0022-538X/93/105939-09$02.00/0
Copyright© 1993, American Society for
Microbiology
Cofactor
Requirement for
Human
Immunodeficiency Virus
Type 1 Entry
into
a
CD4-Expressing
Human
Cell Line
ROBERT D. HARRINGTON* ANDADAMP. GEBALLE
Department of Molecular Medicineand Division of Clinical Research, Fred Hutchinson CancerResearch Center, 1124Columbia Street, Seattle, Washington 98104-2092
Received 14April 1993/Accepted 28 June 1993
Expressionof the human immunodeficiency virus type 1 (HIV-1) receptor CD4 on many nonhuman and some human cell lines is not sufficient to permit HIV-1 infection. We describe a human glioblastoma cell line (U373-MG) which remains resistanttoHIV-1despite the added expression of an authentic CD4 molecule. The block toHIV-1infection of these cellsisstrainindependent and appearstobe at viralentry. Heterokaryonsof
CD4-expressingU373-MG(U373-CD4) cells fusedtoHeLa cells allow HIV-1entry.AU373-CD4/HeLahybrid clone allows efficient HIV-1 replication. These results suggest that HeLa cells express a factor(s) that can complement the viralentrydefect of U373-CD4cells andisnecessaryfor efficient CD4-mediatedHIV-1 infection.
Entry of human immunodeficiencyvirus type 1 (HIV-1) into cells is a complex process involving the viral envelope protein (gpl2O/gp4l), theviral receptor CD4, and possibly other unidentified cellular factors. Entry begins when a domain located in thecarboxy-terminal half ofgpl20 binds to aregion within the first(V1)extracellulardomainof CD4
(3, 14, 16, 18, 19). Fusion of the virus and cell membranes is ultimately mediated by the gp4l subunit of the HIV-1
envelope protein (9, 18). However, many reports have suggested the presence ofintermediate steps involving the hypervariable region (V3 loop) ofgpl20 (7, 21, 31); regions within the first, second, and third
(Vi
toV3) extracellular domains of CD4(2,14, 20,25, 26,28);and,conceivably, cell surface cofactors. Thenatureof these intermediate steps is unclear but mayinvolve cleavage of the V3 loop, release ofgpl20(2, 7, 13, 23, 24, 29,33), or receptor-envelope confor-mational changes which ultimately permitgp4ltofusevirus and cellmembranes (29).
Although CD4 is clearly a receptor for HIV-1, many
CD4-expressing cells are resistant to HIV-1 infection (6).
Murine cellsexpressinghuman CD4canbind HIV-1 but do not fuse with the viral membrane (22). Avariety of other
nonprimate mammalian cell lines that express human CD4 canbind recombinantgpl20withoutallowingviral entry and areincapable offusingwith cells which express the HIV-1
envelope (6). Additionally, the human cell lines U87 and
SCL1, when made to express CD4, remain resistant to HIV-1 infectiondespite bindingthe virus(4).
Even cells permissive for HIV-1 are often limited to infection withonly certain viral strains(5, 15). Many reports have demonstrated theimportanceofthe V3 loop ofgpl20in
determining the tropism ofparticularviral isolates (15, 27,
30), but few studies have addressed the cellular factors
(otherthanCD4)thatmustbeinvolved. Variable expression of these cofactors may explain the distinct tropisms of individual HIV-1 strains. Similarly, the absence of these cofactors mayexplain the resistance of certain CD4+ cells for all strains of HIV-1. Inthis reportwedescribe ahuman cell line made to express CD4 (U373-CD4), which remains resistant to HIV-1. The block to infection in these cells appears tobeatviral entry. Complementation of this entry
* Correspondingauthor.
block occurs following fusion with HeLa cells, suggesting
that U373-CD4 cells lack a cofactor necessary for CD4-mediated HIV-1 entry.
MATERUILSAND METHODS
Cell lines. Celllineswere propagatedinDulbecco's
mod-ifiedEagle's medium(DMEM) supplementedwithpenicillin, streptomycin, glutamine, and 10%Nu-serum(Collaborative
Biomedical Products, Bedford, Mass.). U373-MG cells (a
humanglioblastoma cellline)wereobtained from the Amer-ican Type Culture Collection, Rockville, Md. HeLa cells were obtained from R. E. K. Fournier, Fred Hutchinson Cancer Research Center (FHCRC). U373-CD4 cells were constructedbyinfectingU373-MGcells with the
amphotro-pic retroviralvectorPA317\LT4SN containingahuman CD4 cDNA and neo under the control of the retroviral long
terminal repeat (LTR) and the simian virus 40 promoters,
respectively (provided by V. Garcia and A. D. Miller, FHCRC). Cells were selected by growth in DMEM plus 0.8 mgof (active) G418 (GIBCO, Grand Island, N.Y.) per ml. HeLa-CD4 cells, constructed by using the same retroviral
vector
(PA317\LT4SN),
were provided by M. Emerman, FHCRC (17). Hygromycin-resistant U373-MG and HeLa cloneswereconstructedby calciumphosphate transfection(10) of U373-MG and HeLa cells with plasmid pCMVhph (provided by M. Linial, FHCRC [1]), which encodes a
hygromycin resistance gene under the control of the
cyto-megalovirus immediate-earlypromoter. Cellswere selected
by growthinmedium with 100 U ofhygromycin (Sigma,St.
Louis, Mo.)perml,andindividual cloneswereisolated.
HeLa-CD4-LTR/3-gal
cells, provided by M. Emerman and described in detail elsewhere (17), express CD4 and containasingle integratedP-galactosidase (P-gal)
geneunder the control ofatruncated HIV-1 LTR.Expressionoftatin these cellsactivatestranscriptionfrom theLTR, resultinginI3-gal activity,
which can then beassayed by
5-bromo-4-chloro-3-indolyl-,-D-galactopyranoside (X-Gal) staining (17).
