0022-538X/91/031414-06$02.00/0
Copyright©D 1991,AmericanSociety for Microbiology
Variable Role
of the
Long
Terminal
Repeat
Spl-Binding
Sites
in
Human
Immunodeficiency
Virus
Replication
in T
Lymphocytes
CARMENPARROTT,1,2TODDSEIDNER,12 ELIADUH,1JOHN
LEONARD,lt
THEODORE S. THEODORE,' ALICIABUCKLER-WHITE,3 MALCOLM A. MARTIN,' AND ARNOLD B. RABSONlt*Laboratory of MolecularMicrobiology, NationalInstitute ofAllergyandInfectiousDiseases, Bethesda, Maryland
208921;
HowardHughes MedicalInstitute, Bethesda, Maryland208142;andDivisionofMolecular Virology andImmunology, Georgetown University, Rockville, Maryland 208523
Received 17 September 1990/Accepted 21 November 1990
Thelongterminal repeat(LTR)of thehumanimmunodeficiencyvirus(HIV)containsthreebindingsitesfor thetranscriptional factor Spl. Inorder toinvestigatethe role that theSpl-binding sitesplayinregulation of HIV replication, we have introduceda deletion of all threeSpl-binding sites into the LTRofaninfectious
molecularclone of HIV. Viralstocks have beenpreparedfrom this mutantvirus, designateddl-Sp,andthese stocks havebeen usedtostudy its replicativeabilityin human T cells. Thedl-Spvirusreplicatedefficientlyin MT4cellsand inphytohemagglutinin-stimulated humanperipheralbloodlymphocytes,but itreplicated poorly and with delayedkinetics inA3.01(CEM)Tcells unless those cells had beentreatedwith thecytokinetumor necrosis factor a. Gel retardation assays to study the levels ofDNA-binding proteins present inthese cells showedthat NF-KBactivitycould be detected in the nuclei of MT4 cells but not inA3.01cells unlesstheyhad beentreated with tumor necrosis factora.Thus, thepresenceof NF-KB activity appearedtoberequiredfor efficientreplication ofan HIVwhose LTRSpl-bindingsites had been deleted. This suggests that NF-K.Bcan
functionally compensate for Spl in activating HIV replication. The HIV LTR is therefore similar to the promoter-enhancer units of other viruses in that it is composed ofmultiple functional elements that may
contributedifferentlytoviralreplicationdependingonthe levels ofDNA-binding proteinspresent in thetarget cells.
Thelong terminal repeat(LTR) ofthehuman immunode-ficiency virus (HIV) contains a variety of cis-acting
regula-torysequences responsibleformodulatingtheexpressionof
HIVRNA.These includetheresponseregion, TAR(39), for
the viral transactivating protein, Tat, as well as promoter andenhancerelements that interact with host transcriptional factors (for reviews, seereferences 4, 21, and 47). The TAR
element lies3' tothe startsite of RNA transcription, and it has been hypothesized that this element acts as an RNA
enhancer element (41). In a structural arrangement
analo-goustothatobserved forothercellular and viralgenes(10),
the enhancer-promoter region of the HIV LTR contains a
modular arrangement ofa series of elements that interact
with corresponding cellular DNA-binding proteins to
pro-vide efficient regulation ofRNA synthesis. Well-character-izedelements includeaTATAboxat-24bpwithrespect to
the site ofinitiation ofRNAsynthesis(45), aswell asthree
binding sites for the transcriptional factor Spl (22) located between -45 and -78 bp. An enhancer element (7, 23, 32, 36, 46) is present at -80 to -105 bp and consists of a
tandemly repeated unit to which several cellular proteins (14, 24, 26, 32, 51) including NF-KB have been reported to
bind. Thefunction of these enhancerandpromoterunits in theregulation ofHIV LTR expression was initially studied
inthecontext ofreportergene constructions which
demon-strated the potent effect of the Tat-TAR interaction in augmentinggeneexpression directedby the HIVLTR (1, 5,
* Corresponding author.
t Present address: G. H. Besselaar Associates, Princeton, NJ
08540.
tPresentaddress:CenterforAdvancedBiotechnology and
Med-icine,Piscataway, NJ 08854.
31,37, 43, 50)anddelineated the role of the
NF-KB-binding
sites in mediating activation of the HIV LTR by mitogenic
stimuli and cytokines (9, 23, 32, 35, 42, 46, 52). Similar studiesonthe role oftheSpl-bindingsitessuggested that the
presenceofatleastoneSpl-binding sitewasimportant for in
vitro transcription of the HIV LTR (22) orfor significant
activation of LTR function by the tatgene in transfection
assays inHeLa cells (18).
