JOURNALOFVIROLOGY, Jan. 1993,p.178-188 Vol. 67,No. 1 0022-538X/93/010178-11$02.00/0
Copyright © 1993, AmericanSocietyforMicrobiology
Avian
Retroviral
RNA
Encapsidation: Reexamination of
Functional 5'
RNA
Sequences and the Role
of
Nucleocapsid Cys-His
Motifs
RACHELARONOFF,1'2tADELINE M. HAJJAR,1'2'3ANDMAXINE L. LINIAL12* Divisionof Basic Sciences, Fred Hutchinson CancerResearch Center, 1124 ColumbiaStreet,
Seattle, Washington 98104,1* andDepartments
of Microbiology2
and ComparativeMedicine,3 Universityof Washington, Seattle, Washington 98195
Received 14 August 1992/Accepted 20 October 1992
RNApackaging signals
(*)
from the5' ends of murine and avian retroviralgenomeshavepreviously
been shown to directencapsidationofheterologousmRNA into the retroviralvirion.The avian5'packagingregionhasnow beenfurther characterized, and we have defined a 270-nucleotide sequence,
A*,
which is sufficienttodirect packaging of heterologous RNA. Identification oftheA*
sequence suggests that several retroviral cis-acting sequences containedin*+
(theprimer binding site, the putativedimerlinkage sequence, and thesplicedonorsite)aredispensable forspecific RNA encapsidation. SubgenomicenvmRNA isnotefficientlyencapsidatedinto particles, even thoughtheA1 sequenceis presentinthis RNA.In contrast,spliced heterologous
*-containing
RNA is packaged into virions as
efficiently
asunspliced species; thus splicing perse is notresponsibleforthe failure ofenv mRNA to be encapsidated. We also found that an avian retroviral mutant deleted for bothnucleocapsid Cys-His boxes retains thecapacityto encapsidate RNA containing
*
sequences,althoughthis RNAis unstable andis thusdifficult to detectinmature particles. Electron microscopy reveals that virions produced bythis mutant lack a condensedcore,whichmayallow the RNA to be accessible to nucleases.
Packaging signals are the cis-acting retroviral sequences required forspecific encapsidation of genomic RNA into the virion. These signals were first identified by analysis of a
quailcell linecontainingaspontaneous Roussarcomavirus
(RSV) mutant provirus, SE21Q1b (29), which has 179 bp deleted from the 5' leader of the recombinant proviral genome(3, 42). Analysesof thismutant and of other avian and mammalian retroviral mutants have localized RNA
packaging signals
(*)
to the 5' ends of retroviral genomes(25, 26, 32, 48). Assayshave also been developed todefine sequences which are required for efficient packaging of nonviral RNAs. Such assays have shown that about 800 nucleotides (nt)of5' sequences containing part of the gag
gene(called
*+)
candirectpackagingofheterologousRNA,equivalent to that of genomic RNA, into murine leukemia
virus (MLV) particles (1, 5). Sequences from the 5' end of the avian retroviral genome (avian leukosis virus
[ALV])
have also been tested for the ability to induce specificpackagingof nonviral RNA(4).A683-base
*
+ sequencehasbeen foundtodirectefficient encapsidation ofaheterologous mRNA into avian retroviral virions. However, unlike the
case of MLV, this packaging was reduced about 10-fold
relative to that ofRNAencoded bya retroviral genome or complete retroviralvectors (4).
Subgenomicenv mRNAs are notefficiently encapsidated
bywild-type avianormurine retroviruses. The 'I sequences in MLV and spleen necrosis virus are located between the splice donorand acceptor sites; thus,
*
is not contained in the subgenomic env mRNA. In contrast, the defined 5'*
signal in avian viruses is upstream of the splice donor and thus isincluded in both the genomic and the splicedmRNAs.Nevertheless, env RNA is inefficiently encapsidated into
* Correspondingauthor.
tPresent address: Laboratory of Molecular Biology, Medical
ResearchCouncil, HillsRoad, Cambridge CB2 2QH, England.
ALVvirions. Anothercis-actingpackagingsignal, the 115-nt direct repeat(DR)sequence,originallyfoundflankingthesrc
gene of RSV, has been implicated in the encapsidation of avian retroviral RNA into virions (43). However, because the DR is present in bothsplicedsubgenomicandunspliced
genomicRNAs, itcannotexplainthe difference inpackaging
between these RNAs.
The retroviral genome encodes threepolyprotein precur-sors, Gag, Pol, and Env. In thecaseof avianretroviruses, Gag is proteolytically cleaved to yield five polypeptides: matrix (MA), a protein of unknown function (plO), capsid
(CA), nucleocapsid (NC), and protease (PR). Only the gag
geneproductisrequiredforproductionof retroviralparticles
containing RNA. Viruses with mutations in the PR active
site in which no processing of Gag occurs form particles which containwild-type amounts of RNA (13, 44). These and other data(38)supportthe idea that thefull-length Gag precursoris bothrequiredand sufficient for RNA packaging. AfterproteolysisofGag, the NCpolypeptidebinds RNA in vitrowithhigh affinitybut withno apparentspecificity(17).
Proteolytic cleavage also coincides with the formation of
mature C-type virions containing ahighly condensed elec-tron-densecore(24).
Analyses of virions produced by mutated proviruses have indicated that a 14-amino-acid Cys-His motif, CX2CX3
(G)HX4C (whereXrepresents anyaminoacid)found within
the NCdomain ofGag, is required forproduction of particles
containing genomic RNA (34, 35). Cys-His boxes have
generallybeenthoughttoform metalbinding domains
some-what similar to zinc finger metal binding domains. It was reported that retroviruses donotcontain zinc(21), but more
recent work has detected zinc in association with Gag proteins (for example, see reference 7). Although zinc fin-gers are generally associated with DNA binding activity,
transcriptionfactor IIIA bindstoRNAaswellasDNAand
containsmultiplezincfingers (6).
178
on November 9, 2019 by guest
http://jvi.asm.org/
A
'TI SI SV40PA
Sa=
Length PackagingS.D.
683 ....
S.D.
AS.D.
5'ST
3S T
B
C(rTA) (A)n
ACGAACCACTGAATTCCGCATCGCAGAGATATTGTATTTAAGTGCCTAGCTCGATACAAT
AAACGCCATTTTACCATTCACCACATTGGTGTGCACCTGGGTTGATGGCCGGACCGTCGA
PBS
TTCCCTAACGATTGCGAACACCTGAATGAAGCAGAAGGCTTCATTTGGTGACCCCGACGT
LIP A'T 5'Sv
GATAGTTAGGGAATAGTGGTCGGCCACAGACGGCGTGGCGATCCTGCCCTCATCCGTCTC
GCTTATTCGGGGAGCGGACGATGACCCTAGTAGAGGGGGCTGCGGCTTAGGAGGGCAGAA
3'5JS
GCTGAGTGGCGTCGGAG§GAGTACTGCAGGGAGCCCAGATACCCTACCGAGAACTCA
GAGAGTCGTTGGAAGACGGGAAGGAAGCCCGACGACTGAGCAGTCCACCCCAGGCGTGAT
Gag S.D.
TCTGGTCGCCCGGTGGATCAAGCATGGAAGCCGTCATAAAGGTGATTTCGTCCGCGTGTA
4
GCT-AT
AAACCTATTGCGGGAAAACCTCTCCTTCTAAGAAGGAAATAGGGGCCATGTTGTCCCTCT
(dLs)
TACAAAAGGAAGGGTTGCTTATGTCTCCCTCAGACTTATATTCCCCGGGGTCCTGGGATC
4-L¶' B'S'!