HeLa-Rev-Tat-Env cells expressing the HIV-1 envelope,
Tat, andRev wereprovided byM. Emerman.
U373-CD4-LTR/,B-gal indicator cellswereconstructed
by
electroporating U373-CD4 cells with
plasmid pEQ447.
pEQ447wasconstructedbyinserting aStuIIBglII
fragment
of pCMVhph(containing
thehygromycin
gene under the 5939on November 9, 2019 by guest
http://jvi.asm.org/
control of the cytomegalovirus major immediate-early pro-moter) into the StuI-BamHI sites of pJK2 (provided by M. Emerman [17]). The
,-gal
protein controlled by the HIV-1 LTR in pJK2 and pEQ447 contains the simian virus 40 nuclear localization signal (17). U373-CD4 cells into which pEQ447 was electroporated were selected by growth in DMEM plus hygromycin (100U/ml). Clones were screened for low basal n-galexpression that was inducible by Tat.Viruses and infections. Viral stocks of the macrophage-tropic isolate (NFN-SX [27]) and the primary isolate
(NRO3143) were prepared as described previously (17). To obtain high-titer stock of the lymphocyte-tropic, laboratory-adapted (LAI) strain, transfected cell supernatants were used to infect CEM cells and cell-free virus was collected 1 week later (19a). PA317\LtatSN, an amphotropic retroviral vector encoding tat, was provided by V. Garcia and A. D. Miller. Infections with HIV-1 were performed in the pres-ence of DEAE-dextran (20
p,g/ml),
andPA317\LtatSN infec-tion was performed with Polybrene at a concentrainfec-tion of 2,ug/ml.
X-Gal staining for,-gal activity is described elsewhere (17). Cell-free supernatant p249ag levels were determined by
enzyme-linked immunosorbent assay (ELISA) (Coulter Im-munology) as described by the manufacturer.
FACSanalysis. Fluorescence-activated cell sorter (FACS) analysis for CD4 expression was performed by using the fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody leu-3A (3,ug/ml; Becton Dickinson). Between
105
and 106 cells were detached from the monolayer by scraping in the presence of Versene. After being washed once in
phosphate-buffered saline (PBS) with 5% Nu-serum, cells were pelleted, resuspended in 40 ,u of PBS plus 5% Nu-serum with 10,u ofleu-3A, and incubated at
4°C
for 30min.
The cells were washed three times and resuspended in 300,u of PBS plus 5% Nu-serum. FACS analysis was performed on aFACSCAN (Becton Dickinson).FACS analysis for HIV-1 envelope binding was done by using FITC-gpl20. Recombinant gpl20 (strain SF2; AIDS Research and Reference Reagent Program, National Insti-tutes of Health) was conjugated to fluorescein, column
purified (11), and incubated with cell types (100 ng of
FITC-gpl20/105
to 106 cells in a volume of 50RI)
as de-scribed above.Westernimmunoblot. Protein preparations were obtained bylysing cell monolayers with 50 mMTris-HCl (pH8.0)-150
mM NaCl-1% Nonidet P-40 (Sigma)-1% deoxycholic acid
(Sigma). Equal volumes of the lysate supernatants were boiled for 5 min, and proteins were separated by electro-phoresis through 10% polyacrylamide gels with 0.1% sodium dodecyl sulfate (SDS). Approximately 2 x
105
cell equiva-lents were loaded into each well. Proteins were transferred to nitrocellulose in 25 mM Tris-HCl (pH 8.5)-195 mM glycine-20% methanol at 1 A for 2.5 h. The blots were then soaked twice (20 min each) in 10 mM NaPO4 (pH 7.5)-0.9%NaCl-0.5% Tween 20 (Sigma)-5% dry skim milk (T-PBS) before being incubated with 10
RI
of a rabbit polyclonal anti-CD4 antibody (American Biotechnologies, Cambridge, Mass.) in 10 ml of T-PBS for 90 min. The blots were then washed three times in T-PBS before being incubated with 15 ,ul ofalkalinephosphatase-conjugated
goat anti-rabbit serum(Sigma)in 10 ml ofT-PBS for 60min. After being washed in T-PBS, the blots were rinsed in 0.1 MTris-HCl (pH9.5)-0.1
MNaCl-50 mMMgCl2 (reaction solution) and developed in reaction solution with 5-bromo-4-chloro-3-indolylphosphate toluidinium (BCIP) and Nitro Blue Tetrazolium (NBT) (Be-thesda ResearchLaboratories, Gaithersburg, Md.) as
chro-mogenic substrates for alkaline phosphatase as specified by the manufacturer.
PCR. Polymerase chainreaction(PCR) analysis wasused todetect HIV-1 reversetranscription products1day
postin-fection. Approximately105
cells ofeach type wereinfectedfor 2 h at
37°C
at a multiplicity of infection of 1 U(deter-mined by the MAGI assay
[17])
per cell with HIV-1 (labora-tory-adapted isolate LAI). Virus had beenpretreatedwith 27 U of DNase I(Worthington, Freehold,N.J.)perml for 1hat room temperature. HIV-1 was heat inactivated byincuba-tion at
56°C
for 30min.
The cells were washed, trypsinized, and replated to remove any adherent virus. At 1day follow-ing infection, DNA was purified from the cells bydigestion with proteinase K and SDS and was then subjected to phenol-chloroform extraction and ethanolprecipitation.
PCR amplification was done in the presence of[3
P]dATP
with primers (5'-AGTGGGGGGACATCAAGCAGCCATG CAAAT-3' and
5'-TGCTATGTCACTTCCCCTTGGTTC
TCTC-3') spanning a 142-bp sequence within the gagregion (positions 1366 to 1507) of HIV-1. Each sample was also amplified in a separate reaction with primers (5'-ACACAACTGTGTGTTCACTAGC-3' and 5'-CAAC1TCATC
CACGTTCACC-3')spanning a 114-bp sequence of the human
1
globin
gene asaninternal
control. For eachreaction,
1,ul
of sample DNA was amplified in a 30
,ul
reaction at94°C
for 4min
followed by 30 cycles of94°C
for 1min,
60°C
for 30 s, and72°C
for 1min.