Recently, we have examined the functional roles of the
HIVpromoterandenhancer elementsby devising asystem torapidly study the effects of mutations in these elementson
the replication of an infectious HIV provirus (27). These studies have demonstrated that theNF-KB-binding sitesare notrequiredfor the replication of HIV in human T lympho-cytes as long asthe three Spl-binding sites areretained in
the LTR. A functional TAR region was required for HIV
replication,aresultconsistentwiththe previous demonstra-tionthat Tatwas anessentialgeneproduct for HIV replica-tion. We havenow extendedthese analysesto examine the requirement of the LTR Spl sites for HIV replication in different T-cell lines. These studies indicate that the Spl sites are notrequired for efficient HIV infection of human peripheral bloodlymphocytes (PBLs) or ofMT4cells with
proviralconstructions containingtwoNF-KBelements. De-letion of the Spl motifs, however, markedly reduces HIV replicationin A3.01cells, acell line derived from the CEM
T-cell line that is usually permissive for viral replication. Treatment of A3.01 cells with tumor necrosis factor a
(TNF-cx), an inducer ofNF-KB-binding activity(9, 28, 35), results in efficient infection of A3.01 cells by HIV with deleted Spl sites, suggesting that the NF-KB-binding sites
canfunctionallysubstitute fortheSpl sites indirecting HIV
geneexpression.
1414
on November 10, 2019 by guest
http://jvi.asm.org/
+1 mRNA
U3 NFkB RF
TAR
-108 NF-kB NF-kB Spi Spi SpI 38
pILIC TACAAGGGACTTTCCGCTGGGCXTYTCAAGGCGTGV TGGC GG CT=AGTGGCGAGCCCTC
pdl-Sp TACAAGGGACTTTCCGCTGGGGACTTTCCAG---CTC
FIG. 1. Nucleotide sequence of theSpl sitedeletion mutation introduced into the HIV-1 LTR.Thestructure of theHIV LTR isshown schematically at the top,and the nucleotide sequences of the NF-KB andSplsites (positions -108 to -38 withrespect to the startofRNA transcription) of the wild-type (pILIC) and dl-Sp (pdl-Sp) viruses are shown. U3, 3' unique region; R, repeated region; U5, 5' uniqueregion; NRE, negativeregulatory element.
MATERIALS AND METHODS
Plasmids and viral stocks. Deletion mutations of the Spl-binding sites in the HIV LTR were constructed by using
procedures described previously (27). Briefly, oligonucle-otide-directed site-specific mutagenesis (54) as modified by
Kunkel (25) was employed to generate the mutations by using the Muta-gen kit from Bio-Rad (Richmond, Calif.). These mutationswereintroduced intoa2.0-kb BamHI-SphI
segment of HIV containing a single reconstructed LTR
flanked by 5' nef and 3' gag sequences cloned in M13.
Followingconfirmation of thepresenceof the desired
muta-tionbyDNAsequencing(40),theHIVsegmentwas
reintro-duced intoacircularly permuted infectious molecular clone
of HIV, pILIC (27), to generate pdl-Sp. Wild-type and mutagenized HIV proviral DNAs wereexcised from vector
sequences by BamHI digestion, ligated to themselves to
form concatemers, and transfected into the A3.01 CD4+ T-cell line(11) by usingamodified DEAE-dextran procedure
(8). Transfected cells were cocultivated with MT4 cells to
allow rapid and efficient recovery of progeny virus (27). Virusgenerated from thetransfection-cocultivation protocol
was subjectedtoone ortwofurtherpassagesinMT4cellsto
generate cell-free viral stocks used for subsequent infec-tions. A partial molecular clone of the dl-Sp virus was
obtained by cloning EcoRI fragments of infected MT4 cell DNA in K Wes B arms (Life Sciences-Bethesda Research
Laboratories, Gaithersburg, Md.), and this clone was
sub-jectedtoDNAsequencing.
Cells and viral infections. A3.01 and MT4 cells were
maintained in RPMI 1640 supplemented with 10% fetal bovine serum and with penicillin, streptomycin, and glu-tamine. Human PBLs were activatedfor3 daysby incuba-tion with phytohemagglutinin at a concentration of 0.25
pLg/ml(Burroughs Wellcome, Research Triangle Park, N.C.) and then were maintained in RPMI 1640 with 10% fetal
bovine serum, penicillin, streptomycin, andglutamine with 10% interleukin-2 (Electro-nucleonics, Silver Spring, Md.). Infections were performed in 24-well cluster plates. Cells
wereplatedat250,000perwell andwereinfected with5,000
cpm ofreverse transcriptase (RT) activity ofwild-type or
Spl-deleted viruses for MT4 cells or with 500,000 cpmfor A3.01 cells and PBLs. The kinetics of viral infection were
monitored with a 32P-based assay for RT activity that has been previously described (49). For experiments involving TNF-a treatment, TNF-ct (Genzyme, Boston, Mass.) was
added tothe culturesat 100 U/ml once aweekfor48 h. Nuclear extracts and gel retardation assay. Nuclear
ex-tractswereprepared byamodification(38)of theprocedure of Dignam et al. (6). Gel retardation assays employing
32P-labeled, double-stranded synthetic oligonucleotides con-taining either both of the HIV NF-KB sites or all three
Spl-binding sites were performed as described previously (16). The ST competitor oligonucleotide was a 24-mer con-taining sequences mapping between the Spl-binding sites and theTATAbox in theHIVLTRand has beenpreviously described (9).