CCATTACCGCGGCGCTATCCCAGCGGGCTATGATACTTGGGAAATCGGGAGAGTTAAAAA
CCTGGGGATTGTTTTGGGGGCATTGAAGGCGGCTCGAGAGGAACA I+
In thisreport,we demonstrate thatasequenceof 270nt,
A*,
can direct heterologous neo RNA into ALV. We alsoshow that incontrast tosplicedenvmRNA inthe retroviral
context, both spliced and unspliced heterologous mRNAs
containing
*
are efficiently packaged. Inaddition,reexami-nation of the role of the ALV Cys-His motifs in RNA
packaging suggests that these are dispensable for initial
interactionsofGagwith 4.AnRSVCys-Hisdeletionmutant
produces particles containing RNA, but this RNA is not
intact and thusis more difficult to detect in mutant than in
wild-typevirion RNApreparations.
MATERIALS ANDMETHODS
Cell culture and transfections. ThequailcelllinesSE21Q1b (29), Q2bn-4D (46),andQT35 (36) andQT35 containingthe
398 ....
270 ....
135 +
263
FIG. 1. (A) Constructs usedtoassay5'regions of thegenomefor
4ifunction. Portionsof the 5' endof the RSVgenometobe tested
werecloned into the plasmidpCMVneo asdescribed in Materials and Methods.S.D., splice donor site ingag; AS.D.,splice donor site mutated inconstruct;S.A., spliceacceptorsite inSV40sequences. (B) Sequences of*+,L+,A*,and
S*.
Arrowsdenote the limits of the 683-base *+, 398-baseL+, 270-base A+, 135-base 5'S*, and 270-base 3'S4 sequences.The PBSis denoted byaline above the sequence. The promoter element (TATA), the polyadenylation [(A)n] signal, thestartof Gagsynthesis, the major 5' splice donor site (S.D.), and the putative dimer linkage sequence (dls) are indicated above thesequence. Thelocation of theSacl restriction endonuclease site(GAGCI7C) is indicated byabox.The splice site is mutated inA*toaHindIII restriction siteasindicated by GCTatthe arrowhead.
dell,2 provirus (35)weregrowninGM+D+CK(Ham'sF10
medium containing 10% tryptose phosphate broth [Difco], 5% calf serum, 1% heat-inactivated chick serum, and 1% dimethyl sulfoxide). The dell,2 producer cell line was
de-rivedby transfecting QT35cellswith theRSV dell,2 proviral plasmid (see below) andthepCMVneoL4, plasmidataratio
of 10:1. Cellsweregrownat37°Cina6% CO2 atmosphere.
Transfections were performed by the modified calcium
phosphatemethod ofChenandOkayama (10). All cellswere
seeded in Dulbecco modified Eagle medium with 10% calf
serum prior to the addition of plasmid DNA. Cells were
selected for drug resistance in CM (Ham's F10 medium
containing 10% tryptose phosphate broth, 12% calf serum,
4% chick serum, and 1% nonessential vitamins [GIBCO]). Hygromycin (Sigma) was used at 0.1 mg/ml and G418 (Sigma) was used at 0.25 mg/ml. Mass cultures of
drug-resistant cellswereobtainedafter 1to 3 weeksofselection. Individualdrug-resistantcell cloneswerepickedwithfinely
pulledoutPasteurpipetsintofresh selectivemedia. Plasmids. Plasmids were constructed by standard
tech-niques (31). pCMVneo and
pCMVhph4,'
have been previ-ouslydescribed(4, 27). testsequences(A4,L*4,
5'S+, and3'S+)wereinserted into the SmaI site ofpCMVneoin either
thesense orantisense
(QI)
orientation. Figure 1Acomparesthe
Ali,
L+, andS*
sequences relative to the previouslydescribed 4+ sequence (4). To construct Lx4, the 5'-most
BstEII-XhoI fragment of pATV8R was cloned into vector pSL1180 (9), creating pSL1180. This was the template
for amplification with the following oligonucleotides: 5' primer (5'--3'), CGAGTCGACGGGAATAGTGGTC, and 3'primer, CGATCATGAGCCTTCAATGCC.Theamplified productwas isolated, digestedwithHincIIandBamHI,and
CMVIF
zx %.PIVI V IL- VW I
I A
A T
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.83.519.107.259.2]180 ARONOFF ET AL.
1
2
3
-dii lZ
P+
LT'
aLTPFIG. 2. Primer extension ofanalysisof virion RNA todetermine
*+-versusLq,-directed encapsidation. Q2bn-4D cellswere
trans-fected with pCMVneo*+ (q+), pCMVneoL* (sense) (L+), or
pCMVneoLqi (antisense) (aLqi), andpoolsof G418-resistant clones
were selected. Equivalent amounts of viral supernatants were pelleted, and virions were extracted. RNA was hybridized to a
22-baseneo-specificantisenseoligonucleotide,andprimerextension assayswereperformed.
inserted into vectorpSL1180DNApreviously digestedwith BamHIand StuI.Theresultant subclone
(pSL1180L*)
wasthendigested withPvuII and SmaI and ligated into SmaI-linearizedpCMVneo. The A4 fragmentwas amplifiedfrom
pSL1180L4
by polymerase chain reaction with the5'AqI
oligonucleotide primer, CGAGTCGACGGGAATAGTGG TC, and the 3'A primer, ATCAAQllATGACGGCT TCC. The 3'A primerintroducesamutation inthe retrovi-ralsplice donor site thatcreates aHindlIl restriction endo-nuclease site (underlined). The amplified fragmentwas iso-lated anddigestedwithHindIII andHincIl; the endswere
repairedwithKlenowpolymeraseandligatedintopCMVneo linearized with SmaI.
pCMVneo5'S*
and pCMVneo3'S4i plasmidswereconstructedby digestingtheL*
fragmentwith Ec1136II (anisoschizomer ofSacI) andligatingthe 5' or 3' fragment into the SmaI site of pCMVneo. pVZLti wasconstructed for generation of RNase protection probes by insertingtheNsiI-BamHIfragmentfrom
pSL1180L*4
into thePstI-BamiHI-digested pVZ1 (pBluescribe [Stratagene, La
Jolla, Calif.] containingadditionalrestrictionenzymesitesin the polylinker [20]). When pVZLqi is digested withEcoRI and transcribed by T3 polymerase, a sense
L*
RNA isgenerated;when
pVZL*
is digestedwithHindlIl and tran-scribedbyT7polymerase, an antisense RNAisgenerated. The RSV PRactivesite mutant(pgag.neo.D37N) and dele-tion(pgag.neo.APR)constructswerekindly provided byL. Stewart and V. Vogt (45). D37N also contains a secondmutation in PR (D--Y at amino acid 41 [47a]). These plasmids express only the appropriately mutated retroviral gag gene (not pol or env) and a neo gene (that is expressed from a spliced mRNA). The RSV NC mutant proviral construct, pAPrCdell,2, deleted for both Cys-His boxes, was kindly provided by P.-F. Spahr (35; the detailed con-struction is given in thisreference).