Amplified products were detected by autoradiography after electrophoretic separation on 8% polyacrylamide gels.Cell fusions. Cell hybrids were generated by plating 4 x
105
cells of both fusion partners onto25-cm2
flasks toobtain a subconfluent monolayer. The following day cells were washed three times with serum-free DMEM and overlaid with 1 ml of prewarmed polyethylene glycol (PEG) (molec-ular weight, 1,500; NBS Biologicals, Haverhill, England) in serum-free medium (50%,wt/vol).
After 1min
the PEG was aspirated and the cells were washed three times with serum-free DMEM and fed with DMEM plus 10% Nu-serum. At 1 day later G418 (500jig/ml,
active weight) and hygromycin (100U/ml)
were added to select hybrid clones.Heterokaryons were formed by plating 5 x
104
CD4-LTR/,B-gal
cells and 2.5 x105
fusion partners onto wells of a 24-well plate. At 1 day later, the cells were fused with PEG as described above. At 1 day following fusion, the cells were exposed to HIV-1; they were stained with X-Gal (17) 2 days postinfection.RESULTS
CD4 expression on U373-CD4 cells. We introduced CD4 into U373-MG cells, by using the retroviral vector PA317\LT4SN, with the intent of creating a human neural cell line that would be highly permissive for HIV-1. The same vector was used previously to transduce CD4 into HeLa cells, rendering them susceptible to HIV-1 (17). The level of CD4 expression by U373-CD4 cells was determined byFACSanalysis with the anti-CD4 antibody leu-3A. Figure 1A demonstrates that leu-3A binding to U373-CD4 cells was clearly greater than to their CD4- parent cells (U373-MG) and was similar to the binding of leu-3A to HeLa-CD4 cells (Fig. 1B).
Since leu-3A binds to CD4 at a different site than HIV-1 does, we next compared the binding of the HIV-1 envelope protein,
gpl20,
to HeLa-CD4 and U373-CD4 cells.FACS
analysis with
FITC-gpl20
demonstrated similargpl20
bind-ing to both HeLa-CD4 and U373-CD4 cells that was greateron November 9, 2019 by guest
http://jvi.asm.org/
E .
z
n.
z
00
A
aS
25
to
to
a
io~ B
0s
0
0
So t t smbo b sob t1
Relative Fluorescence
FIG. 1. CD4 expressiononU373-CD4 (A and C) and HeLa-CD4 (B and D) cells. U373-CD4orHeLa-CD4 (thick lines) and their
CD4-parental cells (thin lines)wereanalyzed by FACS with FITC-leu-3A (A and B)orFITC-gpl20 (C and D).
than to their respective CD4- controls (Fig. 1C and D). Histograms of HeLa-CD4 and U373-CD4cells, labeled with
a 10-fold dilution of FITC-gpl20, were no different from
those of theirrespective CD4- parents,suggesting that the affinities ofCD4 forgpl20aresimilaronboth celltypes(data notshown).
Western blot analysis with a rabbit polyclonal anti-CD4
antibody demonstrated comigration of CD4 from U373-CD4 and HeLa-CD4 cells (Fig. 2). As expected, no CD4 was
detectedonHeLa cells. These data confirm that U373-CD4
cells express a CD4 molecule of the same size and with
similargpl20-binding propertiestothatonHeLa-CD4 cells,
whicharehighly permissive for HIV-1.
HIV-1 infection of U373-CD4 cells. Some HIV-1 isolates
9 D
CD4-0
MW
- 205
- 116 - 97
68 45
29
FIG. 2. Western blot analysis ofCD4. Cellular proteinswere
separated by SDS-polyacrylamide gel electrophoresis, transferred
tonitrocellulose, and probedfirst with rabbit polyclonal anti-CD4
serum and then with alkaline phosphatase-conjugated goat
anti-rabbitserum. Thepositionof CD4(55 kDa)isindicatedonthe left
(arrow), and molecularweightmarkers(MW) areindicatedonthe
right (in thousands).
infectonlyasubset ofCD4-expressing cells. Strainsthat do not infect T-cell lines will infect primary macrophages or
lymphocytesin cell culture (5, 15). We tested the suscepti-bility of U373-CD4 cells to several HIV-1 strains with different cell preferences. Supernatant p24fag levels ob-tained 2to3 weekspostinfectionwith themacrophage-tropic strain (NFN-SX),thelaboratory-adaptedstrain(LAI),anda
primary isolate ofHIV-1 (NRO3143)revealed that none of
the strains infected U373-CD4 cells or their CD4- parents (Table 1). On the otherhand, HeLa-CD4 cellswere highly permissive for the lymphocyte-tropic strain (LAI) and the primary isolate. The low p24&ag levels detected in the supernatantfrom U373-MG and U373-CD4cells may have
representedresidualinputvirusor averylimited infection of these cells mediated by a non-CD4 receptor. Indeed, U373-MG cells reportedly express galactosyl ceramide (GalC), a surface glycoprotein which can function as an
inefficient HIV-1receptor(12). Regardlessof thereasonsfor these low p249ag values, it is noteworthy that the added expression of CD4 did not enhance HIV-1 production in U373-MG cells. Furthermore, the resistance of U373-CD4 cells toHIV-1 infectionwasstrainindependent.
