aged
DNA orconversion
of the procapsid
to abacteriophage-like capsid;
nodetectable
conver-described in the text, in the presence of the indicated
sion occurred (data
notshown). In
contrast, sucrose, maltose,dextran
10,dextran
40, anddextran
500all stimulated (i) production of
infec-tive P22
(Table 2), (ii) production
of particles
with packaged
DNA(data
notshown), and(iii)
conversion of the
procapsid
to abacteriophage-like
capsid (data
notshown). Stimulation
oc-curred
only when the compound added
was adisaccharide
orlarger compound. Smaller,
relat-ed
compounds
werenotstimulatory.
Thesmaller, nonstimulatory compounds used
above did
notinhibit
stimulation of infectious
particle assembly by
dextran 40(Table 3).
Thisobservation
suggests thatthese smaller
com-pounds do
notirreversibly
inactivate
anextract component necessaryfor
DNApackaging.
The above-mentionedcompounds
thatdidstimulate
assembly
arechemically closely
related to thecompounds
thatdid
notstimulate
assembly.
Therefore,
itis
unlikely
thatbinding of
stimula-torycompounds
to an extract componentacces-sible
toallcompounds is
the causeof
thestimu-lation. Concentrations of mannitol
and glucosesufficient
tolower
P tolevels
stimulatory for
on November 10, 2019 by guest
http://jvi.asm.org/
P22 IN VITRO DNA PACKAGING 101
PC 5 10 15 0
I,I t~ Ii
*a
\e}-PC
I
b.
-PC
C.
0! 27.9xii
-| we t
~~~A:14.6x
6-B: 5.84x 106
.-C 4.05
x
10
D: 2.67
x106
-E:1.40x
106
- F: 21x106
FIG.2.DNAentryas afunctionof time. Several
15-,ul samples (=8 ,ug) of 2- P22procapsids, isolated as
described in the legend to Fig. 1, were diluted with
DNA donor extracts and incubated at 35°C in the
presenceof 20% dextran 40,as described in thetext.
Atthe indicated times after incubationwasbegun,the
reaction was terminatedbychillingand the additionof
DNase. After theaddition of 10±lofsample buffer A,
30 ,ul of each sample was subjected to agarose gel
electrophoresis for fractionation of undenatured
parti-cles. Of the remainder, 5 Ill, diluted as indicated
below,wastreatedtoinhibit DNase and release DNA
fromcapsids (denatured) before electrophoresis in a
0.90% agarose gel. (a) Undenatured, stained with
Coomassie brilliant blue (contrast reversed). (b)
Un-denatured, stained with ethidium bromide. (c)
Dena-tured(DNAreleased), stained with ethidium bromide.
Thetimes of incubation (inminutes) and theyield of
infective particles (PFU x 106/ml, background
sub-tracted)were(time, yield): (1) 0, 0; (2) 0.25, 0; (3) 0.50,
0; (4)0.75,0; (5)1.0, 0; (6)1.25,0; (7)1.50, 0;(8)1.75,
0;(9) 2.0, 7.5; (10) 2.25, 10.1;(11) 3.0, 12.0 (12) 5.0, 15.0; (13) 10.0, 22.0; (14) 15.0, 24.0; (15) 25.0, 24.1;
(16) 60.0,24.5. On(a)and(b), the lanemarked4)has
matureP22assembled invivo(16,ug); the lane marked
PC has 2- proheads, unreacted (=8 ,g). On (c), the
lane markedHhasaHindIIIdigest ofbacteriophageX
DNA, and the lane marked 4) has mature P22 DNA
frombacteriophages assembled in vivo. Themolecular
weights of theHindIllfragments, obtained from
refer-ence 43, are indicated. For (c), the samples were
dilutedasfollows inNETbuffer before
electrophore-sis: (1) through (6), nodilution; (7) and (8), 1:10; (9)
[image:6.491.58.234.77.353.2]and(10),1:12.5;(11) 1:16;(12)through (16),1:20. The
TABLE 3. Effects onassembly of mixing dextran 40
with smaller compoundsa
Yield of infectious
particles
(X107) None... 2.6
Ethylene glycol ... 2.4
Glycerol
... 2.5Ribitol... 2.6
Sorbitol... 2.7
Inositol... 2.7
Methylglucopyranoside ... ... 2.5
Mannitol... 2.5
Glucose... 2.8
a In vitro assembly was conducted for 60 min at
35°C, as described in the text, in the presence of20%
dextran and 20% of the indicated
compound.
sucrose were nonstimulatory (Table 2). There-fore, it is unlikely that lowering P isa sufficient cause for stimulation. Because of its apparent dependence on compound size, stimulation probablydepends on the nonpenetration by the compound ofsomeparticle, probably acapsid.