RNApreparations. Viral supematants were collected from
virus-producingcell line cultures every 12 h and clarified of
cell debris by low-speed centrifugation (2,500 rpm for 10 min) in an American Scientific Products Omnifuge. Virions
werepelleted througha2-ml20%sucrosecushion in isotonic buffer(SB) (0.1MNaCl, 1 mM EDTA, 10 mMTris-HCl,pH
7.4) by centrifugation at 23,000 rpm for 2 h in a Beckman
SW28rotor.Viralpelletswereresuspended in 100 ,ul of SB. The protein concentration was determined by Bradford analysis of resuspended pellets (Bio-Rad, Richmond, Calif.). Virions were lysed in 0.5% sodium dodecyl sulfate (SDS), and RNA was extracted by using two or three phenol-chloroform extractions followedbyachloroform extraction and ethanolprecipitation. In someexperiments, virion prep-arationswereincubated in the presence of0.5%SDS and 200
,ug of proteinase K (VWR Scientific) perml for 30 min at
37°Cpriortothephenol-chloroformextractions. Viral RNA
pelletswereresuspendedsothat each microliter of RNAwas
equivalentto1ml oforiginalviral supernatant. Total cellular
RNA was extracted by the guanidinium isothiocyanate method ofChomczynski and Sacchi (11) in 0.5 ml of solution D per10-cm2plate oftissue culture cells. RNAintegritywas
monitoredby ensuring the presence of distinct rRNA bands after ethidium bromide stainingof total cellular RNA elec-trophoresed in0.8% agaroseminigels.
RNAanalysis. Methods forprimer extension and RNase
protection have been previously described (4, 28). The
sequence of the neo-specific oligonucleotide (neo 22-mer) primer is (5'-3') GCAGCCCTlTGCGCCCTGAGTGC.
Sam-ples were resuspended inloading dye and electrophoresed
through 8% (primer extensions)or 6% (RNaseprotections)
polyacrylamide gels containing 8M urea.
Protein analysis. SDS-polyacrylamide gel electrophoresis
and techniques for cell labelling and immunoprecipitation
have beenpreviouslydescribed(41).Aftergel
electrophore-sis, proteinswere electroblotted ontonitrocellulose or
Im-mobilon P (Millipore) with a Semi-phor apparatus (Hoefer Scientific Instruments). Western immunoblots were per-formed withone ofthefollowing primaryantibodies (Abs); rabbit otRSV Pr-C (used at adilution of 1:500) and
mono-clonal Abs aCA and aMA (used at a dilution of 1:1,000) obtained from V. Vogt (Cornell University). To prevent
nonspecific Ab binding, filters were incubated for 2 h in
phosphate-bufferedsaline-0.5% Tween-5% driedmilk. The
filterswere then incubated with the primary Ab diluted in
phosphate-buffered saline-0.5% Tween-5%dried milkfor 1
hatroomtemperaturewithshaking and then washed for 10 minthreetimes with thesamebuffer. Secondary Abs
(Am-ersham, Arlington Heights, Ill.), either horseradish
peroxi-dase-conjugated donkey a-rabbit Abs (1:6,000) for
rabbit-derived antisera or horseradish peroxidase-conjugated a-mouse Abs (1:2,000) for reaction with monoclonal Abs,
wereincubated with the filter in phosphate-buffered
saline-0.5% Tween-5% dried milk for 1 h at room temperature.
Filters were washed as described above, and signal was detectedby usingthe enhanced chemiluminescence system (Amersham).
Electron microscopy. Culture supernatantswere clarified ofcell debrisbylow-speedcentrifugation, and viruses were J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.126.230.70.361.2]A.
B.
C.
D.
1 2 3 4 5 6 7 8 16 17 18 19 20 21 22
9 10 11 12 13
-112 nt
A
Al
5'S5'SI 3'Svirus
14 15
-112 nt- _ _4 w~
_L-A AI
cell
v none L A AL 5'S S'S 3'S 1Js
primer
neo L CH- A AIS'S 5'S 3'S
FIG. 3. Primerextension analysis ofviralRNAs containing
neo*
sequences. (A) RNAs were extracted from virion RNAs, and primerextensionanalysiswith the neo 22-meroligonucleotidewasperformedasdescribedinMaterials and Methods. Equivalentamountsofviral supernatantswereloaded in each lane.Q2bn-4Dcells weretransfected with pCMVneo with the indicated test*sequence. Lanes: 1, none
(neo); 2,L+; 3, NC mutantdell,2;4, A+; 5,AI, (antisense or inverted); 6,5'S4;7,5'SI; 8,
3'S*.
Thegel wasexposed for 18 h with twointensifyingscreens. (B)Exposureof thegelcontaining lanes 4 to 8 (panel A) for 3 days withintensifying screens; samples were as indicated
forpanelA. (C) CellularRNAextractedfrompCMVneoAe4iconstructswithAu,in the sense(lane14)orantisense (lane15)orientation. (D)
Westernblot analysis ofvirionsproduced by G418-resistant mass cultures. Test j sequencestransfected into each Q2bn-4D line arelisted
below each lane. The bandscorrespondingtothe Gag proteins CA (27 kDa) and MA (19kDa) areindicated. Lane 21 containsone-fifththe amountofsamplethatlane 20 contains.
pelleted through a 20% sucrose cushion. The viral pellets
were fixed in a 1:1 dilution of SB and half-strength Kar-novsky's fixative (22) for4 to6 h and thendehydrated in a graded series of ethanols. The pellet was then infiltrated with
amixture ofpropyleneoxide andLuft's Epon 812(30),with
gradual increasing of the proportion of Epon 812 to 100%.
Afterbeing infiltrated overnight, the pellet was embedded in Epon 812 andpolymerized ina60°Covenfor 48 h. Sections
were cut at athickness of 70to 80 nm, stained withuranyl
acetateandlead tartrate, and examined withaJEOL100SX transmission electronmicroscope at80 kV.
RESULTS
Packaging of heterologousRNAcontainingavianretroviral 4 sequences.Wepreviouslyfound thata683-base sequence called 4+ can directspecific encapsidation of hygromycin
(hph) RNA into the ALVvirion when inserted 3' of the
codingsequences(4).Inordertofurtherdefinefunctional
*
domains, sequences containedwithin
*+
were tested in asimilar heterologous system. These
*
test fragments wereinserted atthe 3' end ofpCMVneo in sense and antisense
(inverted)orientations (Fig. 1A). The sequencesof theA+,
L+,,
5'S+, and3'S*
test fragments relative to that of thepreviouslydefined
4i+
are shown in Fig. 1B.Lls
lackstworetroviralcis-actingelements, theprimer binding site(PBS) anda sequence postulatedtobe involved ingenomic RNA
dimerization, the dimer linkage sequence (dls) (8, 12). The
A4,
sequence also lacks the PBS but ends at the major 5'splice site,whichwasmutatedduringthecloningprocedure
sothat it isnolonger functional (Fig. 1B). Deletion analysis hadpreviously shown the regionsurroundingthe SacI site in theavian leader regiontobeimportant for RNApackaging
(25). Cleavage of
L*
atthe SacI site creates the5'S*
and3'S*
test fragments (Fig. 1B). ThepCMVneo vectorscon-tainingeach of these 4 testsequencesweretransfected into
theRous-associated virus type 1-derivedpackagingcell line Q2bn-4D (46). Virionsproducedby the cell lineswerethen
assayed for the presence ofheterologousneoRNA.
Wefirst directlycomparedthepackagingmediatedby
uP+
tothatdirected by thelongesttestfragment, L+. As shown
in Fig. 2,packagingdirectedby
L*
in thesenseorientation(lane2)isequivalenttothat directedby
4.+
(lane 1),whereas in the antisense orientation(aL4.;
lane 3),this packaging is decreased by more than 100-fold. Because there is nodifference between
L+.-
and*+-directed
packaging, weconclude that neither the PBS nor the dls contributes to
specificencapsidation.