HIV-1 infectionofU373-CD4 cells is blocked atentry.We constructed the indicator cellline, U373-CD4-LTR/,B-gal, by stably transfectingU373-CD4 cells withaplasmid encoding
the HIV-LTRdrivingthereportergenelacZ(see Materials andMethods).TheseU373-CD4-LTR/,B-galcells,like
HeLa-TABLE 1. HIV-1infection of cell lines
Supernatantp240ag levels(pg/ml)ain HIV-1 strain:
Celltype LAJb I.MLNNSXARY13
(expt1) (expt2) NFN-SXb NR03143'
U373-MG 24 NDd 7 ND
U373-CD4 30 10 8 1
HeLa-CD4 >320 >4,000 6 3,200
ap249aglevelsweredeterminedbyELISA(seeMaterialsandMethods).
bAssayed2 weekspostinfection.
cAssayed3 weekspostinfection.
dND,notdone.
11 so tab 1mb 21
500
uso-c
o
100
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.144.468.81.282.2] [image:3.612.111.246.488.662.2]FIG. 3. Activation of
n-gal
expression inHeLa-CD4-LTR/,B-galindicator (A to D) andU373-CD4-LTR/P-gal(E toH) cells.Mock-(panels Aand E),PA317\LtatSN- (panelsB andF),and HIV-1 (LAI) (panels C and G)-infectedcells were fixed and stained with X-Gal2days postinfection.CD4-LTR/3-gal
cells coculturedwithHeLa-Rev-Tat-Envwereassayed1dayafterplating(panelsDandH).Magnification,x100.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.98.530.98.669.2]A 7 HIV-1 infected
-oV O
12 'E -1
., 0~
E 25 Co-0 J 9 7E
EoI I0 D I X
-M.X .z
-354 -176
1 2 3 4 5 6
B
ia
I
I
HIV-1 .*ctd
L I
-176
is
FIG. 4. PCR analysis of HIV-1 infected cells. At 1 day after HIV-1 (LAI) infection ofU373 (lane 3), U373-CD4 (lane 4), and HeLa-CD4(lane5) cells,total cellular DNAwaspurifiedandPCR
amplifiedwithprimers spanninga 142-bpsequencewithin thegag
region of HIV-1 (A) and (in a separate reaction) with primers spanning a 114-bp sequence of the human globin gene (B). Heat-inactivated HIV-1 (lane 2)wasincubatedat56°Cfor30min. The control plasmid (panel A, lane 6) encoded the HIV-1 (LAI) provirus.
CD4-LTR/,B-gal indicatorcells, expressed little or no basal
1-gal activity, as measured by X-Galstaining (Fig. 3A and
E). Transduction of tat into U373-CD4-LTRI3-gal and HeLa-CD4-LTR/1-gal indicatorcells withthe amphotropic retroviral vector, PA317\LtatSN (titer, 7 x 104 by MAGI
assay [17]), yielded numerous blue foci after 1 h ofX-Gal staining (Fig.3B andF).In contrast,HIV-1(LAI) (titer,1 x
105
byMAGIassay)infection ofboth celllinesyieldedmanyblue
HeLa-CD4-LTR/P1-gal
indicator cell foci (Fig. 3C) butno blue U373-CD4-LTR/,B-gal cells (Fig. 3G). These data
suggestedthat HIV-1infection ofU373-CD4-LTR/I-galcells didnotleadtotheexpression oftat,consistent withablock earlyinthe virallife cycle.
Toexamine stages inthe HIV-1 infectious cycle priorto
tatexpression,weinvestigatedwhetherproductsofreverse
transcriptionwerepresent inU373-CD4 cells afterexposure
toHIV-1.Cellular DNAwasharvested 1dayafter infection
with HIV-1 and PCR amplifiedwith oligonucleotides
span-ninga142-bpsequencewithinthegagregion(positions1366
to 1507) as described in Materials and Methods. A band
corresponding to the amplifiedgag sequence was evident onlyafteramplificationof DNA fromHIV-1-infected HeLa-CD4 cells and ofplasmidcontrol DNA(Fig.4A,lanes 5 and 6). Amplificationwith 13globin primers yieldedintensebands inalllanes, suggestingthat thosesampleswithout detectable HIV-1sequencesdid have DNA and didnothave inhibitors of the PCR reaction (Fig. 4B). These data suggested that HIV-1 infection of U373-CD4 cells was blocked at either
entry or reverse transcription. PCR analysis with primers spanning a region in the HIV LTR (positions 496 to 616) yieldedsimilar results (data not shown).
Tospecificallyexamine membrane fusion and viral entry, we cocultured HeLa-Rev-Tat-Env cells with
HeLa-CD4-LTR/1-gal indicatorcells and U373-CD4-LTR/,B-gal indica-tor cells (Fig. 3D and H). Since HeLa-Rev-Tat-Env cells express Tat and Env constitutively, the formation of blue syncytia requires only Env-CD4-mediated fusion of the HeLa-Rev-Tat-Env cells with a
CD4-LTR/0-gal
indicator cell followedby entry of Tat into the nucleus of the indicator cell andactivation of the HIV LTR. Bluesyncytiaareclearly visible after coculture ofHeLa-CD4-LTR/1-gal
indicator cells with HeLa-Rev-Tat-Env cells(Fig.3D). The absence of blue U373-CD4-LTR/I-gal cells on coculture with HeLa-Rev-Tat-Env cells (Fig. 3H) therefore suggests that CD4-viral envelope mediated fusion didnot occurand indicates thatablocktoHIV-1 infection ofU373-CD4 cellsoccurs at membrane fusion.HIV-1infection of
U373-CD4-LTR/j3-gal/HeLa
heterokary-ons.Thefailure of HIV-1 entry into U373-CD4 cells suggests that these cells may lack a cofactor required for CD4-mediated viral entry. To investigate this hypothesis, we constructedheterokaryons from
U373-CD4-LTRIP-gal