Theefficiency of infective particle formation increased when the sucrose or dextran concen-trationwas increased from5 to20% but did not further increase at 40% of either sucrose or dextrans (Table 2).
Sucrose
presentin
the ex-tractoriginally used for in vitro P22 DNApack-aging (24)
wasapparently
necessaryfor
the packaging observed. This was not discussed previously (24). The maximum efficiency has beenobtained withdextrans 10 and 40 (Table 2). Temporal kinetics of DNA entry.When DNA
entry was observed as a
function
of time after the initiation of in vitroassembly,
DNA pack-aged in capsids fractionated by agarosegel
elec-trophoresis first appeared at 1.5 min (Fig. 2b, lane7). The amount of capsid-associated DNAincreased from 1.5
to 5.0 min(Fig.
2,lanes
7 through 14), and all packaged DNA was in bacteriophage-like capsids, notprocapsids.
The procapsid began to convert to abacteriophage-migrating capsid
at1.5min
(Fig. 2a, lane 7),
the sametime thatpackaged DNAbegantoappear and 0.5 min before infectious P22 began to appear (legend toFig. 2).
The appearance of infectious P22 at 2 min is in agreement with datapreviously obtained
(24).Portions of
samples
from
theexperiment
(Fig.
2a and b) were also treated with DNase and
subjected
to agarosegel
electrophoresis
afterorigin of electrophoresis in (c) is indicated with an arrowhead; the origin of electrophoresis in (a) and (b)
is not shown. The direction of electrophoresis is
indicated withanarrow.
VOL.47,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.491.253.446.96.223.2]disruption of capsids
andrelease of
DNA asdescribed
above. It was known from previousexperiments
that the amountof
matureP22-sized DNA became large enough
atthe later
times
to cause theband
distortions described
previously
(30) if amountsoflysate
necessary todetect smaller
DNAfragments
atthe
earlier
times
wereused.
Thus,
asdescribed in the
legend to
Fig.
2, the amountof sample
usedin
Fig. 2c was decreased with the
time
atwhich the
sample
was taken. Aband
at theposition of
DNA
from
maturebacteriophage
P22first
ap-peared at 1.5min after the
startof the
incubation
(Fig.
2c, lane7).
DNAforming this band also
comigrated with
mature P22 DNAduring the
procedure
of
comparatively
high-resolution
agarose
gel
electrophoresis described above,
indicating
amolecular
weight
differing by
no more than2% from
themolecular
weight of
mature P22 DNA
(data
notshown).
The
sample taken
at1.25 min after
infection
had
nodetectable
DNAeither shorter than
or the samelength
as mature P22 DNA(Fig.
2c,
lane6), even
though the
amountof
the1.25-min
sample used
was 10times
the amountof the
1.50-min
sample
used.This
was truefor
a1.8%
agarose
gel
aswell
asfor the
0.9%
gel
usedin
Fig.
2c(data
notshown).
Inaddition,
noevi-dence of
capsid-associated
DNA
wasfound
at 1.25min
(Fig. 2b, lane 6). The failure
toobserve
partially
packaged
DNA at 1.25min
indicates
that
either
(i)
entryof
DNAinto
capsids
occursin less than
0.25min,
(ii)
partially
packaged
DNA
empties from
capsids before completion of
the
agarosegel
electrophoresis
in
Fig.
2b and
before the
completion
of the DNase
digestion
performed for Fig. 2c,
or(iii)
DNases entercapsids
withpartially packaged
DNAand
digest
this
DNA;
endogenous
DNaseswould
do this in
the
experiment of Fig. 2b. Although the
correctalternative
has notbeen
rigorously determined,
possibilities
(ii)
and(iii)
seemless
likely
than(i)
for
thefollowing
reasons.The
conditions of in
vitro
packaging
aredesigned
tostabilize the
packaged
stateof
DNA, and dextrans have
adramatic
stabilizing
effect on thepackaged
stateof
P22 DNA(36).
Therefore,
it
seemsunlikely
thatpartially
packaged
DNAemptied from
cap-sidsbefore
thecompletion
of
DNasedigestion
performed
forFig.
lc.The datapresented
above suggestcapsid
impermeability
to disaccharidesduring
DNApackaging
(see
alsothediscussion
below). Thus,
larger
molecules
suchas DNasesprobably
also could notenterthecapsid during
packaging.
