Copyright © 2000, American Society for Microbiology. All Rights Reserved.
RNA Binding Properties of Bunyamwera Virus Nucleocapsid
Protein and Selective Binding to an Element in the
5
⬘
Terminus of the Negative-Sense S Segment
JANE C. OSBORNEANDRICHARD M. ELLIOTT*
Division of Virology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow G11 5JR, Scotland
Received 27 March 2000/Accepted 28 July 2000
The genome ofBunyamwera virus(BUN) (familyBunyaviridae, genusBunyavirus) comprises three negative-sense RNA segments which act as transcriptional templates for the viral polymerase only when encapsidated by the nucleocapsid protein (N). Previous studies have suggested that the encapsidation signal may reside within the 5ⴕ terminus of each segment. The BUN N protein was expressed as a 6-histidine-tagged fusion protein in Escherichia coli and purified by metal chelate chromatography. An RNA probe containing the 5ⴕ-terminal 32 and 3ⴕ-terminal 33 bases of the BUN S (small) genome segment was used to investigate binding by the N protein in vitro using gel mobility shift and filter binding assays. On acrylamide gels a number of discrete RNA-N complexes were resolved, and analysis of filter binding data indicated a degree of cooperativity in N protein binding. RNA-N complexes were resistant to digestion with up to 1 g of RNase A per ml. Competition assays with a variety of viral and nonviral RNAs identified a region within the 5ⴕterminus of the BUN S segment for which N had a high preference for binding. This site may constitute the signal for initiation of encapsidation by N.
Bunyamwera virus (BUN) is the prototype of the genus
Bunyavirusand the familyBunyaviridaeand possesses a
single-stranded negative-sense tripartite RNA genome. The three RNA segments, termed L (large), M (medium), and S (small), encode six proteins. The L segment codes for the L protein, the viral RNA-dependent RNA polymerase, which is responsible for both transcribing and replicating the genome RNAs. The M segment encodes the two virion glycoproteins, G1 and G2, and a nonstructural protein, NSm, as a polyprotein which is probably cotranslationally cleaved by host proteases. The S segment encodes the nucleocapsid (N) protein and, in an over-lapping reading frame, a second nonstructural protein called NSs (reviewed in reference 8).
In common with other negative-sense RNA viruses, the bun-yavirus genome RNA segments are replicated via full-length cRNAs termed antigenomes. Both the negative-sense genome and positive-sense antigenome RNAs are encapsidated by the N protein and are associated with the viral polymerase in ribonucleoprotein complexes called nucleocapsids. It is only within the nucleocapsid that the RNA is transcriptionally ac-tive. Bunyavirus genome and antigenome RNAs contain highly conserved, complementary terminal sequences that may form panhandle structures in vivo and are probably responsible for the circular appearance of isolated nucleocapsids (19, 20, 22, 26, 28).
Full-length genome and antigenome segments are usually the only RNAs that are encapsidated in the infected cell. Viral mRNAs, which are not encapsidated, are truncated at the 3⬘ end and contain a nontemplated capped primer on the 5⬘ terminus (2, 4, 9, 14, 21). It is therefore likely that the terminal sequences of the genome and antigenome RNAs are involved in the encapsidation process. This theory is supported by the observation that an antisense chloramphenicol
acetyltrans-ferase gene flanked by BUN terminal sequences was success-fully encapsidated by N and transcribed by L in an in vivo system (7). Raju and Kolakofsky (25) reported that in infected cells a minority of encapsidated bunyavirus transcripts either had the 3⬘-terminal truncation similar to mRNA but not the 5⬘ primer sequence or were full-length transcripts containing a capped primer on the 5⬘terminus (26). Taken together, these data suggest that the encapsidation signal is probably within the 5⬘terminal sequence of genome and antigenome RNAs.
Investigations concerning the N proteins of viruses in the
Hantavirus(11, 29) andTospovirus(27) genera of the
Bunya-viridae have shown that binding of the N proteins to RNA in
in vitro assays is essentially sequence nonspecific. However, Severson et al. (29) reported hantavirus N protein to have a preference for full-length hantavirus S segment RNA over RNA comprising only internal sequences, and Go¨tt et al. (11) reported hantavirus N to have a preference for double-strand-ed RNA.