cells and HeLa cells and exposed them to HIV-1. In theseexperiments HIV-1 infection could occur and be detected onlyinheterokaryonsexpressingboth CD4 and1-gal. Thus, any cell staining blue must have been derived from a
U373-CD4-LTR/P-gal
cell. The increase in the number of3-gal-staining U373-CD4-LTR/,B-gal
cells after fusion withHeLa cells and incubation with HIV-1indicated that HeLa cells couldcomplement the viral entry defect of
U373-CD4-LTR/0-gal
cells (Fig. 5). On the other hand, fusion of U373-MG cells to U373-CD4-LTR/I-gal cells followed byexposure to HIV-1 yielded only two blue foci, suggesting
that enhanced viral entry into
U373-CD4-LTRI/-gal
cells required aHeLacell factorandwasnotduetothe nonspe-cific effect of PEG-mediated cell fusion. The two cellsexpressing
1-gal
in infectedU373-MG/U373-CD4-LTR/PI-gal
fusionsmay represent non-CD4-mediated(e.g., GalC)viral infection of
U373-CD4-LTRI/-gal
cells. No blue fociwere detected inuninfected cell fusions.HIV-1 infection of U373-CD4/HeLa and HeLa-CD4/ U373-MGhybrids.Tofurtherinvestigatethehypothesisthat U373-CD4 cells lackacofactor necessary for CD4-mediated viral entry, we established hybrid cell clones after PEG-mediated fusion of U373-CD4 cells with HeLa cells and fusion of HeLa-CD4 with U373-MG cells(seeMaterials and
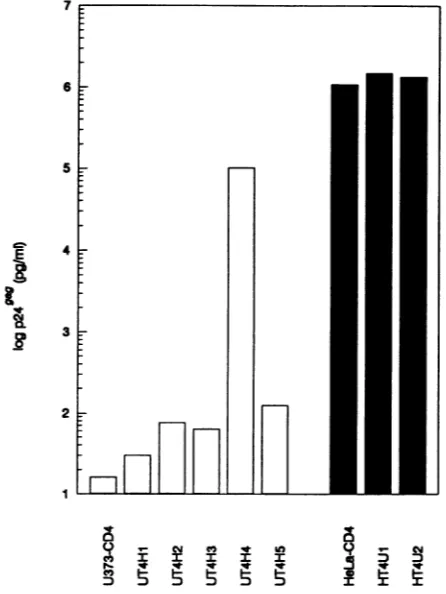
Methods).FACSanalysis with the anti-CD4antibodyleu-3A confirmed that all clonesexpressed CD4 althoughto differ-ent degrees (Fig. 6). The hybrid clones were exposed to HIV-1 andsupernatantp249aglevelsweremeasured upto2 weeks postinfection. Both HeLa-CD4/U373-MG clones tested were permissive for HIV-1 whereas one of five
U373-CD4/HeLa hybrids (UT4H4) supportedHIV-1
replica-tion(Fig.7). Later passages of the UT4H4clonedeveloped
resistancetoHIV-1(datanotshown), suggestingthelossor extinction of the complementing HeLa gene(s). Consistent with the results of the heterokaryon experiments (Fig. 5),
these data suggest that HeLa cells express a
factor(s)
thatcomplementsthe viral entry defect of U373-CD4 cells. DISCUSSION
We havedescribed ahuman cell
line, U373-CD4,
which expressesan authentic CD4 molecule yet remains resistant0) I.-0)
D
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.95.270.76.335.2]A
B Nutme ofblueoeM
U373-CD4-LTR/I,-gal fusionpartner HeLacells U373-MGceMs
HIV-1infection 28 2
[image:6.612.67.298.75.341.2]Mockintection 0 0
FIG. 5. Heterokaryons infectedwith HIV-1. U373-CD4-LTR/,3-gal cells were fused to HeLa cells orparental U373-MG cells by using PEGand werethen infected withHIV-1(LAI) and stained(2 days postinfection) for
1-gal
expression asdescribed in Materials and Methods. (A) InfectedU373-CD4-LTR/13-gal/HeLa
hetero-karyon.Magnification, x320.(B) Results ofarepresentative exper-imentshowingthe numberofblue cells per 17-mm well of HIV-1-ormock-infected heterokaryons.
to HIV-1 entryin astrain-independent manner. This entry blockcanbeovercomebyfusingU373-CD4 cells with HeLa
cells, creating.either heterokaryons or cell hybrids. These data suggest thatacellularfactor(s), expressedin HeLacells butnotinU373-MGcells, is necessary, inconjunctionwith
CD4, for HIV-1 infection.
PA317\LtatSN
infection ofU373-CD4-LTR/13-gal
cells demonstrated that expression oftatin these cells activates the HIV LTR (Fig. 3F). The failure ofHIV-1 infection ofU373-CD4-LTR/0-gal
cells to activate,3-gal
expression lo-calizes a block in theviral life cycle to stagesprior to tatexpression.The absence ofreversetranscriptionproducts in U373-CD4 cells exposed toHIV-1 further localizes ablock toviral entryor reverse transcription. Onemodel of HIV-1
latency in restingT lymphocytes proposes that the HIV-1 genome isonlypartially reverse transcribed and that com-pletion of reverse transcription and subsequent steps in viral replication occur only after T-cell activation (34). However, this model is not a likely explanation of our data, since U373-CD4 is a continually dividing transformed cell line. Furthermore, since coculture of HeLa-Rev-Tat-Env cells with
U373-CD4-LTR/0-gal
cells failed to activate the HIV LTR (Fig.3H), adefect in gpl60-CD4-mediatedmembrane fusion must exist. Indeed, all our data are consistent with a blockat fusion of the virus to the cell membrane and viral entry.Previous reports have shown that U373-MG cells express the surfaceglycoproteinGalC,which can serve as an HIV-1 receptorwith anaffinityfor the viral envelope protein similar
tothat of CD4(12). However,infection ofU373-MG cells is inefficient and detection of HIV-1replication requiresnested PCR followedby Southernblotting or coculture of
HIV-1-exposed U373-MG cells with a highly permissive cell line.