Attempts to slowDNApackaging. The results in the
previous section
suggest that the invitro
rate of DNAentry intoaP22
capsid
istoohigh
tobe measuredby
theprocedures
used here. Tomeasurethis rate and the effects of
compounds
such
asdextrans
and ATP onthis
rate,attempts
were made to slow down P22 DNA packaging in
vitro.
In such an attempt, useof
25°C, instead
of35°C, resulted in first
appearanceof infective
particles at the time of appearance in
Fig.
2 (2min). However,
the amount ofinfectiveparticles
produced at 25°C was two to three orders of
magnitude
lower than the amount produced at35°C
(data
notshown).
At 5and 15°C,
nopro-duction
of infectious particles
wasobserved.
Thus, lowering the
temperature appeared not to succeed in slowing packaging.Inafurther attempt to slow DNA packaging,
the temporal
kinetics
of DNA entry were mea-sured in the presence of 2, 3, 5, 10, and 20%dextran
10. Noalteration in the time of the first
appearance
of
packaged DNA
wasobserved,
and
nopartially packaged
DNA wasdetected.
The
efficiency of
DNApackaging did, however,
decrease with decreasing
dextran 10concentra-tion (Table
2).Thus, all
attempts atobserving
partially packaged
DNAby slowing
DNApack-aging
have, thusfar, been unsuccessful.
Further characterization of particles assem-bled.
The data presented above indicate that the
length of
alldetectable
DNApackaged invitro in
the
bacteriophage-like capsids of Fig.
1is
the sameasthe
length of
matureP22 DNA.Quanti-tative
densitometry and fluorimetry, performed
as
described above, revealed
that theratio of
packaged
DNA tocapsid
protein is
lessfor
particles forming the band in Fig. lb
and c thanfor
purified
bacteriophage P22.
Atthe
comple-tion of
packaging
(15 minafter starting),
theratio
of
packaged
DNA tocapsid
protein for particles
assembled in vitro
had avalue
0.8times its
valuefor
purified bacteriophage
P22 assembledin
vivo.
Theseobservations
indicate
that20% of
the
bacteriophage-like capsids
did not package DNA.Because
packaging
was nolonger
occur-ring
at 15min, these
"empty"
bacteriophage-like capsids
areabortive end products.
Thecomigration
of
emptyand
DNA-containing
cap-sids
during
agarosegel
electrophoresis has also
beenobserved for
bacteriophage
T7(37). Fromquantitative densitometry of ethidium
bromide-stained
bands(Fig.
lc), it wasalso
found that
the numberof infectious
particles
perpackaged
DNA is 1 x10-4
to 3 x10-4.
Thisratio
is 3 x10-1
to 4 x10-1
for
P22assembled invivo.
The reason for thecomparatively large
number
of uninfective
particles with packaged
DNA
formed
invitro
is not known. However, twopossible
reasons have beeneliminated.
Thefirst
reasonis packaging of
host DNA. HindIIIrestriction
enzymedigests
of DNApackaged
invitro
areindistinguishable from HindIII digests
of DNA
packaged
invivo.
Thesecond reasonis
on November 10, 2019 by guest
http://jvi.asm.org/
breaks
in single
DNAstrands.
Moststrands
have the
length of the strands
of DNA packaged
in vivo,
determined by
electrophoresis in
the alkaline agarose gelsdescribed
previously (20) (data not shown).DISCUSSION
TheP22
procapsid, like
the T7procapsid,
wasfound
tohave
anegative
RA
higher in magnitudethan
thenegative
po
of
the maturebacteriophage
capsid. The capsids of the
maturebacterio-phages T7 and
P22 aredifferent serologically
(35),
and
the arrangementsof
genesin
the ge-nomesof these
twobacteriophages
aredifferent
(cf. references
6and
40). Thus,it is unlikely
thatthe
above
similarity is explained by
common ancestryand
maybe
a response to aselective
evolutionary
pressureexperienced
independent-ly by
T7 and P22.Analysis of
the>0's
of otherbacteriophages
and their
procapsids is currently
being performed.
Agarose
gel electrophoresis
appears tobe
the mostreliable and
efficient procedure for
deter-mining
whether or not a P22capsid is in
theprocapsid
stateduring
orafter in vitro
DNApackaging. Velocity
sedimentation is of
limited valuefor this
purpose (seethe discussion for429
in reference 4).