The BUN N protein is a 26.7-kDa basic protein of 233 amino acids that does not show any sequence similarity to the hanta-virus (ca. 50-kDa) or tospohanta-virus (ca. 29-kDa) N proteins. Like the analogous nucleocapsid proteins of all members of the
Bunyaviridae family, it contains no RNA binding consensus
sequence (8). In this paper we describe investigations into the binding of bacterially expressed BUN N protein to negative-sense BUN S segment RNA sequences in vitro. We also pro-vide an analysis of the relative selectivity of the binding of N to such sequences with the intent to construct a model for the selective encapsidation of full-length viral genome and antige-nome RNA.
MATERIALS AND METHODS
Expression and purification of BUN N protein.The BUN N open reading frame (ORF), from codon 2 to stop codon 234, was amplified by PCR using primers 5⬘GCCGCGGATCCATCGAGGGAAGGATTGAGTTGGAATTT and 5⬘GCCGCGTCGACTTACATGTTGATTCCGAA, which also contain, re-spectively,BamHI andSalI restriction enzyme sites (underlined). The DNA amplicon was digested withBamHI andSalI and cloned between theBamHI and
* Corresponding author. Mailing address: Institute of Virology, Uni-versity of Glasgow, Church St., Glasgow G11 5JR, Scotland. Phone: 44 141 330 4024. Fax: 44 141 337 2236. E-mail: [email protected].
9946
on November 9, 2019 by guest
http://jvi.asm.org/
SalI sites of pQE30 (Qiagen). Recombinant N was expressed as an N-terminally 6-histidine-tagged protein inEscherichia colistrain M15 by induction with 1 mM isopropyl--D-thiogalactopyranoside (IPTG). The bacteria were lysed by freeze-thawing and sonication, and N protein was purified under native conditions (23) by Ni-nitrilotriacetic acid (NTA) column chromatography (Bio-Rad Econosys-tem). Protein immobilized on the Ni-NTA column was washed with 50 mM sodium phosphate buffer (pH 6.0) containing 300 mM NaCl and 10% glycerol, and N was eluted with a linear gradient of 0 to 500 mM imidazole in wash buffer. Fractions containing large amounts of N were eluted at around 250 mM imida-zole, pooled, and dialyzed at 4°C against 10 mM NaCl in 10 mM Tris-HCl (pH 8.0). The dialyzed protein was concentrated using a Vivaspin concentrator (mo-lecular weight cutoff, 10,000) (Vivascience) and stored at 4°C.
RNA transcription plasmids.pT7BUNS5⬘(32)/3⬘(33) contains cDNA to the precise 32 5⬘-terminal bases and 33 3⬘-terminal bases of the negative-sense S segment, linked by a 5-base sequence which creates aSmaI site, under control of a T7 promoter in pUC119. BUNS5⬘(32)/3⬘(33) RNA was transcribed following linearization withBbsI, and BUNS-5⬘(32) RNA was transcribed following lin-earization withSmaI.
pTZBUNS3⬘(33/22) contains cDNA to the precise 33 3⬘terminal bases of the negative-sense BUN S segment under control of a T7 promoter and was con-structed by inserting a 63-bpSmaI-XbaI fragment from pT7BUNS5⬘(32)/3⬘(33) into pTZ18. pT7BUNS3⬘(33/22) RNA was transcribed following linearization of the plasmid withBbsI.
pT7BUNSCAT contains a negative-sense chloramphenicol acetyltransferase gene flanked by the entire untranslated regions of the BUN S segment in pUC119 (7). BUNS5⬘(65) RNA was transcribed following linearization of this plasmid withFauI, and BUNS5⬘(135) RNA was transcribed following lineariza-tion withTsp509I.
The template for transcription of BUNS5⬘SL2 RNA was generated by anneal-ing two oligonucleotides, 5⬘CTAATACGACTCACTATA (modified T7 pro-moter) and 5⬘CTAAATCAACATTATATTGTTAATGGTATTTTAATATAG TGAGTCGTATTAG, representing bases 23 to 56 of the BUN S 5⬘terminus.