(Inthe present studyweusedonlyasingle PCRto
amplify
HIV-1reversetranscriptionproductsand therefore maynot have detected alowlevel ofnon-CD4-mediatedviral infec-tion.) More to the point, however, we have demonstrated that the expression of CD4 on U373-MG cells does not enhance their susceptibility to HIV-1 entry, regardless of any low background level of infection. This suggests that U373-MG cells, whether they express one (GalC) or two(GalC and CD4) high-affinity receptors for HIV-1, remain resistant to viral entry because efficient postbinding virus-cell membrane fusion doesnotoccur.
One explanation for the block to CD4-mediated HIV-1 entryinto U373-CD4 cells is thatone or morefusion domains of CD4 are not preserved. We have shown that epitopes recognizedby the anti-CD4 antibodyleu-3A andby recom-binantgpl20(bothcontained in thefirst extracellulardomain ofCD4,V1) arepresent onU373-CD4 cells (Fig. 1).
Addi-tionally, CD4 was introduced into both U373-CD4 and HeLa-CD4 cells by using the same retroviral vector, and Western blot analysis suggested that CD4 from both cell types is the same size (Fig. 2). Nevertheless, subtle differ-ences in the posttranslational processing of CD4 in U373-CD4 andHeLa-CD4cellscould leadtodifferences in any of the CD4extracellular domains
(Vl,
V2, orV3)
which have beenimplicatedasimportantmediators of membrane fusion(2, 14, 20, 25, 26,
28).
Amorelikely explanationfor the resistance ofU373-CD4
cellstoHIV-1infection isthat
they
lackanecessarycofactor forCD4-mediatedviral entry. To determine whether HeLa cells express suchacofactor whichcancomplementthe viral entrydefect ofU373-CD4-LTRI,B-gal
cells, weconstructed heterokaryons fromU373-CD4-LTR/P-gal
and HeLa cells andinfected them with HIV-1.U373-CD4-LTR/I-gal/HeLa
cell heterokaryons allowed enhanced viral entry and tat
expression,whereas U373-CD4-LTR/,B-gal/U373-MG
heter-okaryons permitted only background HIV-1 infection
(Fig.
5). Furthermore, the isolation ofa
U373-CD4/HeLa
hybrid
clone(UT4H4)thatwaspermissivefor HIV-1supportedtheheterokaryonstudies suggestingthat U373-CD4 cells lack a CD4 cofactor necessary for viral infection. Since this UT4H4 clonewasalmostas
permissive
for HIV-1as HeLa-CD4 cells were, any blocks in the virallifecycle
other than entrymustalso have beencomplemented bythe HeLacells.Alternatively,there may benoblockstoHIV-1replicationin U373-CD4 cells once entry is achieved.
U373-CD4/HeLa
clones thatwere notpermissivefor HIV-1 may have lostthe HeLa chromosome(s) encoding the factor(s) necessary forcomplementation
of the viral entrydefect.Athirdexplanationfor the fusion defectis that U373-CD4 cells expressaninhibitorof viral entry. Itwas not
possible
to rule this outfrom the heterokaryon experiments, since not everyHeLa-CD4-LTR/I-gal
indicator cell was fused to a U373-MG cell after cell fusion(data
notshown).
Cellsexpressing
1-gal
inthiscrossmayhaverepresented HIV-1-infectedHeLa-CD4-LTR/,B-gal
indicator cells thatwere not fusedor wereself-fused rather thanheterokaryonsofHeLa-CD4-LTR/P-gal
indicator cells and U373-MG cells that al-lowed HIV-1 entry. However, the observation that bothHeLa-CD4/U373-MG hybridclonesremainedhighly
permis-sive for HIV-1 suggests that U373-MG cells donotexpress an inhibitor ofHIV-1 infection (Fig. 7). Additionally, later passages of the permissive UT4H4 hybrid clone lost their
on November 9, 2019 by guest
http://jvi.asm.org/
0 50 100bb105
E
Cm
D
sm
o.
smz
4100 too
0 a sb 50 100 isub
.ui
a
-E
200 ms
0 0o
[image:7.612.148.468.75.378.2]Relative Fluorescence
FIG. 6. CD4expressionof cellhybrids. CD4- parental cells(thin lines), CD4-expressing parentalcells(thick lines), and individual cell hybrids (dashed lines)wereanalyzed by FACSwith theFITC-anti-CD4 antibodyleu-3A.(A) Histograms of thetwoHeLa-CD4/U373-MG (HT4U1 andHT4U2) hybrid clones; (B through F) histograms of the five U373-CD4/HeLa(UT4H1toUT4H5,respectively)hybridclones.
7 susceptibilitytoHIV-1(datanotshown),consistent with the
lossorextinctionof acomplementing HeLa factor. Had the
susceptibilityto HIV-1ofearly-passage UT4H4 cells been due to the loss of an inhibitor that was expressed in the
6 . | parental U373-CD4 cells, the development of a resistant
phenotype in subsequent cell passages would have been
unlikely. Therefore, the most likely explanation for the
susceptibilitytoHIV-1ofourheterokaryonsand cellhybrid
5 is that HeLa cells supplied a cofactor required for
CD4-mediated HIV-1 infection.
Thereare conflictingdata regarding the ability of human cellstocomplementthe viral entry defect ofCD4-expressing
E 4 murine cells.A recent study demonstrated that
heterokary-ons made from human cells and CD4+ murine cells could fuse withcells expressing the HIV-1 envelope (8). In con-trast, another study described four human-murine hybrids
3 thatwere resistant to HIV-1 infection despite containinga
complete (one hybrid) or nearly complete (three hybrids) complement of human chromosomes (32). The hybrids in this latter studywith anincompleteset of human
chromo-2 somes may simply have lost the gene(s) essential for
com-plementation
of thephenotype.