Wenextexaminedtheabilityof all of thetestfragmentsto
direct RNA packaging by using primerextension analyses
(Fig. 3A). Both
L*
andA*4
sequences direct equivalentlevels ofpackagingof theneoRNA(comparelanes 2and4). The
5'Sf
retains some abilityto inducepackaging (lane
6).
Thisismoreclearlyseeninalongerexposureof thisgel
(Fig.
3B, lane 11). The
3'SI
sequence does not directspecific
encapsidation of the heterologous mRNA
(Fig.
3A, lanes 8and13),nordo any of theinverted
*4
sequences([denoted by
subscriptI]lanes5, 7,10,and
12)
relativetoA*
(lane
9).
neomRNAis present in all cell culturesatsimilar levels
(Fig. 3C,
V- fo,tw
AL. mmdo -cA
,x7ft",
kw MAon November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.75.534.78.327.2]182 ARONOFF ET AL.
Sac
S.D.
I~~~~~~~~~I
le:thnI(nt)
420 Pro'ected species
328 270
410
B
V1i-ion RNA
MI 1 2 3 4 2' 5
527
--404 4--4 -U
309 -
-242 W -s
2429
spliced L 'V
[--C
Cell RNA 1 2 3 4 5
527 -404-
:w.5
YIP309--s Q
242 - _
275 260
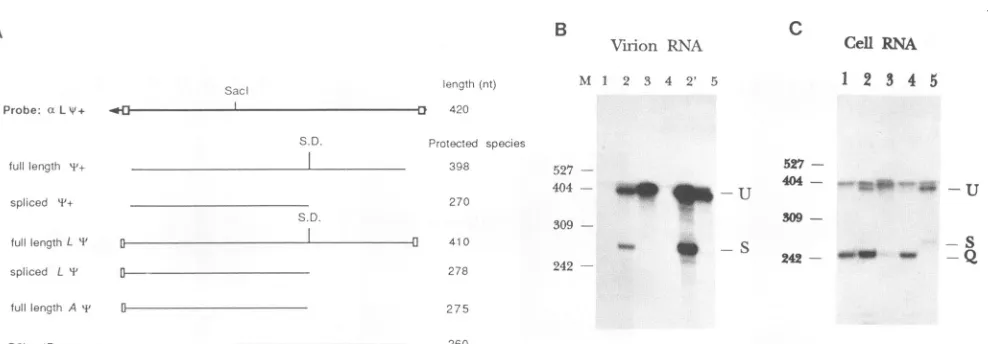
FIG. 4. RNase protection analysistodeterminepackaging of spliced and unspliced+-containingRNAs.(A)Location and size ofexpected protection products forthetest*sequences.The antisenseL++ probeis shownonthetopline,with theexpected protected specieslisted below. The small boxes indicateplasmid-derived polylinkersequencesshared withsomeof the RNAs. (B)RNaseprotection analyseswere performedasdescribed in Materials and Methods. Virion RNAswereisolated fromG418-resistantmasscultures ofQ2bn-4Dcellstransfected with plasmids as follows (per lane): 1, pCMVneo; 2and 2', pCMVneoqi+ (virusobtained after transfections withtwo differentplasmid preparations); 3,pCMVneoL*;4,pCMVneoL*(antisense).Lane5containsQT35cellsinfected withreplication-competentALV butcontains
noneoplasmid. Unspliced and splicedRNAsaredenoted U and S. M indicates molecularweightmarkers of the indicated nucleotidelengths. The faint band above the unsplicedprotected product,bestseenin lane 1,is causedbyresidualundigested full-lengthaLI probe. (C)Total guanidinium isothiocyanate-extracted cellular RNAwasexamined by usingRNaseprotectionassays asdescribed forpanelB. Each lane contains RNA extracted from the cellsusedtoproducethe virions inpanelB. Lanes:1,pCMVneo; 2,pCMVneoqi+;3,pCMVneoL*;4,
pCMVneoL*(antisense); 5, QT35cellsinfected with ALV. The location of theQ2bn-4D packagingvectorband is denoted(Q).There isno
sequencemismatch betweentheprobeandthisvector.
lanes 14 and 15 for A4, and
A*,,
and data not shown), regardless of the orientation of the test insert. As a measure of viral particle production, the level of viralproteins was examined by Western blot analysis. Similar
levels of particles (within threefold) are produced byeach
Q2bn-4D culture (Fig. 3D). When the primer extension
experimentshown in Fig. 3was quantitated by
densitome-try, after correction for theamount of RNAloaded, itwas
found that A4, directs encapsidation that is greater than
100-fold-more efficient than the level seen with no
A*
sequence orwithA4I,. Although5'S4 directssome
encapsi-dation ofneoRNA,the level isincreasedonly byabout5-to
10-fold abovebackground (eitherneo aloneorneo
contain-inginverted sequences).
Spliced RNA is efficiently packaged in the heterologous vector system. Although both genomic and spliced
subge-nomic avian retroviral RNAs contain sequences, only genomic RNA is efficiently encapsidated. Because the
Lq4
and 4+ sequencescontain the retroviral 5'splicesite and the simian virus 40 (SV40) sequences included in theheterolo-gous vector contain a 3' splice site (Fig. 1A), the neo plasmidscould be usedtodeterminewhethersplicingaffects
packaging ofheterologous RNAs. We previously detected several different 4,+ RNA bands by Northern (RNA) blot
analysis, indicating that the splice sites are used in the
pCMVneo4+ construct(datanotshown). In orderto deter-mine whether spliced heterologous RNA is packaged into virions, an antisenseL4 probewasused for RNase protec-tion analysiswith cell and virion RNA from Q2bn-4D cell
linestransfected withneo4, vectors.
Figure4AdepictstheRNase-protected products expected
for each 4-containing RNA species. The results of the RNase protection assays with virion RNAs (Fig. 4B) and
cellular RNAs (Fig. 4C) are shown. Control cells were
transfected with plasmid encoding ALV. As expected for
wild-type virus, both unspliced and spliced ALV RNA is detected in cell RNA(Fig. 4C, lane 5) but only unspliced
RNAcanbereadilydetected in virion RNA(Fig. 4B,lane5).
We found that the retroviralsplicesite is usedin thecaseof
the
pCMVneo*+
construct butnot theLq,
construct (Fig.4C, lanes 2 and 3). Both the spliced and unspliced
*+-containingpCMVneoq,+
RNAspeciesareefficientlyencap-sidated into virions (Fig. 4B, lanes 2 and 2'). Since the identicalsplice sites arenotused in transcriptsencodedby
the
pCMVneoL*
construct,nospliced productis detected invirions (Fig. 4B, lane 3). In the case of the
L*
protected species,theunsplicedRNAwasdifficulttoseparate fromthe freeprobe on this gel,but these could be distinguished byusing gradient gels (datanotshown). The RNA encodedby
theQ2bn-4Dpackagingcellvectorcanclearlybeseeninall of theQ2bncell RNAs(band QinFig. 4C,lanes1 to4),but this +-deleted RNA ispackagedveryinefficiently (Fig. 4B,
lanes 1 to4).