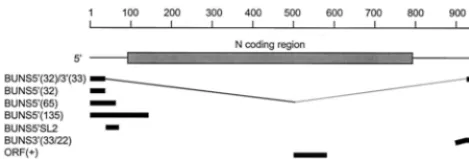
ORF(⫺) RNA was generated from a PCR product produced by amplification of the N ORF from nucleotides 577 to 86 of the cDNA to which a T7 promoter was incorporated, using primers 5⬘CTAATACGACTCACTATAGATCCCGA TTGCTAAGGG and 5⬘CTGCGAATTCATGATTGAGTTGGAATTTCACG. The product was linearized withMnlI and made blunt ended using T4 DNA polymerase before being added to a transcription reaction mixture to generate an 87-base RNA. ORF(⫹) RNA, also 87 bases in length, was generated from a PCR product by amplification of the N ORF from nucleotides 491 to 784 of the cDNA to which a T7 promoter was incorporated, using primers 5⬘CTAATACGACTC ACTATAGATGGAGAGGAAG and 5⬘CTGCGGATCCTTACATGTTGATT CCGAATTTAGC. The DNA was linearized withBstYI before being added to a transcription reaction mixture. To generate double-stranded ORF (dsORF) RNA, ORF(⫺) and ORF(⫹) RNAs were heated together at 95°C for 5 min and 65°C for 10 min, and then annealed by cooling to 25°C over 40 min. Figure 1 shows the regions of the BUN S segment represented by the in vitro-transcribed RNAs.
In vitro transcription reactions.RNA transcripts were generated using the Megashortscript T7 in vitro transcription kit (Ambion). BUNS5⬘(32) RNA was made under the standard reaction conditions recommended by the manufac-turer. All other unlabeled transcripts were synthesized in 20-l reaction volumes containing 3.75 mM each nucleoside triphosphate, 5 g of plasmid template DNA or 8g of oligonucleotide template DNA, 1⫻transcription buffer, and 1
l of enzyme mix (both Ambion) and incubated at 37°C for 4 to 6 h.32P-labeled
riboprobes were synthesized in similar reaction mixtures containing 3.75 mM each ATP, GTP, and UTP, 3.75M CTP, and 17 pmol of [␣-32P]CTP (3,000
Ci/mmol). All reaction mixtures were then treated with 1l of DNase I (Am-bion) for 15 min at 37°C, and unincorporated NTPs removed with RNA Mini Quick spin columns (Roche Molecular Diagnostics). The transcripts were puri-fied by acid phenol (pH 4.5)-chloroform extraction and ethanol precipitation in the presence of ammonium acetate on dry ice.
RNA-protein binding reactions.32P-labeled riboprobes were heated at 90°C
for 2 min and cooled on ice for 5 min before being incubated with purified N
protein in a 10-l volume binding reaction based on that of Go¨tt et al. (11) containing 1⫻binding buffer (10 mM HEPES [pH 7.3], 150 mM NaCl, 20 mM KCl, 5 mM MgCl2, 1 mM EDTA, 2 mM dithiothreitol [DTT], 10 U of rRNasin
[Promega]) for 20 min at 30°C; N protein was the last component to be added. Control reaction mixtures lacking N contained an equivalent volume of 1⫻
dialysis buffer. In competitive binding reactions the competitor RNA was mixed with the riboprobe in the reaction prior to the addition of N. For gel mobility shift analysis, glycerol was added to the binding-reaction mixtures to 12.5% and the reaction products were separated by electrophoresis on a 6% polyacrylamide gel plus 5% glycerol and 0.5⫻Tris-borate-EDTA (TBE) at 200 V for 2 h at 4°C or on a 1% agarose gel plus 0.5⫻TBE at 200 V for 1 h at 4°C. After drying, the gels were analyzed by autoradiography or by using a PhosphorImager (Bio-Rad). For filter-binding assays, 90l of 1⫻binding buffer was added and the reaction products were passed under vacuum through a BA85 nitrocellulose membrane filter (Schleicher & Schuell) that had been presoaked with 1⫻binding buffer. The filter was washed with 500l of 1⫻binding buffer under vacuum and dried, and radioactivity was determined by Cerenkov counting.