RESULTS
Recovery of HIV containing a deletion of the LTR Spl-binding sites. Adeletion of the three Spl-binding sites was introduced into the LTRofacircularlypermuted infectious molecularcloneofHIV,pILIC, bysite-specificmutagenesis
of the 2.0-kb BamHI-SphI segmentcontaininga single LTR aspreviouslydescribed (27). The mutagenized LTR segment was transferred back into pILIC (in pUC8) cleaved with BamHI andSphI, and thereconstructed proviruscontaining
the deletion of the three Spl sites was designated pdl-Sp. Thenucleotide sequences of the enhancer-promoterregions ofthis clone and of the wild-type virusare shown in Fig. 1. The pILIC and pdl-SpDNAs were cleaved withBamHI to excise the HIV proviral DNA sequences, were ligated to themselvestoform concatemers, andwerethentransfected
into A3.01 lymphocytes (27). Transfected cells were cocul-tivated withMT4 cells2days following transfection. Cocul-tivation with MT4 cells, which are exquisitely sensitive to HIV infection, was usedto amplify any
replication-compe-tentvirus generatedfrom the initial transfection. RT activ-ity, indicative of HIV replication, was detected in the
dl-Sp-transfected A3.01-MT4 cocultures by 10 days follow-ing the initial transfection. In contrast, RT activity was detected4 to 5daysafter transfection in culturestransfected
with thewild-type pILIC DNA.
Following detection ofRT activity from the A3.01-MT4 cocultures, viral stocks of the ILIC and dl-Spviruses were prepared by passage of the recovered virusthroughthe MT4 cell line.UnintegratedDNA(20)was also
prepared
from the MT4 cells infected by dl-Sp for molecularcloning
andsequencing ofthedl-Sp
provirus
toconfirmthat thedeletionof the three Spl sites had been maintained
following
thetransfection-cocultivation procedure. This DNA was cleaved with EcoRI andligatedintoX Wes Barms, and then the 5' LTR from a phage plaque
hybridizing
to an internal 6.5-kbHindlIl HIVprobe, pBenn6 (11),wassequenced.
The clonecontainedanSpl
site deletion identicaltothatpresentinpdl-Spandexhibited nootherchanges over aspanof396
sequenced bases from -359 to +37,which containedall of theknownupstream
positive regulatory
elements in theHIV promoter(datanot shown).on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.104.519.74.173.2]A
0
v-E
a.
I-.
B
5000
4000
-3
CL
I-20
r1
3000
-2000
-1000
- 0-Days
- ILIC
* di-Sp
- mock
4 6 8 1 0
Days
C
12000
10000
E a.
'U
:c
8000
6000
4000
2000
0 10 20
days
Replicative ability of Spl-deletedHIV. The RTactivitywas
determinedfor the viral stocks of dl-Spand ILIC prepared from MT4 cells. Equal amounts ofRT activity were then
used insubsequent infections of human T-cell linesto study the relativerates of replication of the Spl deletion virus as
comparedwiththose of the wild-typeparent.The results of thesekinetic analysesareshown in Fig. 2. Figure 2A shows
the replication of the dl-Sp virus in the MT4 cell line as
measured by RT activity in the culture supernatant. The dl-Sp virus replicates efficiently in these cells with kinetics thataresimilartothose of the wild-type virus. As isseenin
Fig. 2B, dl-Sp also replicates well in phytohemagglutinin-stimulated humanPBLs, again with replication kineticsand levels ofvirus production that are similarto those of the wild-type control virus. In contrast, Fig. 2C shows that deletion of the Spl sites markedly delays productive HIV infection of the A3.01 cell line, a derivative of CEM cells.
While thepeakofwild-type virus production occurredat12 days following infection in these cells, only low levels of replication were observed 24days following infection with
the dl-Sp virus. Treatment ofA3.01 cells with TNF-ot, an
inducerofHIVLTR function(9, 12, 33, 35) that accelerates HIV replication in some T-cell lines (30), led to a marked
FIG. 2. Replication of HIV containing a deletion of the LTR Spl-binding sites in human T lymphocytes. Replication of dl-Sp virus was assessed by measuring RT activity in MT4 cells (A), phytohemagglutinin-stimulated PBLs (B), and A3.01 cells (C). In-fectionswereperformedin 24-wellplates; 250,000cellswereplated perwelland infectedwith5,000cpmof RTactivityof virus for MT4 cellsor500,000cpmforA3.01cellsandPBLs.Thekinetics of viral replication were monitored by using a 32P-based RT assay as previously described (49). For panel C, TNF-a (Genzyme) was
added toduplicate cultures at100 U/mlondays 0, 7, and 13 and maintained intheculture for48huntilthenextchangeofmedium. Intermittent TNFtreatment was employed toreduce drug-associ-atedcellulartoxicity.
increase in the replication of the dl-Sp virus. Peak virus
replicationwasdetectedat16to18days followinginfection. Evenin the presence ofTNF-ot, replicationof thedl-Spvirus was delayed compared with that of the wild-type virus, suggesting that while induction of NF-KB permitted the
replication of virus deleted in the Spl-binding sites, itwas not sufficient to completely compensate for the defects intrinsic tothis deletion.