Acomparison of the ratio ofunspliced to spliced (U/S)
RNAs in thecell and virion RNAsprovides an estimateof
theefficiency ofencapsidationof each RNA. In thecase of
pCMVneo4,+,the total intracellularU/Sratio is 8.3(Fig. 4C,
lane2),while intwodifferent virion RNApreparations, the
U/S ratios are2.9 and 1.4 (Fig. 4B, lanes 2 and 2').These numbersarenotcorrected for the difference in the numberof radioactive residues protected by each RNA species and thus overrepresent unspliced RNA. We conclude that
splicedneo RNAs arenotexcluded from thevirion, unlike thecasefor thesplicedALVenvmRNA(whichisthesame
sizeasthesplicedvectorproducts; Fig.4B andC,lanes5).
The ratio ofU/SALV RNAs inthe cell is9.8; however, in the virus it ismorethan65(atthe limit ofquantitationofthis
gel). As the 270-base
A*
lacking a splice donor site ispackaged, theseresultssuggestthatsequenceswhichare3'
A
Probe: {x L-v+
fIulllenq,h k'' spliced 41
fulllength L4'
full lencrih A 'v
02bn-4Dgenome
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.68.562.66.238.2]virus: as
PL
- - 410
-- 270
-, *- 260
--u
1%
4.
-7 8
0p
*:
1 2 3456
FIG. 5. Effect of proteinase K treatment onvirion RNAs de-tected by using RNase protection. (A) RNA was extracted as described in Materials and Methods from cellscontainingthedell,2 provirus and transfected with pCMVneoL4i (lane 1) or from Q2bn-4D cells transfected with pCMVneoA* (lane 2). Virus was isolated from these cultures, and RNAwasextracted either after digestion (lanes4and6)with 200 pLgofproteinaseKpermlorwith noaddition ofproteinaseK(lanes3 and5).Viruswasfromdell,2 cells (lanes 3 and 4) and Q2bn-4D cells (lanes 5 and 6). RNA
extracted fromequivalentvolumes of viralsupernatantwereloaded
ineach lane.(B)RNAwasextracted from cellscontainingthe APR provirus (lane 7)orthe PR active sitepointmutantprovirusD37N (lane 8). Virion RNAwas extracted after proteinaseKtreatment
(lanes9 and 10).
of the splice site and the splicingprocessitselfare
dispens-able forspecific RNAencapsidation.
Because the antisense
L*
probe is alsoprotected by the+-deleted RNA derived from the Q2bn-4D packaging line
vector, it was possible to directly compare packaging of RNAscontaining and lacking
*
in the same virion popula-tion. The ratio of the Q2bn-4D vector-derived RNA to unsplicedneo*+
RNA in the cell is 6.4 (Fig. 4C, lane 2), while this ratio is0.007in thevirion(Fig. 4B,lanes2and2'; theQ2bn-4D-protected productof260ntisbarelydetectableatthisexposureof thegel). Thus, packagingof the heterol-ogous +-containing RNA is about 1,000-fold more efficient than that of thevector RNAlacking Ji.
NCCys-Hismotifsarenotrequiredfor RNApackaging.An intactGag polyprotein precursor appearstoberequiredfor avian retroviral genomic RNA encapsidation (38), although
processing of the precursor is not required (44). It has been suggested that the NC Cys-His motifs are an essential component of the packaging machinery, as deletion of both of the Cys-His motifs from the NC protein of RSV (dell,2) leads to production of virions in which RNA cannot be detected by Northern blot analysis (35). We confirmed this resultby using denaturing Northern blots (data not shown). However, Northern blot analysis will detect only intact RNA molecules and is thus not a very sensitive assay. During the course of these studies, we obtained evidence that RNA was present in dell,2 viral particles, albeit in a degraded form. Primer extension analysis with virion RNA obtained from cells containing both a dell,2 provirus and pCMVneoL4 sometimes showed a smear of neo extension products, possibly indicative of degraded RNA (for exam-ple,seeFig. 3A, lane3). In this experiment, only the dell,2
RNA yielded this result, although all virion RNAs were prepared at the same time and with the same reagents. In other experiments, we could sometimes detect a faint band ofthe expected size (data not shown).
This prompted us to reexamine whether
*-containing
RNAis packaged into the NC dell,2 virions by using RNase protection assays and a variation of our standard virion RNA extraction protocol. Intact virion RNA is readily obtained from wild-type virions by using phenol-chloroform extrac-tioninthe presence of0.5%SDS. RNA can also be extracted fromvirion suspensions after digestion with proteinase K in the presence of0.5% SDS (38, 44), although we have not found this treatment to affect RNA integrity when using wild-type virus. We reasoned, however, that if RNA in thedell,2 particleswas moresusceptibletoRNasedegradation,
aproteinase Ktreatment might inhibit such nuclease activ-ity. To examine whether proteinase K treatment would allow detection of dell,2 virion RNA, RNase protection assays wereperformed with the
aLLI
probe and RNA obtained from virions which had been extracted either by our usual methodor after proteinase K treatment (Fig. 5). We compared cellularand virion RNAs from dell,2 cells transfected with
pCMVneoL*
and Q2bn-4D cells transfected with pCMVneoA,4. When total cellular RNA from dell,2 cells
trans-fected with
pCMVneoLfi
was analyzed, the expectedLqi
band at410nt(Fig. 4A) is readily detectable (Fig. 5A, lane
1). Aprotected species of 270 ntis expected from hybrid-ization of theodL14probe
toA*
RNA(Fig. 4A),and both the 270-baseband and the Q2bn-4D +-deleted provirusbandat260 nt are evident in the Q2bn-4D cellular RNA (Fig. 5A, lane 2).
Aqi
RNA, as expected, is enriched in virion RNA relative to the +-deleted packaging vector RNA, although the apparent ratio islessenedbythelongexposureof thegeltofilm. Furthermore, proteinaseK treatment hasnoeffect
onthe amount of A or Q2bn-4D RNA detectable (lanes 5 and 6). In contrast, proteinase Ktreatment hasa profound effectontheamountof RNA detectable in thedell,2virions
(lanes3and4).Inadditiontothemajor protected species at
410bases, there are many shorterprotected species which
are notfound in the cellular RNA(comparelanes 1and4).In contrast, most of the shorter species found in Q2bn-4D virionsareattributabletosimilarspeciesin the cellularRNA
(lanes 2, 5, and 6). These results are consistent with the
presenceofpartially degradedRNAinthedell,2virions.In
thisexperiment,RNAs extracted fromequalvolumes of cell
supernatant were loaded in each lane. The dell,2 QT35
culture produces about 40% of the number of
particles
produced by the Q2bn-4D cultures as detected byWestern
blot analysis(data notshown).
We also examined the PRmutantAPR and D37N virions
A. B.
cell: b C; neoI
neo RNA L A
,irus: dell,2 Q2bn
RENA. L L A A
PK - + - +
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.60.295.77.408.2]184 ARONOFF ET AL.
RSV
SE
21Qlb
X
40K
Q2bn
del
1,2
[image:7.612.151.476.73.411.2]X
50K
FIG. 6. Analysis of pelleted virions by electron microscopy. Preparationwas as described in Materialsand Methods. Magnifications: x40,000(top panels) andx50,000(bottom panels).
in the presence of proteinase K as controls. It has been shown by using assays other than RNase protection that APR, which has a Gag polyprotein deleted for the PR
domain,doesnotpackageRNA(38),while the PR active site
point mutant D37Nvirions do contain wild-type levels of
RNA(44).Theexpectedresultswereobtained in bothcases.
The active site mutant D37N packageswild-type levels of
unspliced genomicRNAbutnotsplicedRNA(Fig. 5B,lanes
8 and10).APRvirions haveaseverepackagingdefect(Fig.