In thesingle hybrid
with aFIG. 7. HIV-1(LAI)infection of cellhybrids.U373-CD4 cellsor
hybridsof U373-CD4 and HeLa cells(openbars)andHeLa-CD4or
9 _ Cc e * to hybrid clonesofHeLa-CD4 andU373-MG cells (solid bars)were
.5
; infectedwith HIV-1(LAI),andcellsupernatantswereassayedfor DDDDDDI t Y p249'9 10 days postinfection.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.67.290.428.725.2]complete set of human chromosomes, extinction of the essentialgene(s) mayhaveoccurred. Our results are consis-tent with those of the former report; however, our study shows that CD4+ human cells, defective for fusion with HIV-1,canbecomplemented byfactors expressed inother human cells and, furthermore, that once viral entry is
achieved, complete viralreplication can occur. These find-ings agreewith those of Dragic and Alizon, who recently
reported complementation of two other fusion-defective humancell linesbyHeLa cells inheterokaryon studies (7a). It ispossible that the entry blockof HIV-1into U373-CD4 cells is mechanistically related to the resistance of CD4+
nonprimate cells for all strains of HIV-1 and thetropism of individual isolates for only certain CD4+ human cells.
In-deed, essential cofactors for CD4-mediated entry may be
lacking in each case in which HIV-1 cannot infect a
CD4-expressing cell. In this regard U373-CD4-LTR/4-gal cells shouldproveausefulreagentin futureexperiments designed to identify the cofactor(s) necessary for CD4-mediated HIV-1 infection.
ACKNOWLEDGMENTS
We thank Dusty Miller and Victor Garcia for providing the PA317\LT4SNandPA317\LtatSNretroviral vectors, MaxineLinial fordonating plasmid pCMVhph,and Paul Lewis for his assistance in growing high-titer HIV-1 stock. We also thankMichael Emerman forproviding the HeLa-CD4-LTR/p-gal cells, the HeLa-Rev-Tat-Env cells, and plasmid pJK2 and for his critical review of the manuscript.
Thisworkwassupported byPublic Health Service grant AI27291 from the NationalInstitutes of Health.
REFERENCES
1. Aronoff, R.,and M.Linial. 1991. Specificityof retroviral RNA packaging. J.Virol. 65:71-80.
2. Berger,E.A., J.D.Lifson,and L. E.Eiden. 1991. Stimulation of glycoprotein gpl20dissociation from theenvelope glycoprotein complexof human immunodeficiency virus type 1by soluble CD4 and CD4peptide derivatives:implications for the role of thecomplementarity-determining region 3-like regionin
mem-branefusion.Proc. Natl. Acad. Sci. USA 88:8082-8086. 3. Camerini, D.,andB. Seed. 1990. ACD4 domainimportantfor
HIV-mediatedsyncytiumformation lies outside the virus bind-ingsite. Cell 60:747-754.
4. Chesebro, B.,R.Buller, J. Portis,and K.Wehrly.1990.Failure ofhuman immunodeficiencyvirus entry and infection in CD4-positivehuman brain and skincells. J. Virol. 64:215-221. 5. Chesebro, B., J. Nishio, S. Perryman, A. Cann, W. O'Brien,
I. S. Y. Chen, and K. Wehrly. 1991. Identification of human immunodeficiency virus envelope gene sequences influencing viral entry intoCD4-positiveHeLacells, T-leukemia cells, and macrophages.J. Virol. 65:5782-5789.
6. Clapham,P.R., D.Blanc,and R.A. Weiss. 1991. Specificcell surfacerequirementsfor the infectionofCD4-positive cells by human immunodeficiency virus types 1 and 2 and by simian immunodeficiencyvirus. Virology181:703-715.
7. Clements, G.J., M.J. Price-Jones, P. E. Stephens, C. Sutton, T. F.Schulz,P. R.Clapham,J. A. McKeating, M.0.McClure, S.Thomson,M. Marsh, J.Kay,R.A.Weiss, and J. P. Moore. 1991. The V3loopsofthe HIV-1 and HIV-2 surface glycopro-teins containproteolytic cleavage sites: a possible function in viralfusion? AIDS Res. Hum. Retroviruses 1:3-16.
7a.Dragic, T., and M. Alizon. 1993. Different requirements for membrane fusionmediatedbytheenvelopes of human immu-nodeficiencyvirus types 1 and 2. J. Virol. 67:2355-2359. 8. Dragic, T., P. Charneau, F. Clavel, and M. Alizon. 1992.
Complementationof murine cells for humanimmunodeficiency virus envelope/CD4-mediated fusion in human/murine heter-okaryons.J. Virol.66:4794-4802.
9. Gallaher, W. R.1987. Detection ofafusion peptide sequence in the transmembraneprotein of human immunodeficiencyvirus. Cell 50:327-328.
10. Graham,F.L., and A.J.vander Eb.1973. Anewtechniquefor the assay ofinfectivityofhuman adenovirus 5 DNA. Virology 52:456-467.
11. Harlow,E., and E. Lane. 1988.Antibodies,alaboratorymanual, p.354-355. Cold Spring Harbor Laboratory, Cold Spring Har-bor,N.Y.
12. Harouse, J. M., S. Bhat, S. L. Spitalnik, M. Laughlin, K. Stefano, D. H.Silberberg, and F. Gonzalez-Scarano. 1991. Inhi-bition of entry of HIV-1inneural cell lines by antibodiesagainst galactosyl ceramide. Science253:320-323.
13. Hart, T. K., R. Kirsh, H. Ellens, R. W. Sweet, D. M. Lambert, S. R. Petteway, Jr., J.Leary,and P. J. Bugelski. 1991. Binding of solubleCD4proteinstohuman immunodeficiency virus type 1 and infected cells induces release of envelope glycoprotein gpl20. Proc. Natl.Acad. Sci. USA 88:2189-2193.
14. Healey, D., L. Dianda, J. P. Moore, J. S. McDougal, M. J. Moore, P. Estess, D. Buck, P. D. Kwong, P. C. L. Beverley, and Q. J. Sattentau. 1990. Novel anti-CD4 monoclonal antibodies separatehumanimmunodeficiencyvirusinfection and fusion of CD4+ cells from virusbinding. J. Exp.Med. 172:1233-1242. 15. Hwang, S.S., T. J. Boyle, H. K. Lyerly, and B. R. Cullen. 1991.