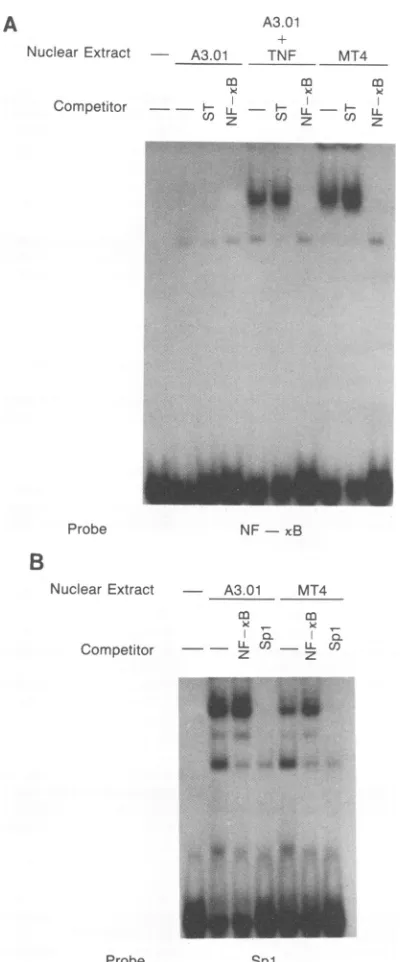
Presence of Spl- and NF-KB-binding activities in human T-cell lines. In order to examine the potential mechanisms underlying the reduced capacity ofthe Spl-deleted viruses toinfect A3.01 cells butnototherT-celllines suchasMT4,
we investigated the presence oftranscriptional factors im-portantinactivatingthe HIV LTRin these cellsby perform-ing gel retardation assays. MT4 cells contained abundant amounts ofspecific NF-KB-binding activity, whereas none was detected in A3.01 cells unless they were treated with TNF-ax,whichhad beenpreviously shown to induce NF-KB in human T cells(9, 28, 35)(Fig. 3A). BothA3.01 and MT4 cells constitutively synthesize readily detectable Spl-bind-ing activity (Fig. 3B); A3.01 cells appeartocontain slightly more of thisactivity than do MT4cells.
DISCUSSION
The results presented here show that the HIV LTR
Spl-binding sites are not requiredfor infection of
phytohe-magglutinin-stimulated normal humanlymphocytesorMT4 cells. This was surprising in view of in vitro transcription studies(22)and transfectionanalyses (18)thatsuggestedthat
0 IF a
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.70.297.71.483.2] [image:3.612.324.555.74.277.2]A
A3.01Nuclear Extract A3.01 TNF MT4
m m m
Competitor - - L - u. -o
ln z U) z Cl,
Probe NF-xB
B
Nuclear Extract - A3.01 MT4
m m
CL Q0
Competitor - co
z 1
."4
Probe Spi
FIG. 3. Presence ofnuclearfactors bindingto HIV LTR DNA
segmentsinhuman T-cell lines. Nuclearextractswerepreparedby
the method of Dignametal.(6, 38)fromMT4cells,A3.01cells,and
A3.01 cells 12 haftertreatment with 100 U of TNF-a perml. Gel
retardationassayswereperformedaspreviously described (16)with
32P-labeled, double-stranded synthetic oligonucleotides
correspond-ing to the HIV LTR NF-KB sites (A) or Spl-binding sites (B).
Competition experiments were performed by addition of 10 ng
(greater than 10-fold molar excess) of unlabeled oligonucleotides.
The ST oligonucleotide-containing sequence mapping between the
Spl sites and the TATA box of the HIV LTR (9) wasused as the
nonspecificcompetitor.
at least one Spl-binding site was required for significant
levels of HIV LTR-directed gene expression. These latter
studies were performed in HeLa cells, which do not have significant levels ofNF-KB activity in the uninduced state.
Similarly, inourstudies,HIVfromwhich Spl-binding sites
hadbeen deletedreplicated only poorly in A3.01 cells, which
donotcontain detectable NF-KB-binding activity; induction of NF-KB binding in these cells resulted in their becoming
permissiveforreplication of dl-Sp. MT4 cells and phytohe-magglutinin-stimulated human PBLs, both of which
sup-portedthe replication ofthe dl-Sp virus,contain high levels
of NF-KB-binding proteins (Fig. 3). These results suggest that NF-KB, which hasbeen previously shown to be
induc-ible byTNF-a (9, 28, 35), mayfunctionally substitute for Spl in activating transcription of the HIV LTR in certain cell types. An additional role of other inducible nuclearproteins that bind the HIV enhancer, suchas HIVen86A (14), cannot
be excluded at this time.