Western blotting.Purified N was subjected to sodium dodecyl sulfate-poly-acrylamide gel electrophoresis (SDS-PAGE) on a 15% polysulfate-poly-acrylamide gel and blotted onto a nitrocellulose membrane (Amersham Life Science) by using a semidry electrophoretic transfer apparatus. The membrane was blocked for 16 h with blocking buffer (phosphate-buffered saline [PBS] containing 10% nonfat milk and 0.1% Tween 20) and then incubated in blocking buffer for 1 h with rabbit serum raised against purified BUN (1:500 dilution). The membrane was washed four times with PBS–0.1% Tween 20, incubated with protein A-horse-radish peroxidase conjugate (1:1,000 dilution) in blocking buffer for 1 h, washed four times as before, and incubated twice in 50 mM Tris-HCl (pH 7.5) for 15 min each. Visualization was performed using enhanced chemiluminescence (Amer-sham Pharmacia Biotech).
Northwestern blotting.Purified N was subjected to SDS-PAGE and blotting as described above. The membrane was washed twice with PBS and blocked with PBS plus 5% nonfat milk and 1 mM DTT for 16 h, four times with 1⫻HBB (25 mM HEPES [pH 7.5], 25 mM NaCl, 5 mM MgCl2, 10 mM DTT) for 15 min each,
and once with hybridization buffer (RNA-protein binding buffer plus 0.5% Non-idet P-40) for 15 min. The membrane was then probed with 5 ml of hybridization buffer plus 5⫻105cpm of32P-labeled riboprobe and 40 U of rRNasin
(Pro-mega) at room temperature for 2 h, washed three times with hybridization buffer, and analyzed by autoradiography.
RNase treatment.32P-labeled BUNS5⬘(32)/3⬘(33) riboprobe was incubated in
the presence or absence of 800 ng of N protein in a standard binding reaction. RNase A was added to the concentrations specified, and the reaction mixtures were incubated at 37°C for 10 min. RNA was extracted with acid phenol (pH 4.5)-chloroform and precipitated with ethanol in the presence of ammonium acetate on dry ice. The RNA pellets were washed with 70% ethanol, resuspended in 5l of water, and boiled in 50% formamide, and the products were separated by PAGE on a 6% polyacrylamide sequencing-type gel at 300 V. After being dried, the gel was analyzed by autoradiography.
RESULTS
[image:2.612.57.292.74.153.2]Expression and purification of recombinant BUN N protein. The BUN N protein was expressed as an N-terminally 6-histi-dine-tagged protein in E. coliand purified under native con-ditions by nickel affinity chromatography. The bulk of N was eluted by approximately 250 mM imidazole. Eluted fractions were dialyzed, concentrated, and stored at 4°C, at which tem-perature N was typically stable for approximately 3 weeks. Analysis of the preparation by SDS-PAGE with subsequent staining with Coomassie blue showed a single band at the expected size for His-tagged N protein (data not shown). Fur-ther analysis by Western blotting using a rabbit polyclonal antiserum raised against purified BUN virus confirmed the identity of the protein as N (Fig. 2, lane 1). Analysis of the highly concentrated eluate from bacteria containing empty parent plasmid and purified under similar conditions did not yield any bands (lane 2). Western blot analysis of 10-fold-more N than that in lane 1 yielded two additional bands of higher molecular mass (lane 3). The intensity of these bands was unaffected following treatment of the protein with 5 mg of RNase A per ml prior to electrophoresis (lane 4). This con-centration of RNase A is more than sufficient to digest RNA in bunyavirus nucleocapsids. Thus, we suggest that these bands represent homodimers and homotrimers of N and were not the result of two or three N molecules binding to the same RNA molecule.