Determination of
the presenceof
or
absence of
p8 is also
notreliable. Some
T7capsids that have bacteriophage capsid-like
out-erenvelopes
as seenby electron
microscopy
and
agarosegel
electrophoresis contain T7
p9
(32), the
counterpart to P22p8; the p9
appar-ently
had been
dislodged
from its normal
posi-tion in the procapsid. Electron microscopy
re-quires
acomparatively large
amountof time.
Inaddition, electron microscopy depends
on accu-ratesubjective appraisal of the shape of capsids
and absence of
selective washing of capsids
from
grids.
Theseconditions
cansometimes
bedifficult
toachieve.
To assay
for
packaged
DNAduring
in vitro
assembly,
agarosegel electrophoresis of (i)
DNA
packaged in intact capsids
and(ii)
DNase-resistant
DNAreleased
from capsids
were used.The data
indicate that it takes 1.5
minfor
achievement of all
stepsin the
initiation of
packaging
and the
entryinto
a capsid of thefirst DNA
tobe
packaged. If capsids with
par-tially packaged
DNA are stableduring thepro-cedures of
analysis
used here, the data further suggestthat
entryinto
the capsid of the first DNA to bepackaged
occurs between 1.25 and 1.50 minafter
the startof
assembly. No informa-tion wasobtained
concerning
events occurringbefore
1.25min,
although
thesepresumably in-cludebinding of
theprocapsid
to DNA. Theconversion of the
P22procapsid
to abacterio-phage-like
capsid
occurred at atime
indistin-guishable from the
time of
appearanceof
a maturelength
of packaged
DNA.Because the
procapsid expands
during this
transition and
because the
procapsid
(before expansion) is
probably not
big enough
topackage
allof the
P22DNA (9),
it is likely
that the P22procapsid
expands
to abacteriophage-like capsid before
completion of
DNApackaging.
Dataobtained
with
T4 (13),429
(4),and T7 (32)
further indicate
that
this
conversion
occursbefore
mostDNAis
packaged.
If
so,the
temporal coincidence of the
P22
capsid conversion and the
appearanceof
a maturelength of
packaged
DNA isfurther
evi-dence
that entryof the first DNA
packaged
occurs
in
atime
spanless than the
difference in
sampling times, 0.25 min. Attempts
toslow
DNA
packaging
tobetter
measurethe
entry ratehave,
thusfar, been unsuccessful. These results
are
in
contrast toresults
in
astudy of
bacterio-phage 429 in
which
partially packaged
DNA wasdetected
by
velocity sedimentation (3, 4).
Be-cause
the
procedure shown in
Fig.
2c involved
no
fractionation
during which
DNAcould
emptyfrom P22
capsids, it is concluded that either
entry
of P22
DNAinto
capsids
occurs morerapidly
than entryof
429
DNA orloss
of
partial-ly packaged
P22 DNA(by
themechanisms
de-scribed above)
occurs to a greater extent thanloss
of
partially packaged
429
DNA. Asdis-cussed
above, the former alternative is
probably
correct.
P22
assembly, like
T7assembly,
wasfound
tobe
stimulated
by
sucroseand dextrans in
ex-tracts.Maltose
wasalso stimulatory, but
sedo-heptulose and all smaller
sugarsand
polyols did
not
stimulate
packaging. The correlation of
stim-ulatory effect with
compound size
suggeststhat
nonpenetration
of
someparticle, probably
acapsid,
is
required for
stimulatory
activity.
Incontrast to the results
obtained here with
P22,
sorbitol and
glucose
(but
notglycerol)
stimulat-ed
production
of
infective
bacteriophage
T7(36).
This latter observation
suggests thatthe
smallest
hole in the
T7"nonpenetrated"
particle is
small-erthan
thesmallest
hole in
the P22"nonpene-trated"
particle.
Thestimulation by dextran of
T7
infective
particle formation decreases above
14%
dextran
10(36). This is
asecond difference
in the results
obtained with
T7 and P22 (Table 2).
The nonstimulatory compounds glycerol and ribitol were
previously
shown to stabilizepack-aged
DNAin mature P22(36). Thisobservation
suggests that
stabilization of
packaged
DNA(after
entryinto
acapsid) by
thestimulatory
compounds (possibly
tofacilitate tail
addition)
is not asufficient
explanation of the stimulatory
effect.
Thatis,
atleast somestimulation
occursduring
orbefore
entry of DNA into thecapsid.