The mechanisms by which NF-KB may functionally sub-stitute for Splin activating HIV expression remain undeter-mined. The results of DNase I footprinting performed on LTRscontaining point mutations of the Spl-binding sitesare
of interest in this regard (18). Such mutations resulted in decreased bindingtothe NF-KB sites, suggesting thatoneof
the effects ofSpl maybeto promote the binding ofcellular
factors tothe HIV enhancer. In cells containing high levels of enhancer-binding proteins, this function of Spl might be less important. Alternatively, NF-KB might be able to di-rectly substitute for Spl in mediating activation of the transcriptional apparatus by mechanisms similar to those used by Spl. In the dl-Sp deletion mutant, the NF-KB sites have been moved 38 bp closer to the promoter element, potentially increasing their ability to act as a promoter-proximal transcriptional activation domain in addition to
actingas anenhancer. Recentstudies have tendedtoblurthe distinction between promoter-proximal factors and enhanc-ing factors workenhanc-ing at adistance; the glutamine-rich region ofSpl can activate transcription even at adistance (3).
It should be noted that the induction of NF-KB-binding activity in A3.01 cells did not fully restore wild-type repli-cation kinetics to the dl-Sp virus in those cells. This is in contrast to results for PBLs and MT4 cells, in which dl-Sp showed only slight delays in replication kinetics compared with wild-type virus. Therefore, it is possible that other factors in additionto NF-KBthat interacteitherdirectly with the LTR or with the NF-KB bound to the LTR may be present in PBLs and MT4 cells. Both stimulated PBLs and MT4cells are notable for their high degree ofsensitivity to HIV replication; wild-type virus replicates much more rap-idly inthese cells thaninA3.01 cells (Fig. 2), aphenomenon thatmaybe the resultofmanydifferences in the various cell types, including levels of NF-KB-binding activity.
Our previous studies had suggested that the ability of viruses containing LTR mutations to replicate in human T cells correlatedwith the abilityofthose LTRstobe activated by the HIV tat gene. Of interest in this regard is our recent observation that a mutant LTR with deletions of the Spl-binding sites can still be transactivated by Tat (2). This suggests that NF-KB can mediate promoter-proximal func-tions required forTat transactivation. An LTR from which the NF-KB sites had also been deleted wascapable ofbeing transactivated by Tat; however, replacement of both the NF-KB and Spl sites by analternative enhancer unitgreatly reduced Tattransactivation. Thus, both theNF-KB and Spl sites appear to be able to provide the promoter-proximal signals required to mediate efficient transactivation of viral geneexpression.Transactivation maythen, inturn,result in expression of sufficient levels of viral RNAtoallow produc-tive infectionto proceed.
Interestingly, the Spl deletionvirus exhibits aphenotype
.:i.'.
P.:.:
.im
.: gi.:I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.84.284.64.545.2]very similartothat ofSpC (27), a virus thatcontains
point
mutations in the two 5'
Spl-binding
sites. These results suggestthat invivo, the 3'-mostSpl
site(site
I)maynotbecritically important
fortranscriptional
activation. This sitehas
previously
been shown to have a lower affinity forpurified Spl
than the most 5' site(22) and,
in our hands, binds to factors present in crude nuclear extracts fromlymphocyte
celllines withaloweraffinity
than either site II or III, asanalyzed
incompetition gel
retardation assays(40a).
The observation that the
promoter-enhancer
in the HIV LTR contains multipleprotein-binding
sites that may have variableimportancein different celllines and may becapable
of functional
complementation
is similartoresults obtainedfor otherviral and cellular enhancer and promoter units. The enhancers of murine retroviruses contain
multiple
protein-binding
sites that contribute to thespecificity
oflymphoid
versuserythroid leukemogenesis in acomplex and cooper-ative manner (17, 29, 44). In
particular,
the concept thatdifferent
binding
sites may be able tofunctionally
comple-mentoneanother hasbeen wellstudiedin thecontextof the
simian virus 40 enhancer. Mutations in different elements present within the simian virus 40 enhancer can affect
enhancerfunctionand theability of simian virus 40togrow in different celllines(15, 19, 34, 53).Furthermore,theability ofmutantvirusesto
replicate
in thesevariouslines has been shown to bedependent
on the nature oftheDNA-binding
proteins
present in these lines. Revertantsarising
after themutation of enhancer elements contain
duplications
of theremaining
elements, implyingthat the different parts of the enhancer canfunctionally
substitute for each other (19).Thesekinds of studieshave allowedthecharacterization of enhansons (10, 34), discrete units within an enhancer that when present in multiple copies result in transcriptional
activation. In the HIV LTR,both the NF-KB andSpl sites
function asdiscrete but complementary units that activate
transcription
and may thus serve asenhansons.Delayed replication ofthedl-Spvirus in non-TNF-treated A3.01 cells, such as that shown in Fig. 2C, has been
observed in two of five separate infections of A3.01 cells
(40a);
in three cases, we did not observe evidence of viralreplication.