FIG. 1. Location of RNA probes used in binding assays relative to the BUN S genome segment. All of the transcripts were initiated with a truncated T7 promoter and did not contain additional, nonviral nucleotides at their 5⬘ends, except for BUNS3⬘(33/22) which had 22 nucleotides of vector sequence (bent line).
on November 9, 2019 by guest
http://jvi.asm.org/
RNA binding by N protein.The terminal sequences of each of the three bunyavirus genome segments have been impli-cated in encapsidation and are proposed to contain the site for nucleocapsid assembly (25). A 69-base riboprobe designated BUNS5⬘(32)/3⬘(33) was generated to investigate whether the recombinant N protein would bind the S-segment termini. This RNA corresponds to the exact terminal 32 bases of the 5⬘end and 33 bases of the 3⬘end of the genome-sense S segment (Fig. 1). Samples (100 pg) of radiolabeled BUNS5⬘(32)/3⬘(33) ribo-probe were incubated with increasing concentrations of N un-der reaction conditions based on those described by Go¨tt et al. (11) and then analyzed by polyacrylamide and agarose gel electrophoretic mobility shift assays (GEMSA), filter binding assays, and Northwestern blotting.
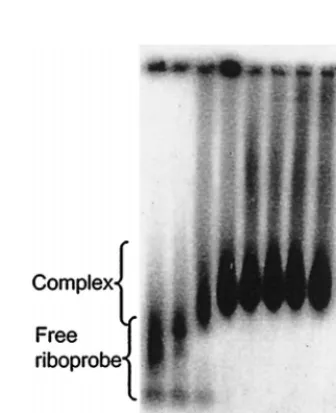
When GEMSA was performed using an acrylamide gel (Fig. 3), at low concentrations of N a small proportion of riboprobe was shifted into a single band of higher molecular mass. At higher concentrations of N the riboprobe was shifted into mul-tiple higher complexes, forming a ladder-like profile, which suggests sequential filling of binding sites on the RNA (3). The multiple bands are interpreted as the result of riboprobe being bound by discrete numbers of N molecules. Further addition of protein resulted in saturation of the riboprobe, which was shifted toward the top of the gel. To investigate the saturated complexes further, identical reactions were analyzed by aga-rose GEMSA (Fig. 4). In this assay the saturated complexes ran into the gel and could be seen to reach a finite maximum size. To confirm that the mobility shifts of the riboprobe were due to interaction with N, the protein was shown to bind BUNS5⬘(32)/3⬘(33) RNA directly by Northwestern blot anal-ysis (Fig. 2, lane 5). No binding of the riboprobe was observed in a mock expression lane in which bacteria containing the empty parent vector had been subjected to an identical induc-tion and purificainduc-tion regime (lane 6), suggesting that the shifts observed in the GEMSAs were not attributable to interaction with a native bacterial protein. When N was replaced by bovine serum albumen in binding reactions, no shift of the riboprobe was observed, indicating that the mobility shift was specific for the N protein (Fig. 4).
Effect of ionic concentration on binding.Binding reaction mixtures were assembled in the presence of different concen-trations of NaCl (0.15 to 2 M) and of MgCl2(0 to 20 mM), and
[image:3.612.348.516.453.659.2]the products were analyzed by agarose GEMSA (Fig. 5). Com-plex formation was not impaired by up to 0.5 M NaCl; at higher salt concentrations there was no evidence of dissociation of the complex, indicated by the lack of free riboprobe, but radioac-tivity was smeared further up the gel. We assume that this is an electrophoresis artifact caused by the high salt concentration in the sample. Binding of N to the riboprobe was unaffected at FIG. 2. Characterization of expressed, histidine-tagged BUN N protein. A
220-ng (lanes 1 and 5) or 2.2-mg (lanes 3 and 4) sample of purified N protein was blotted onto a nitrocellulose membrane following SDS-PAGE; extracts of bac-teria containing parent plasmid and subjected to the same purification protocol were run in lanes 2 and 6. The protein preparation in lane 4 was incubated with 5 mg of RNase A per ml before being loaded on the gel. Filters of lanes 1 to 4 were reacted with a polyclonal rabbit antiserum prepared against purified BUN and detected monomeric N (lane 1) and presumed oligomeric forms of N (lanes 3 and 4). No reaction with control lysate was observed (lane 2), and prior treatment with RNase A had no effect (lane 4). The filter of lanes 5 and 6 was reacted with32P-labeled BUNS5⬘(32)/3⬘(33) RNA, and the N band was revealed
following autoradiography (lane 5); no bands were detected on the blot of the extract from control bacteria (lane 6).