Thisstimulation could
resultfrom
therequire-VOL.47,1983
on November 10, 2019 by guest
http://jvi.asm.org/
mentforan
osmotic
pressure across capsids toassist DNAentry(32). Becausethe in vitroentry rate
of
P22 DNAinto
capsids could not be measured here, it has not yet been possible to test theeffect of external osmotic
pressure onthe DNA entry rate.
ACKNOWLEDGMENTS
Fortechnical assistance, wethank Elena T. Moreno. For
secretarialassistance,wethankDonnaScoggins.
This researchwassupported by Public Health Servicegrant GM24365from theNational Institutes ofHealth andgrant AQ-764from the RobertA.WelchFoundation.
LITERATURECITED
1. Benchimol, S.,H.Lucko, and A.Becker.1982. Bacterio-phage A DNA packaginginvitro;theinvolvement of the A Fl gene product, single-stranded DNA, and anovel
X-directed protein in thepackaging reaction.J.Biol.Chem. 257:5201-5210.
2. Bjornsti, M.-A.,B. E. Reily, and D. L. Anderson.1981. In vitro assembly oftheBacillus subtilis bacteriophage4)29. Proc. Nati.Acad. Sci.U.S.A. 78:5861-5865.
3. Bjornsti, M.-A., B. E. Reilly, and D. L. Anderson. 1982. Morphogenesis of bacteriophage 4)29ofBacillus subtilis: DNA-gp3 intermediateinin vivo and in vitroassembly. J. Virol.41:508-517.
4. Bjornsti, M.-A.,B. E.Reilly,and D. L. Anderson. 1983. Morphogenesis of bacteriophage4)29ofBacillus subtilis: oriented andquantizedinvitropackagingofDNA-protein gp3. J. Virol. 45:383-3%.
5. Black,L.W. 1981. In vitropackagingofbacteriophageT4 DNA.Virology 113:336-344.
6. Botstein, D., C.H. Waddell,andJ. King. 1973. Mecha-nismofhead assemblyandDNAencapsulation in Salmo-nellaphageP22.I.Genes,proteins, structuresandDNA
maturation.J. Mol. Biol. 80:669-695.
7. Casjens, S., and J. King. 1974. P22 morphogenesis I:
catalytic scaffolding proteinincapsid assembly. J. Supra-mol. Struct.2:202-224.
8. Casjens, S.,and J. King. 1975. Virus assembly. Annu. Rev.Biochem.44:555-611.
9. Earnshaw,W.C.,andS.Casjens.1980. DNApackaging by double-stranded DNA bacteriophages. Cell 21:319-331.
10. Earnshaw, W., S. Casjens, and S. C. Harrison. 1976. Assemblyof the head ofbacteriophageP22:X-ray diffrac-tion fromheads, proheadsand relatedstructures.J. Mol. Biol.104:387-410.
11. Fujisawa, H.,and M.Yamagishi.1981.Studiesonfactors involvedin in vitropackagingofphageT3DNA,p. 239-252. In M.Dubow(ed.), Bacteriophage assembly. AlanR. Liss, Inc.,NewYork.
12. Hohn,T.,and I.Katsura.1977. Structure andassembly of bacteriophage lambda. Curr. Top. Microbiol. Immunol. 78:69-110.
13. Hsiao,C. L., and L. W. Black.1977.DNApackagingand the pathway of bacteriophage assembly. Proc. Natl. Acad.Sci.U.S.A.74:3652-3656.
14. Jones,H. C. 1914. The elements of physical chemistry,p.
220-222.The MacMillanCo., New York.
15. King, J.,andS.Casjens.1974.Catalyticheadassembling protein in virus morphogenesis. Nature (London) 251: 112-119.
16. King, J.,E. V.Lenk,andD. Botstein. 1973.Mechanism of head assembly and DNA encapsulation in Salmonella
phage P22. II. Morphogenetic pathway. J. Mol. Biol. 80:697-731.
17. Kuemmerle, N. B., and W. E. Masker. 1977. In vitro packaging of UV radiation-damaged DNA from bacterio-phage T7. J. Virol. 23:509-516.
18. Masker, W. E. 1982. In vitropackaging of bacteriophage T7 DNA requires ATP. J. Virol. 43:365-367.
19. Masker, W. E., and P. Serwer.1982. DNA packaging in vitro by an isolated bacteriophage T7 procapsid.J. Virol. 43:1138-1142.
20. McDonell, M. W., M. N. Simon, and F. W. Studier. 1977. Analysis of restriction fragmentsof T7 DNA and determi-nation of molecularweights by electrophoresis in neutral andalkaline gels. J. Mol. Biol. 110:119-146.