5B,
lanes 7 and9).The fact thatnoRNAwasdetectablebyour techniques with the APR virions further validates the
fact that RNA is encapsidated into dell,2 particles by Gag
lacking Cys-His domains.IfthisRNA wasmerely associated
with theoutside of thevirions, it would have been detectable in all viralpreparations.
Weconclude from these studies that dell,2 virions retain theabilitytoencapsidateRNA.This doesnotcontradict the previous conclusion that dell,2 virions do not contain intact virion RNA (35). However, it demonstrates that other do-mains of the Gag precursor, not the Cys-His boxes of the NC domain, are required for specific
*
recognition and RNA encapsidation.Morphology of dell,2 andpackaging cell line virions.
Viri-onsproducedby packaging cell lines generally contain RNA,
even in the absence of apackageable retroviral vector (4). Our studies withdell,2 ledustopostulate that encapsidated RNA in this mutant is susceptible to degradation because
matureNCbinding is disrupted by theCys-His box deletion. Wewishedto seewhether this differencemightbe reflected in an altered dell,2 virion ultrastructural morphology. In
addition, we wished to determine whether the presence of
+-containing RNAmightcontribute to theformation of the
well-characterizedmorphology of the mature type C virus (for example, see reference 39). Wechose three viruses to
analyzein orderto addressthissecondquestion. Wild-type
RSV encapsidates abundant genomic RNA containing 4
sequences. Q2bn-4D virions contain little or no +-deleted RNA, derived from the packaging vector, and a subset of cellular RNAs suchasglyceraldehyde3-phosphate dehydro-genase (4). SE21Q1bis apackaging mutantwhich encapsi-dates cellular mRNAs, including the 4,-deleted mutant ge-nome, randomly (3, 4, 42). Pelleted virions fromwild-type RSV,Q2bn-4D,SE21Q1b,anddell,2 cellswereanalyzed by electronmicroscopy (Fig. 6).We found that virionsreleased by the Q2bn-4Dpackagingcell line and cellscontaining the
SE21Q1bmutant areindistinguishable fromwild-type RSV
and containanelectron-densematureviralcore.Incontrast,
the dell,2 virions have a diffuse central core, with a
mor-phology resembling neither mature nor immature C-type
particles. The virion proteins are processed in this mutant
(35 and data not shown), so an immature morphologywas notexpected. However, the lack ofawild-type, hexagonal, and condensedmature coremostlikelyreflects theinability J. VIROL.
k'm -;.v,
'.C. ANMN-.
,.I.
ill
ip
I
Vi".ik.,f-v.
on November 9, 2019 by guest
http://jvi.asm.org/
A 3B
x14f+
A
,o
B
i_ C
LXf
A
C
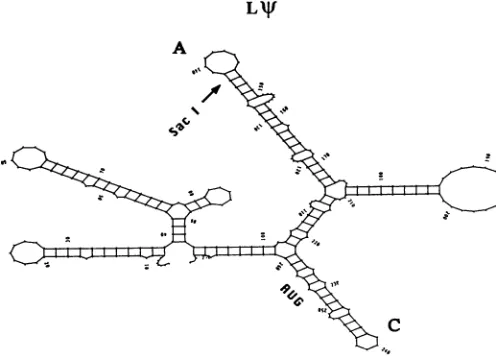
FIG. 7. Predictedsecondarystructuresof*+,L+,and A i.The GeneticsComputerGroup (UniversityofWisconsin) (14)RNAfolding
program,FOLD,basedonthecomputeralgorithmof Zucker(49),and thegraphics outputprogramSQUIGGLESwereused.Folding energies obtained(intenths of kilocalories[1cal= 4.184J])were asfollows: for*+,-204.0; forL+, -134.3; and forA,-90.9.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.176.424.493.673.2]186 ARONOFF ET AL.
of
dell,2
mutantNCprotein
tobindwithhigh affinity
tothepackaged
RNA.DISCUSSION
Inthis
study,
wehave examined thecis-andtrans-acting
determinants ofRNA
encapsidation
into the avian retroviral virion. We show that the functional ALV 5' 4 sequence is 270 nt or less inlength,
thatspliced
RNA can be encapsi-datedefficiently
into retroviralparticles,
and that NCCys-His boxes are not
required
for the initialGag-qij
RNA interaction whichbrings specific
4-linked RNA into theparticle.
Regions
of thecis-acting packaging domain, 4,
from the 5'end ofthe retroviral genomewere tested in a
heterologous
packaging
assay. A270-nt sequence,A+,
was found to be sufficient to directencapsidation
ofheterologous
neo RNA into virionsproduced by
an ALV-derivedpackaging
cell line. Neither Ai nor any of the other avian retroviral sequences testedcan directpackaging
that is asefficient as that ofacomplete
retroviralvectorRNA,
possibly
because of the lack of the 3' DRsequence.However,
we have not been able to show that the addition of DR sequences can either inducepackaging
whenplaced
3' of aheterologous
mRNAoraugmentpackaging
ofheterologous
+-containingRNA
(4a).
These data thus show that the 4 sequence isnecessaryfor
packaging
in aretroviralcontextandsufficientfor
packaging
inaheterologous
context.However,
it shouldbe noted that 4 sequencesdefined assufficient ina heterol-ogous context may be different from those defined
by
deletions within
genomic
RNA,
perhaps
because of differ-ences in RNAfolding
constraints. Forexample, although
MLV sequences in U5 are not
required
forpackaging
ofheterologous
RNA(1),
deletions inU5canaffectencapsida-tion of
genomic
RNA(37).
TheA4, sequencedefined in the
heterologous
assaylacks the retroviralsplice
donorsite,
theputative
dimerlinkage
sequence, and the PBS.
Thus,
none of thesecis-acting
retroviral elements is
required
forspecific
RNAencapsida-tion. In
addition,
the sequences present in the 398-baseL+,
which lacks both the PBS and the
dls,
aresufficienttoallow detection of dimers andhigher-order
complexes
innondena-turing
gel
systems(4a).
Thus,
the dls isunlikely
to be thesole determinant of RNA
multimerization,
an eventwhichprobably
occursonly
afterproteolytic
maturation ofGag
(40), subsequent
to the interactionsdetermining specific
RNA
encapsidation.
What is
required
for sequences to have*4
function in aheterologous
context? It isgenerally
believed that RNAfolds into a
particular
structuralconfiguration
to allowpro-tein
recognition (33).
Wehave shown that 4 sequencescanfunction when
placed
next to bothhph
and neo, and it is reasonable to assume that the functional 4 foldsindepen-dently
of the linkedheterologous
sequences. In order toanalyze potential secondary
structuresthatareconserved in the functional 4 sequences identified in this work, the GeneticsComputer
Group (University
ofWisconsin)
FOLD program(14),
basedonthe Zuckeralgorithm (49),wasused to fold sense,antisense,
and scrambled 4, sequences. Avariety
ofstructuresresulted,
and themoststablestructuresobtained forsense
4,+,
L+,
andAq4
areshown inFig.
7. Each contains three identicalstemloops,
denotedA, B, C,which could constitute a corepackaging
domain. Inverse and scrambled4,
sequences resulted inquite
different predictedsecondary
structureswhich donotcontainA, B,orC(datanot
shown).
Similar computermodeling
coupledwith directbiochemical assays have definedpotential 4, structures for humanimmunodeficiency virus and MLV(2, 19).However, it hasnotbeen determined whether the human immunodefi-ciency virus and MLV sequencesanalyzedactually function biologically in a heterologous context.