FIG. 3. Analysis of N-RNA binding by polyacrylamide GEMSA. A 100-pg sample of32P-labeled BUNS5⬘(32)/3⬘(33) RNA was incubated with different
concentrations of N protein, as indicated, before being subjected to analysis on a polyacrylamide gel. In the presence of increasing concentrations of N, the riboprobe was shifted into multiple higher complexes in a ladder-like profile. At high concentrations of N, the riboprobe was shifted toward the top of the gel.
FIG. 4. Analysis of N-RNA binding by agarose GEMSA. A 100-pg sample of
32P-labeled BUNS5⬘(32)/3⬘(33) RNA was incubated with different
concentra-tions of N protein, as indicated, before being subjected to analysis on an agarose gel. In the presence of N, the riboprobe was shifted into a higher band of finite maximum size. The two bands observed for the free riboprobe are thought to be due to secondary structure in the RNA. Replacement of N by bovine serum albumen (BSA) in the binding-reaction mixture did not result in a mobility shift of the probe.
on November 9, 2019 by guest
http://jvi.asm.org/
the MgCl2concentrations used (Fig. 5). These results suggest
that electrostatic interactions do not play a major role in the binding event. The fact that the N-RNA complexes were stable at high salt concentrations also suggests that the binding is strong.
Complexes are resistant to RNase.The RNA in bunyavirus nucleocapsids is relatively resistant to digestion by “reasonable concentrations” (12, 16) of RNase A but is digested by 100 g of RNase A per ml (19). To investigate whether in vitro-assembled N-RNA complexes were resistant to RNase A, BUNS5⬘(32)/3⬘(33) riboprobe was incubated in binding re-actions either with or without N and then different amounts of RNase A were added and incubation was continued for 10 min at 37°C. All reaction mixtures contained 20 U of RNasin RNase inhibitor (Promega), which is needed to inhibit any RNases present in the N protein stock (and thus would other-wise digest the RNA before N was able to bind); this amount of RNasin was considered to exert a nominal effect on the amount of RNase A added (1 U of RNasin inhibits 5 ng of RNase A by 50%; Promega). After phenol extraction the RNAs were analyzed by electrophoresis on a denaturing, se-quencing-type polyacrylamide gel. As shown in Fig. 6, RNA complexed with N was resistant to 1g of RNase A per ml whereas the naked riboprobe was almost fully digested at this concentration. Neither complexed nor naked riboprobe was resistant to 2.5g of RNase A per ml or higher concentrations.
Kinetics of N binding. The kinetics of binding of N to BUNS5⬘(32)/3⬘(33) RNA was measured by filter binding assays with a wider range of concentrations of N than that used in GEMSA. Binding-reaction mixtures containing 100 pg of ra-diolabeled riboprobe were passed through nitrocellulose mem-branes under vacuum. Whereas free riboprobe passed through the membrane, riboprobe complexed with N was immobilized on it. The degree of binding could thus be measured as the proportion of radiolabeled RNA retained on the membrane by its association with N (Fig. 7a). Maximal binding was obtained with 180 ng of N in the reaction, equivalent to a molar ratio of 1:1,500 (RNA to protein). The dissociation constant (Kd),
which was calculated as half-maximum binding, was approxi-mately 7⫻10⫺8M. Analysis of binding kinetics using a Hill
plot provides a mathematical calculation of the degree of co-operativity in the binding event (3). The gradient of the result-ing line serves as a measure of the number of sites that are bound cooperatively. Analysis of the binding data in Fig. 7a by this technique gave the results shown in Fig. 7c and yielded a gradient of approximately 1.2, indicating that the binding event showed a degree of cooperativity (3).