21. Miyazaki, J.-I., H. Fujisawa, and T. Minagawa. 1978. Biological activity of purified bacteriophage T3 prohead-like structures as precursors for in vitro head assembly. Virology91:283-290.
22. Murialdo, H., and A. Becker. 1978. Head morphogenesis of complexdouble-stranded deoxyribonucleic acid bacter-iophages. Microbiol. Rev. 42:529-576.
23. Poteete, A. R., and D. Botstein. 1979. Purification and properties of proteins essential to DNAencapsulation by phage P22. Virology 95:565-573.
24. Poteete, A.R., V. Jarvik, and D.Botstein. 1979. Encapsu-lation ofphage P22 DNA in vitro. Virology95:550-564. 25. Pruss, G. J., J. C. Wang, and R. Calendar. 1975. Invitro
packaging of covalently closed circular monomers of bacteriophageDNA. J. Mol. Biol. 98:465-478.
26. Rutila, J.,and E. Jackson. 1981. A physical map of the bacteriophage P22 genome.Virology 113:769-775. 27. Sadowski, P.D.1977.Genetic recombination of
bacterio-phageT7 DNA in vitro. II. Further properties of the in vitrorecombination-packaging reaction. Virology 78:192-202.
28. Sadowski, P., A. McGeer, and A. Becker. 1974. Terminal cross-linking of DNA catalyzed by an enzyme system containingDNA ligase,DNA polymerase, and exonucle-ase ofbacteriophage T7. Can. J. Biochem. 52:525-535. 29. Serwer,P. 1976. Internal proteins of bacteriophageT7. J.
Mol. Biol. 107:271-291.
30. Serwer, P. 1980.Electrophoresis of duplex deoxyribonu-cleic acidin multiple-concentration agarose gels: fraction-ation ofmolecules with molecular weights between 2 x 106 and 110 x 106. Biochemistry 19:3001-3004. 31. Serwer, P.1980. A technique for electrophoresis in
multi-ple-concentration agarose gels. Anal. Biochem. 101:154-159.
32. Serwer, P. 1980. A metrizamide-impermeable capsid in the DNApackaging pathway of bacteriophageT7. J. Mol. Biol. 138:65-91.
33. Serwer, P.,and S. J. Hayes. 1982. Agarose gel electropho-resis ofbacteriophagesandrelated particles. I. Avoidance of binding to the gel and recognizing of particles with packaged DNA. Electrophoresis 3:76-80.
34. Serwer, P., and S. J. Hayes. 1982. Agarose gel electropho-resis ofbacteriophagesandrelated particles. II. Correc-tion ofelectrophoretic mobilities for the electro-osmosis of agarose. Electrophoresis3:80-85.
35. Serwer, P.,and S. J., Hayes. 1982. Detection of bacterio-phage-antibody complexes by agarose gel electrophoresis. Electrophoresis 3:315-317.
36. Serwer, P., W. E. Masker, and J. L. Allen. 1983. Stability and in vitro DNA packagingofbacteriophages: effects of dextrans,sugars, and polyols. J. Virol. 45:665-671. 37. Serwer, P., and M. E. Pichler. 1978. Electrophoresis of
bacteriophageT7 and T7 capsids in agarose gels. J. Virol. 28:917-928.
38. Serwer, P., and R. H. Watson. 1981. Electrophoresis in density gradients of metrizamide. Anal. Biochem. 114:342-348.
39. Steven, A. C. 1981. Visualization of virus structure in threedimensions, p. 297-323. In J.Turner (ed.), Methods and perspectives in cell biology, vol. 22. Academic Press, Inc., NewYork.
on November 10, 2019 by guest
http://jvi.asm.org/
IN DNA 105
40. Studier, F. W. 1972. Bacteriophage T7. Science 176:367-376.
41. Studier,F. W. 1973. Analysis ofbacteriophage T7 early RNAs andproteinsonslab gels. J. Mol. Biol. 79:237-248. 42. Tye,B.-K., andD.Botstein. 1974. P22 morphogenesisIl: mechanism of DNA encapsulation. J. Supramol. Struct.
2:225-238.
43. Wellauer, P. K., R. H. Reeder, D.Carroll, D. D. Brown, T.Deutch, T.Higashinaawa,andI.David. 1974. Am-plified ribosomal DNA from Xenopus laevis has
heteroge-neous spacer lengths. Proc. NatI. Acad. Sci. U.S.A. 71:2823-2827.