We have found thatsplicing doesnotaffect theability of a heterologous
4,
RNA to be encapsidated. When the splice donor site is specifically mutated in A+, packaging is notaffected. Furthermore, when neo4,+ RNA is spliced to the
SV40spliceacceptor,the level ofpackaging of this species is
equivalenttothat of the unspliced RNA. Swain and Coffin have also demonstrated efficient packaging of spliced ALV readthrough RNA (47). The packaged spliced mRNAwas
generated by using the gag 5' splice site and the SV40 3'
splicesite so that 4 was at the5' end of the RNA (47). In
contrast, retroviral spliced subgenomic RNAs are not
effi-ciently encapsidated. Together, these results show that the
splice site is not required for packaging and that some
feature other than an interaction with thesplicing machinery
distinguishes unspliced and spliced retroviral RNAs. The
lack ofpackagingof viralenvmRNA could be causedby a
negative effect ofjuxtaposition of 4 andenv sequences in
spliced RNAs,or evenlong-distance interactions with DRor
U3 regions which prevent proper folding. An analysis of packaging directed by heterologousconstructsin which the
3' splice siteandenv sequences ofan avian retrovirus are
inserted inplace of the SV40 pA sequences of pCMVneo4+ could address this issue. Alternatively, sequences within gag orpol could interact positively with 5' 4 and 3' DR sequencesingenomic RNA so that, when these sequences
aresplicedfrom thetranscript,efficientpackagingnolonger
occurs. However, such positive elements have not been identified in avian viruses.
Suggestions have been made that Gag recognition of 4
involves the NC Cys-His domains (15, 17, 18, 35). Studies with chimeric viruses havereaffirmed the importance of the NC domain in
4,
recognition (16).Our results with the RSVmutantlackingbothNCCys-Hisboxesstrongly suggest that
theprimary RNA-proteininteractiondeterminingpackaging
specificityis genetically separable from the ability to form
matureparticles containing intactgenomic RNA. By using RNase protectionassays,wefound that4,-containing RNA
is encapsidated into particles producedby cells containing
the dell,2 provirus but that it is extremely susceptible to
nucleolytic degradation. This is also reflected in the
mor-phologyof the dell,2 particles as seenby electron
micros-copy. The structure of the diffuse core seen in
dell,2
particles is consistent with the idea that packaged RNAis
readilyaccessibletonucleases and thus difficulttodetect in
virion RNApreparations.Nucleolytic degradation of encap-sidated RNA could
begin
immediatelyafter virionbudding. Nevertheless, in the absence of Cys-His boxes the Gagpolyproteiniscapable of specifically recognizing and
pack-aging
4-containing
RNA.TheCys-His boxes are required forbindingofmatureNCto RNA,aninteractionimportant for
completevirionmaturation, protection ofthe encapsidated
RNA from degradation, and subsequent infectivity.
How-ever, it is not known whether the protein domain
encom-passingtheCys-HisboxesdirectlybindsRNA orwhether it
mediates another interaction, perhaps protein
multimeriza-tion,
which is indirectly responsible for RNA binding andmaturevirion corecondensation. Recent work with bovine leukosis virus suggests that theMAdomain may play a role
in specific RNA encapsidation (23). Combined with a
re-quirementforthePRdomain of ALV Gag, it is likely that the
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
interaction of Gag and
*
is complex and involves more than oneprotein domain.ACKNOWLEDGMENTS
This work was supported in part by grant CA18282 from the
National CancerInstitute to M.L.L. andbyComprehensive Cancer
Center core grant 5P30 CA15704. R.A. was supported by the UniversityofWashington Molecular and Cellular Biology training
grant GM07270, and A.M.H. was supported by University of Washington Comparative Medicine Training Program grant RR07019.
We thank L. Caldwell and J. Groombridge of the FHCRC
Electron Microscope Facility for invaluable assistance and the
Pittsburgh Supercomputing Center for help with some RNA fold-ings. We also thank V. Vogt and P.-F. Spahr for their generous contribution of reagents for these studies. We are grateful to M.
Emerman, J. Overbaugh, and L. Sharmeen for providing critical
assessment of the data.
REFERENCES
1. Adam, M., and A. D. Miller. 1988.Identification of asignalina murine retrovirus that is sufficient for packaging ofnonretroviral RNAinto virions. J. Virol. 62:3802-3806.
2. Alford, R. L., S. Honda, C. B.Lawrence, and J. W. Belmont. 1991. RNAsecondary structure analysisofthe packaging signal for Moloneymurine leukemia virus. Virology 183:611-619. 3. Anderson, D. J., P. Lee, K. L.Levine, J. Sang, S. A. Shah,0. 0.
Yang, P. R. Shank, and M. L.Linial. 1992. Molecular cloning
and characterization of the RNA packaging-defective avian retrovirusSE21Q1b. J.Virol. 66:204-216.
4. Aronoff, R., and M. L. Linial. 1991. Specificity of retroviral RNApackaging. J. Virol. 65:71-80.
4a.Aronoff, R., and M. L.Linial.Unpublished data.
5. Bender, M. A., T. D. Palmer, R. E.Gelinas,and A. D. Miller. 1987. Evidence that the packaging signal of Moloney murine
leukemiavirus extends into the gag region. J. Virol. 61:1639-1646.
6. Berg, J. M. 1988. Proposed structure for the zinc-binding domains from transcription factor IIIA and related proteins. Proc. Natl. Acad. Sci. USA 85:99-102.
7. Bess, J. W., Jr., P. J. Powell, H. J. Issaq, L. J. Schumack, M. K. Grimes, L. E. Henderson, and L.0. Arthur. 1992. Tightly bound zinc in human immunodeficiency virus type 1, human T-cell
leukemiavirus type I, and otherretroviruses. J. Virol. 66:840-847.
8. Bieth, E.,C. Gabus, and J.-L. Darlix. 1990. A study of the dimer
formation ofRous sarcomavirusRNA and of itseffect on viral
protein synthesisin vitro. Nucleic Acids Res. 18:119-127. 9. Brosius, J. 1989. Superpolylinkers in cloning and expression
vectors.DNA 8:759-777.
10. Chen, C., and H. Okayama. 1987. High-efficiency transforma-tion of mammalian cells by plasmid DNA. Mol. Cell. Biol.
7:2745-2752.
11. Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNAisolation by acid
guanidinium-thiocyanate-phenol-chloro-formextraction.Anal.Biochem. 162:156-159.
12. Coffin, J. M. 1984. Structure of the retroviral genome, p. 261-368. In R.Weiss, N. Teich, H. Varmus, and J. Coffin (ed.), RNA tumor viruses. Cold Spring Harbor Laboratory, Cold
Spring Harbor,N.Y.
13. Crawford, S., and S. P.Goff.1985. A deletion mutation in the 5' partof the pol gene of Moloney murine leukemia virus blocks
proteolyticprocessing of the gag and polpolyproteins. J. Virol. 53:899-907.
14. Devereux, J., P.Haeberli, and0.Smithies. 1984. A comprehen-sive setof sequence analysis programs for the VAX. Nucleic AcidsRes.12:387-395.
15. Dupraz, P., S. Oertle, C. Meric, P. Damay, and P.-F. Spahr. 1990. Point mutations in the proximal Cys-His box of Rous sarcoma virusnucleocapsid protein. J. Virol. 64:4978-4987. 16. Dupraz, P., and P.-F. Spahr. 1992.Specificityof Rous sarcoma
virusnucleocapsidprotein in genomic RNA packaging. J. Virol.
66:4662-4670.