Competitive binding assays. The encapsidation signal for bunyavirus genome and antigenome segments is proposed to reside within the 5⬘-terminal sequences (12, 16, 25). We used a panel of RNAs in competitive filter binding assays to investi-gate the presence of such a signal. Binding-reaction mixtures were assembled containing radiolabeled BUNS5⬘(32)/3⬘(33) riboprobe in the presence of a 1,000-fold mass excess of unla-beled competitor RNAs, which were mixed prior to the addi-tion of N (Fig. 8). These experiments would therefore provide a measure of the degree of selectivity of N for the competitor FIG. 5. Effect of ionic strength on complex formation. Binding-reaction
mix-tures contained 100 pg of32P-labeled BUNS5⬘(32)/3⬘(33) RNA, 2,100 nM
puri-fied N protein, and different concentrations of NaCl or MgCl2, as indicated,
before being subjected to analysis by agarose GEMSA. Binding of N was evident, and no free riboprobe was observed for the range of NaCl and MgCl2
concen-trations tested. The increase in retardation coinciding with the increased NaCl concentrations is probably an effect of high salt concentrations on the movement of the complex through the gel.
FIG. 6. Effect of RNase on N-RNA complexes. Binding reactions were per-formed in mixtures containing 100 pg of32P-labeled BUNS5⬘(32)/3⬘(33) RNA
with or without 800 ng of N, and then different concentrations of RNase A were added and incubation was continued for 10 min at 37°C. Following phenol extraction and ethanol precipitation, the RNAs were resolved on a denaturing acrylamide gel. Complexes were resistant to 1g of RNase A per ml but not to 2.5g/ml, whereas most of the free riboprobe was digested by 1g of RNase A per ml.
FIG. 7. Binding kinetics of purified N protein to RNA. (a and b) A 100-pg 32P-labeled BUNS5⬘(32)/3⬘(33) (a) or BUNS5⬘(65) (b) RNA was incubated with increasing concentrations of N protein before being passed through a nitrocel-lulose filter. The retained labeled RNA was determined by scintillation counting and corrected by subtracting background counts of riboprobe in the absence of N. The two probes showed similar kinetics and yielded aKdof approximately 7⫻ 10⫺8M, calculated as equal to 1/2Vmax. Error bars indicate the standard devi-ation from three replicates. (c) Hill plot of binding of N to BUNS5⬘(32)/3⬘(33) RNA (solid diamonds) and BUNS-5⬘(65) RNA (open squares). Linear regres-sion was performed on the data points, yielding gradients of 1.2 for BUNS5⬘(32)/ 3⬘(33) RNA and 1.3 for BUNS-5⬘(65) RNA, indicating a low degree of cooper-ativity in both cases.
on November 9, 2019 by guest
http://jvi.asm.org/
RNA over BUNS5⬘(32)/3⬘(33) RNA. The results were ex-pressed as the percentage of competition shown by the com-petitor RNA for binding to N; a low value indicates that most of N is binding the riboprobe, and a high value indicates that there is competition for binding by the unlabeled RNA. To ensure that any competition observed was not due to loss of labeled riboprobe through RNase degradation, the competitor RNAs were tested for RNase contamination by incubation with the riboprobe, under binding conditions, followed by elec-trophoresis on a denaturing polyacrylamide gel and autora-diography. No evidence of RNase degradation of the ribo-probe was observed (data not shown).
A 1,000-fold molar excess of the homologous RNA, BUNS5⬘(32)/3⬘(33), gave about 50% competition, whereas yeast RNA competed to less than 20% (Fig. 8). Higher levels of competition were observed with competitor RNAs contain-ing the 5⬘ end of the BUN S segment. BUNS5⬘(32) and BUNS5⬘(135) RNAs, comprising the terminal 32 and 135 bases, respectively, reduced retention of riboprobe by about 75 to 80%. BUNS5⬘(65) RNA consists of the 5⬘ terminal 65 bases of the BUN negative-sense S segment and is thus a sim-ilar length to BUNS5⬘(32)/3⬘(33). In the presence of a 1,000-fold excess of this competitor, the proportion of riboprobe retained dropped by approximately 97%, indicating a high de-gree of competition. However, an RNA comprising an internal region of the 5⬘end, BUNS5⬘SL2 (bases 23 to 56), showed only 20% competition.