The delayed replicationobserved in these cells may reflect the induction ofNF-KB-binding activity in afractionof the untreated cellsoverseveral weeks of culture. We have
previously
observed induction of NF-KB-bindingactivity
in theUlchronically
infectedmonocytecell line(13)with increased cell density (8a), emphasizing the dynamic natureof theinteractions betweenHIV and thehost cell that modulate viral replication. Another possibility is that the
delayed
replicationseeninsomeA3.01infectionsmaybe the resultofsecond-sitemutationevents;delayedreplication of HIVscontaining
env gene mutations has been previouslyfound to be associated with the appearance of second-site reversion mutations(48). Wearecurrently investigating the
possibility
thatreversioneventsmayhavetaken place in thedl-Sp
LTR, thus allowing the delayed replication in non-TNF-ot-treated A3.01 cells. Identification and characteriza-tion ofrevertants of HIV LTR mutations might allow the further analysis of enhansons present in the HIV LTR in a manneranalogoustothatemployed for simian virus 40 (19).ACKNOWLEDGMENTS
WethankElizabethRossandGeorgeEnglundfor helpful discus-sions and for sharing unpublished data and Kenneth Katz for assistance insubcloning.
REFERENCES
1. Arya, S. K., C. Guo, S. F. Josephs, and F.Wong-Staal. 1986. Trans-activator gene of human T-lymphotropic virus type III (HTLV-lII). Science 229:60-73.
2. Berkhout, B., A.Gatignol,A. B.Rabson, and K.-T. Jeang. 1990. TAR-independentactivation of the HIV-1 LTR: evidence that TATrequires specific regions of the promoter. Cell 62:757-767. 3. Coury, A.J.,D. A.Holtzman,S. P.Jackson,and R.Tjian.1989. Synergisticactivationbytheglutamine rich domains of human transcriptionfactorSpl. Cell 59:827-836.
4. Cullen, B.,and W. Greene. 1989.Regulatorypathways govern-ing HIV-1 replication. Cell 58:423-426.
5. Cullen,B. R. 1986. Trans-activation of the human immunodefi-ciencyvirusoccursviaabimodal mechanism. Cell 46:973-982. 6. Dignam, J.D.,P. L. Martin,B. S. Shastry,and R. F. Roeder. 1983. Eukaryoticgene transcriptionwithpurifiedcomponents. MethodsEnzymol. 101:583-599.
7. Dinter, H., R. Chiu, M.Imagana, M. Karin,and K. A.Jones. 1987. In vitro activation of the HIV-1 enhancer inextracts of cells treated with aphorbol ester tumor promoter. EMBO J. 6:4067-4071.
8. Dorsett, D.L., I. Keshet, and E. Winocour. 1983. Quantitation of a simian virus 40 nonhomologous recombination pathway. J. Virol. 48:218-228.
8a.Duh,E. Unpublisheddata.
9. Duh, E. J., W. J.Maury, T. M.Folks, A. S. Fauci,and A. B. Rabson.1989. Tumor necrosis factorax activates human immu-nodeficiency virus-1 through inductionof nuclear factorbinding tothe NF-KB sites in thelong terminalrepeat. Proc.Natl. Acad. Sci. USA 86:5974-5978.
10. Dynan, W. S. 1989. Modularity in promoters and enhancers. Cell 58:1-4.
11. Folks, T., S.Benn, A. Rabson, T. Theodore, M.D.Hoggan, M. Martin, M.Lightfoote, and K.Sell. 1985. Characterization ofa continuous T-cell linesusceptibletothecytopathiceffects of the acquiredimmunodeficiency syndrome (AIDS)-associated
retro-virus.Proc. Natl.Acad. Sci. USA 82:4539-4543.
12. Folks, T. M., K.Clouse,J. Justement, A. Rabson,E.Duh,J.H. Kehrl, and A. S. Fauci. 1989. Tumor necrosis factor-alpha induces the expression ofthe human immunodeficiency virus fromachronically infectedTcellclone. Proc. Natl. Acad.Sci. USA 86:2365-2368.
13. Folks, T. M., J. Justement, A. Kinter, C. A. Dinarello, and A. S. Fauci. 1987.Cytokine-induced expression ofHIV-1ina chron-ically infectedpromonocyte cellline. Science238:800-802. 14. Franza, B. R., S. F.Josephs, M. Z.Gilman, W.Ryan, and B.