17. Fu, X., R. A. Katz, A. M. Skalka, and J. Leis. 1988.
Site-directed mutagenesis of the avian retrovirus nucleocapsid
pro-tein, ppl2. J.Biol. Chem. 263:2140-2145.
18. Gorelick, R. J., L. E. Henderson, J. P. Hanser, and A. Rein. 1988.Pointmutants of Moloney murine leukemia virus that fail
topackage viral RNA: evidence for specific RNA recognition by
a"zinc finger-like" protein sequence. Proc. Natl. Acad. Sci. USA85:8420-8424.
19. Harrison, G. P., and A. M. L.Lever. 1992. The human immu-nodeficiency virus type 1 packaging signal and major splice donor region have a conserved stable secondary structure. J. Virol. 66:4144 4153.
20. Henikoff, S., and M. Eghtedarzadeh. 1987. Conserved arrange-ment of nested genes at the Drosophila Gart locus. Genetics
117:711-725.
21. Jentoft, J. E., L.M. Smith, X. D. Fu, M. Johnson, and J.Leis.
1988. Conserved cysteine and histidine residues of the avian
myeloblastosisvirus nucleocapsid protein are essential for viral
replicationbutare not "zinc-binding fingers." Proc. Natl. Acad. Sci. USA85:7094-7098.
22. Karnovsky,M.J.1965. A formaldehyde-glutaraldehyde fixative
of highosmolalityfor use in electron microscopy. J. Cell Biol. 27:137A.
23. Katoh,I., H. Kyushiki, Y. Sakamoto, Y. Ikawa, and Y.
Yoshi-naka. 1991. Bovine leukemia virus matrix-associated protein MA(plS): further processing and formation of a specific
com-plex withthedimer ofthe 5'-terminal genomic RNA fragment.
J. Virol. 65:6845-6855.
24. Katoh, I.,Y.Yoshinaka, A. Rein, M. Shibuya, T. Odaka, and S.
Oroszlan. 1985. Murine leukemia virus maturation: protease
region required for conversion from"immature" to "mature" core form and for virus infectivity. Virology 145:280-292.
25. Katz, R. A., R. W.Terry,and A. M. Skalka. 1986. A conserved cis-actingsequencein the 5' leader of avian sarcoma virus RNA
isrequired forpackaging. J.Virol. 59:163-167.
26. Koyama, T., F. Harada,and S.Kawai. 1984. Characterization of
a Rous sarcoma virus mutant defective in packaging its own
genomic RNA: biochemical properties of mutant TK15 and
mutant-inducedtransformants. J. Virol. 51:154-162.
27. Linial, M. 1987. Creation of a processedpseudogene by
retro-viral infection. Cell 49:93-102.
28. Linial, M., and M.Groudine. 1985.Transcription of three c-myc
exons isenhanced in chickenbursallymphoma cell lines. Proc. Natl.Acad. Sci. USA82:53-57.
29. Linial, M., E. Medeiros, and W. S. Hayward. 1978. An avian
oncovirus mutant(SE21Q1b) deficient in genomic RNA:
biolog-ical andbiochemical characterization. Cell 15:1371-1381.
30. Luft, J. H. 1961. Improvements in epoxy resin embedding
methods. J. Biophys. Biochem. Cytol. 9:409.
31. Maniatis, T., E. F. Fritsch, and J.Sambrook. 1982. Molecular
cloning: a laboratory manual. Cold Spring Harbor Laboratory,
Cold Spring Harbor,N.Y.
32. Mann, R., R. C. Mulligan, and D. Baltimore. 1983. Construction of aretrovirus packaging mutant and its use toproduce
helper-freedefective retrovirus. Cell 153:159.
33. Mattaj, I. W. 1990. A selective reviewofRNA-protein
interac-tions ineukaryotes. Mol. Biol. Rep. 14:151-155.
34. Meric, C., and S. P.Goff. 1989. Characterization of Moloney
murineleukemia virus mutants withsingle-amino-acid
substitu-tions in theCys-His box of the nucleocapsidprotein. J. Virol.
63:1558-1568.
35. Meric, C., E. Gouilloud, andP.-F. Spahr. 1988. Mutations in Rous sarcoma virusnucleocapsid proteinp12(NC): deletions of Cys-His boxes. J. Virol.62:3328-3333.
36. Moscovici, C., M. G.Moscovici,H. Jiminez, M. M. C. Lai, M. J. Hayman, and P. K. Vogt. 1977. Continuous tissue culture cell lines derived fromchemically induced tumors of Japanese quail. Cell11:95-103.
37. Murphy, J. E., and S. P. Goff. 1989. Construction and analysis of deletion mutations in the U5 region of Moloney murine
leukemia virus: effects on RNA packaging and reverse
tran-scription. J. Virol. 63:319-327.
on November 9, 2019 by guest
http://jvi.asm.org/
188 ARONOFF ET AL.
38. Oertle, S.,andP.-F. Spahr. 1990. Role of thegagpolyprotein precursorin packaging and maturation of Roussarcomavirus genomic RNA.J. Virol. 64:5757-5763.
39. Oroszlan, S., and R. B. Luftig. 1990. Retroviral proteinases. Curr.Top. Microbiol. Immunol. 157:153-186.
40. Prats, A. C., L. Sarih, C. Gabus, S.Litvak, G. Keith,and J. L. Darlix. 1988. Small finger protein of avian and murine retrovi-ruses has nucleic acid annealing activity and positions the replication primer tRNAontogenomic RNA. EMBO J. 7:1777-1783.
41. Shaikh, R., M. Linial, S. Brown, A. Sen, and R. Eisenman. 1979. Recombinant avian oncoviruses. II. Alterations in the gag proteins and evidence for intragenic recombination. Virology 92:463-481.
42. Shank, P. R., and M. Linial. 1980. Avian oncovirus mutant
(SE21Q1b) deficient in genomic RNA: characterization of a deletion inthe provirus. J. Virol. 36:450-456.
43. Sorge, J., W. Ricci, and S. H. Hughes. 1983. cis-Acting RNA packaging locus in the 115-nucleotide direct repeat of Rous
sarcomavirus. J. Virol.48:667-675.
44. Stewart, L., G. Schatz, and V. M. Vogt. 1990. Properties of avian retrovirusparticles defective in viralprotease.J. Virol. 64:5076-5092.
45. Stewart, L., and V. M. Vogt. 1991. trans-Acting viralproteaseis necessaryand sufficient for activation of avian leukosis virus reversetranscriptase. J. Virol.65:6218-6231.
46. Stoker, A. W., and M. J. Bissell. 1988. Developmentof avian sarcomaand leukosisvirus-based vector-packaging cell lines. J. Virol.62:1008-1015.
47. Swain, A., and J. M. Coffin. 1992. Mechanism of transduction by retroviruses. Science 255:841-845.
47a.Vogt, V. Personal communication.
48. Watanabe, S., and H. M. Temin. 1982. Encapsidationsequences forspleen necrosis virus,anavian retrovirus,arebetween the 5' long terminalrepeatand thestartof thegag gene. Proc. Natl. Acad.Sci. USA 79:5986-5990.
49. Zucker, M. 1989. On finding all suboptimal foldings ofanRNA molecule. Science 244:48-52.
J. VIROL.
![FIG. splice inA* to a HindIII restriction site as indicated by GCTthe[(A)n] function. mutated Sequences 683-base (S.D.), Methods](https://thumb-us.123doks.com/thumbv2/123dok_us/1303731.83545/2.612.83.519.107.259/hindiii-restriction-indicated-gctthe-function-mutated-sequences-methods.webp)