Identification of theenvelope V3loop as the primary determi-nant of celltropism in HIV-1. Science 253:71-74.
16. Jameson, B. A., P. E. Rao, L. I. Kong, B. H. Hahn, G. M. Shaw, L. E. Hood, and S. B. H. Kent. 1988. Location and chemical synthesis of a binding site for HIV-1 on the CD4 protein. Science240:1335-1339.
17. Kimpton, J., and M.Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated13-galactosidasegene. J.Virol. 66:2232-2239. 18. Kowalski, M., J. Potz, L. Basiripour, T. Dorfman, W. C. Goh, E.
Terwilliger, A.Dayton, C. Rosen, W. Haseltine, and J. Sodroski. 1987. Functionalregions of the envelope glycoprotein of human immunodeficiencyvirus type 1. Science 237:1351-1355. 19. Lasky, L. A., G. Nakamura, D. H. Smith, C. Fennie, C.
Shimasaki, E. Patzer, P. Berman, T.Gregory,and D. J. Capon. 1987. Delineation of a region of the human immunodeficiency virus type 1gpl20 glycoprotein critical for interaction with the CD4 receptor. Cell50:975-985.
19a.Lewis, P. (Fred Hutchinson Cancer Research Center). Personal communication.
20. Lifson, J. D., K. M. Hwang, P. L. Nara, B. Fraser, M. Padgett, N. M.Dunlop, and L. E. Eiden. 1988. Synthetic CD4 peptide derivativesthat inhibit HIV infection and cytopathicity. Science 241:712-716.
21. Linsley, P. S., J. A.Ledbetter,E.Kinney-Thomas, and S.-L. Hu. 1988. Effects of anti-gpl20 monoclonal antibodies on CD4 receptorbinding by the env protein of human immunodeficiency virus type 1. J. Virol. 62:3695-3702.
22. Maddon, P. J., A. G. Dalgleish, J. S. McDougal, P. R. Clapham, R.A.Weiss, and R. Axel. 1986.The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain.Cell 47:333-348.
23. Moore, J. P., J. A. McKeating, W. A. Norton, and Q. J. Sattentau. 1991.Direct measurementof soluble CD4 binding to humanimmunodeficiencyvirus type 1 virions: gpl20 dissocia-tionand itsimplicationsforvirus-cell binding and fusion reac-tions and their neutralization by soluble CD4. J. Virol. 65:1133-1140.
24. Moore, J. P., J. A. McKeating, R. A. Weiss, and Q. J. Sattentau. 1990. Dissociation of gpl20 from HIV-1 virions induced by soluble CD4. Science 250:1139-1142.
25. Moore, J. P., Q. J. Sattentau, P. J. Klasse, and L. C. Burkly. 1992. A monoclonal antibody to CD4 domain 2 blocks soluble CD4-induced conformationalchanges in the envelope glycopro-teins of human immunodeficiency virus type 1 (HIV-1) and HIV-1infection of CD4+ cells. J. Virol. 66:4784-4793. 26. Nara, P. L., K. M. Hwang, D. M. Rausch, J. D. Lifson, and L. E.
Eiden. 1989. CD4 antigen-based antireceptor peptides inhibit
on November 9, 2019 by guest
http://jvi.asm.org/
infectivityof humanimmunodeficiencyvirusin vitroatmultiple
stages of the viral life cycle. Proc. Natl. Acad. Sci. USA 86:7139-7143.
27. O'Brien, W. A., Y. Koyanagi, A. Namazie, J.-Q. Zhao, A. Diagne, K Idler, J. A. Zack, and I. S. Y. Chen. 1990. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gpl20 outside the CD4-binding domain. Nature (London)348:69-73.
28. Poulin,L., L. A. Evans, S. Tang, A.Barboza, H. Legg, D.RI
Littman,andJ. A. Levy. 1991. SeveralCD4domainscanplaya
role in human immunodeficiency virus infections ofcells. J. Virol. 65:4893-4901.
29. Sattentau,Q. J.,andJ.P. Moore. 1991.Conformational changes inducedinthehuman immunodeficiencyvirusenvelope glyco-protein by solubleCD4binding.J. Exp.Med.174:407-415. 30. Shioda,T., J.A.Levy,and C.Cheng-Mayer.1991.Macrophage
and Tcell-line tropisms ofHIV-1 are determined by specific
regionsof theenvelopegpl20gene.Nature(London) 349:167-169.
31. Skinner,M. A., A. J. Langlois, C. B. McDanal, J. S. McDougal, D. P. Bolognesi, and T. J. Matthews.1988.Neutralizing antibod-ies toanimmunodominant envelope sequencedonot prevent
gpl20 bindingtoCD4. J. Virol. 62:4195-4200.
32. Tersmette, M., J. J. M.vanDongen,P. R.Clapham, R.E. Y. de
Goede, I. L. M. Volbers-Tettero, A. G. van Kessel, J. G.
Huisman, R. A. Weiss, and F. Miedema.1989.Human immuno-deficiency virus infection studied in CD4-expressing human-murine T-cellhybrids. Virology 168:267-273.
33. Thali, M., C. Furman, E. Helseth, H. Repke, and J. Sodroski. 1992. Lack ofcorrelation between soluble CD4-induced shed-ding of the human immunodeficiency virus type 1 exterior envelopeglycoproteinandsubsequentmembrane fusionevents.
J. Virol.66:5516-5524.
34. Zack,J. A.,S.J. Arrigo,S. R.Weitsman,A. S.Go,A.Haislip, and I. S. Y. Chen. 1990. HIV-1 entryinto quiescent primary lymphocytes: molecular analysis reveals a labile, latentviral
structure.Cell61:213-222.