Clarkson. 1987. Characterizationof cellular proteins recogniz-ingthe HIVenhancerusingamicroscaleDNAaffinity precipi-tation assay. Nature(London)330:391-395.
15. Fromental, C., M. Kanno, H. Nomiyama, and P. Chambon. 1988.Cooperativity andhierarchical levels offunctional orga-nizationinthe SV40enhancer. Cell 54:943-953.
16. Gimble,J. M., E. Duh, J. M. Ostrove, H.E.Gendelman, E. E. Max, and A. B. Rabson. 1988. Activation ofthe human immu-nodeficiency viruslong terminalrepeatby herpes simplex virus type 1isassociatedwithinductionof anuclear factorthatbinds
totheNF-KB/core enhancersequence. J.Virol. 62:4104-4112. 17. Golemis, E., Y.Li, T. N.Fredrickson, J. W. Hartley, and N. Hopkins. 1989. Distinct segments within the enhancer region collaborateto specifythe type ofleukemiainduced by nonde-fectiveFriend andMoloney viruses.J. Virol.63:328-337. 18. Harrich, D., J. Garcia, F. Wu, R. Mitsuyasu, J. Gonzalez, and R.
Gaynor.1989. RoleofSPl-bindingdomains in invivo transcrip-tionregulationof the humanimmunodeficiencyvirus type 1 long terminalrepeat. J. Virol. 63:2585-2591.
19. Herr,W., and J. Clarke. 1986. TheSV40 enhancer is composed ofmultiple functional elements that can compensate for each other.Cell45:461-470.
20. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmousecell cultures. J. Mol. Biol. 26:365-369. 21. Jones, K. 1989. HIVtrans-activation andtranscription control
mechanisms. NewBiol. 1:127-135.
22. Jones,K.A., J.T.Kadonaga, P. A. Luciw, and R. Tjian. 1986.
on November 10, 2019 by guest
http://jvi.asm.org/
Activation of the AIDS retrovirus promoter by the cellular transcriptionfactor, Spl. Science232:755-759.
23. Kaufman, J. D., G. Valandra, G. Roderiquez, G. Bushar, C. Giri, and M. A. Norcross. 1987. Phorbol ester enhances human immunodeficiency virus-promoted gene expression and acts on arepeated 10-base-pair functional enhancer element. Mol. Cell. Biol. 7:3759-3766.
24. Kawakami, K., C. Scheidereit, and R. Roeder. 1988. Identifica-tion and purificaIdentifica-tion of a human immunoglobulin-enhancer-binding protein (NF-KB) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc. Natl. Acad. Sci. USA 85:4700-4704.
25. Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis without phenotype selection. Proc. Nati. Acad. Sci. USA 82:488-492.
26. Lenardo, M. J., A. Kuang, A. Gifford, and D. Baltimore. 1988. NF-KB proteinpurification from bovine spleen: nucleotide stim-ulationand binding site specificity. Proc. Natl. Acad. Sci. USA 85:8825-8829.
27. Leonard, J., C. Parrott, A. J. Buckler-White, W. Turner, E. K. Ross, M. A. Martin, and A. B. Rabson. 1989. The NF-KB binding sites in thehumanimmunodeficiency virus type 1 long terminal repeat are notrequired for virus infectivity. J. Virol. 63:4919-4924.
28. Lowenthal, J. W., D. W. Ballard, E. Bohnlein, and W. C. Greene.1989.TNF-ainducesproteinbindingspecificallytothe KBlikeenhancer elements and regulates interleukin 2 receptor a chaingeneexpressioninprimaryhuman Tlymphocytes. Proc. Natl. Acad. Sci. USA86:2331-2335.
29. Manley, N. R., M. A. O'Connell, P. A. Sharp, and N. Hopkins. 1989. Nuclear factors that bind to the enhancer region of nondefective Friend murine leukemia virus. J. Virol. 63:4210-4223.
30. Matsuyama, T., Y. Hamamoto, G.-I. Soma, D. Mizuno, N. Yamamoto, and N. Kobayashi. 1989. Cytocidal effect oftumor necrosisfactoroncellschronically infected withhuman immu-nodeficiency virus (HIV): enhancement ofHIVreplication. J. Virol. 63:2504-2509.
31. Muesing, M. A., D. H. Smith, and D. J. Capon. 1987.Regulation ofmRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell48:691-701.
32. Nabel, G.,and D. Baltimore.1987. Aninducible factoractivates expression of the human immunodeficiency virus in T cells. Nature (London)326:711-713.
33. Okamoto,T., T. Matsuyama, S. Mori, Y. Hamamoto, N. Koba-yashi, N. Yamamoto, S. F. Josephs, F. Wong-Staal, and K. Shimotohno. 1989. Augmentation ofhuman immunodeficiency virustype 1 geneexpression bytumornecrosisfactor a. AIDS Res. Hum.Retroviruses5:131-138.
34. Ondek, B.,L. Gloss, and W. Herr. 1988. The SV40enhancer contains two distinctlevels oforganization. Nature (London) 333:40-45.
35. Osborn, L.,S. Kunkel,andG.J. Nabel. 1989. Tumornecrosis factoralpha and interleukin-1 stimulate thehuman immunode-ficiency virus enhancer by activation ofthenuclearfactorKB. Proc. Natl. Acad. Sci. USA86:2733-2735.
36. Patarca, R., C. Heath, C. Goldenberg, C. Rosen, J. Sodroski, W. A.Haseltine,andU. Hansen. 1987.Transcriptiondirectedby thehumanTcelllymphotropicvirus type III(HTLV-III/LAV) long terminalrepeat.AIDS Res. Hum. Retroviruses3:41-48. 37. Peterlin, B. M., P. A. Luciw, P. J. Barr, and M. D. Walker.
1986. Elevated levels of mRNA can account for the
trans-activationof human immunodeficiency virus. Proc. Natl. Acad. Sci. USA83:9734-9738.
38. Quinn, J. P., N. Holbrook, and D. Levens. 1987. Binding of a cellular protein to the Gibbon ape leukemia virus enhancer. Mol. Cell. Biol. 7:2735-2744.
39. Rosen, C. A., J. G. Sodroski, and W. A. Haseltine. 1985. Location of cis-acting regulatory sequences in the human T lymphotropic virus type III (HTLV-III/LAV) long terminal repeat.Cell41:813-823.
40. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminatsequenc-ing inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
40a.Seidner, T., and A. B. Rabson. Unpublished data.
41. Sharp, P. A., and R. A. Marciniak. 1989. HIV TAR: an RNA enhancer? Cell 59:229-230.
42. Siekevitz, M., S. F. Josephs, M. Dukovich, N. Peffer, F. Wong-Staal,and W.C. Greene. 1987.Activation of the HIV LTRbyT cellmitogens and the trans-activator protein of HTLV-1. Sci-ence 238:1575-1578.
43. Sodroski, J. G., R. Patarca, C. Rosen, F. Wong-Staal, andW.A. Haseltine. 1985. Location of the trans-activating region of hu-manT-cell lymphotropic virustypeIII. Science 229:74-77. 44. Speck, N. A., B. Renjifo, E. Golemis, T. Fredrickson, J. W.
Hartley, and N. Hopkins. 1990.Mutation of the core or adjacent LVb elements of the Moloney murine leukemia virus alters diseasespecificity. Genes Dev. 4:233-242.
45. Starcich, B. L., L. Ratner, S. F. Josephs, T. Okamoto, R.C. Gallo, and F.Wong-Staal. 1985. Characterization of long termi-nal repeat sequencesof HTLV-III.Science227:538-540. 46. Tong-Starksen, S. E., P. A. Luciw, and B. M. Peterlin. 1987.
Human immunodeficiency viruslong terminal repeat responds toTcellactivationsignals.Proc.Natl.Acad. Sci. USA 84:6845-6849.
47. Varmus, H. 1988. Regulation ofHIV and HTLV gene expres-sion. Genes Dev. 2:1055-1062.
48. Willey,R.L., E. K.Ross, A. J.Buckler-White,T.S.Theodore, and M. A. Martin. 1989. Functionalinteraction ofconstantand variable domains of human immunodeficiency virus type 1 gpl20. J. Virol. 63:3595-3600.
49. Willey, R. L.,D. H.Smith,L. A.Lasky, T. S.Theodore,P. L. Earl, B. Moss, D.J. Capon, andM. A. Martin. 1988. In vitro mutagenesis identifiesaregionwithin theenvelopegene of the humanimmunodeficiencyvirus that is critical forinfectivity. J. Virol.62:139-147.
50. Wright,C.M., B. K. Felber, H. Paskalis, andG. N. Pavlakis. 1986. Expressionand characterization of the trans-activator of HTLV-III/LAV virus. Science 243:988-992.
51. Wu, F., J. Garcia, D. Harrick, and R. B. Gaynor. 1989. Purification of the human immunodeficiency virus type 1 en-hancer and TARbinding proteinsEBP-1 and UBP-1. EMBO J. 7:2117-2129.
52. Wu, F., J.Garcia, R. Mitsuyasu, and R.Gaynor. 1988. Alter-ations inbindingcharacteristics of the humanimmunodeficiency virusenhancerfactor. J. Virol.62:218-225.
53. Zenke, N., T. Grundstrom, H. Matthes, M. Wintzerith, C. Schatz,A.Wildeman,and P. Chambon. 1986.Multiplesequence motifs are involved in SV40 enhancer function. EMBO J. 5:387-397.
54. Zoller, M. J., and M. Smith. 1983. Oligonucleotide-directed mutagenesis: asimplemethodusingtwooligonucleotide
prim-ersandasingle-strandedDNAtemplate.DNA 3:479-488.