The binding kinetics of N to BUNS5⬘(65) RNA were mea-sured by a filter binding assay (Fig. 7b) and shown to be similar to those of BUNS5⬘(32)/3⬘(33). Analysis of the binding data with BUNS5⬘(65) RNA yielded a similar Hill plot to that of BUNS5⬘(32)/3⬘(33) RNA (Fig. 7c).
BUNS3⬘(33/22) consists of the 33 terminal bases at the 3⬘ end of the S segment and some vector sequences, and com-peted to a low level similar to that of yeast RNA. ORF(⫺) and ORF(⫹) RNAs comprise 87-base transcripts, genome and anti-genome sense, respectively, representing a region of the N ORF which encodes a highly conserved domain in the N protein. Neither of these RNAs competed more than yeast RNA. dsORF RNA was generated by annealing the two single-stranded ORF transcripts and was used to compare the preference of N for dsRNA and ssRNA. dsORF RNA
competed only slightly more than the ssRNAs and did not reach the high levels shown by competitors containing 5⬘ ter-minal sequences.
DISCUSSION
We have described experiments to study the RNA binding properties of BUN nucleocapsid protein in vitro. Recombinant BUN N protein was expressed as a His-tagged protein in bac-teria and purified by nickel affinity chromatography. N was reactive to antiserum raised against purified BUN and dem-onstrated binding activity for RNA containing the terminal sequences of the negative-sense S RNA segment, with a dis-sociation constant of 7⫻10⫺8M, similar to the value reported
for hantavirus N protein (29). RNA binding reactions in the presence of increasing concentrations of N, followed by poly-acrylamide GEMSA, indicated the presence of discrete com-plexes until the RNA was fully encapsidated. Although the observed pattern might represent binding of RNA to mis-folded N protein, we think this unlikely since multiple bands of appropriate sizes are seen on Western blots (Fig. 2), suggesting that N is capable of homo-oligomerizing. Analysis of the filter binding assay data by a Hill plot (Fig. 7) indicated a low (non-simultaneous) degree of cooperativity in the binding of N to the BUNS5⬘(32)/3⬘(33) RNA. Richmond et al. (27) reported RNA-binding by recombinant tomato spotted wilt virus N to be a cooperative event, as did Go¨tt et al. (11) for analysis of bacterially expressed hantavirus N protein binding, although neither group presented corroboratory data, for example in the form of a Hill plot.
RNA-N protein complexes showed a finite maximum size when analyzed on an agarose gel (Fig. 4). We interpret this as indicating that N binds along the RNA as opposed to the RNA associating with preformed multimers of N, whose size would be proportional to the concentration of N in the reaction mixture. The RNA-N complexes formed in the binding reac-tions were shown to be resistant to a level of RNase A diges-tion similar to that of authentic nucleocapsids, indicating that they possess similar properties.
We observed a discrepancy between theKdcalculated from
filter binding data and the equivalent degree of binding in GEMSA: the concentration of N required for 50% maximum binding in the filter binding assay was lower than that required in the gel shift assay. Since this was a consistent observation, we assume that it is because the N-RNA complexes were less stable during electrophoresis, particularly through a polyacryl-amide gel. Filter binding was therefore the preferred method for competitive binding assays to investigate the selectivity of binding by N. Interpretation of the results was complicated by the finding that even in the presence of a 1,000-fold excess of homologous unlabeled competitor, the retention of BUNS5⬘(32)/3⬘(33) riboprobe was decreased by only 50%. The unexpected lower level of competition observed with the ho-mologous RNA may be due to interactions between the 5⬘and 3⬘termini of BUNS5⬘(32)/3⬘(33) such that the labeled RNA is able to base-pair with unlabeled homologous RNA to form multimers, thereby reducing the sensitivity of the assay. This phenomenon would occur when the RNA possesses both ter-mini on the same strand.
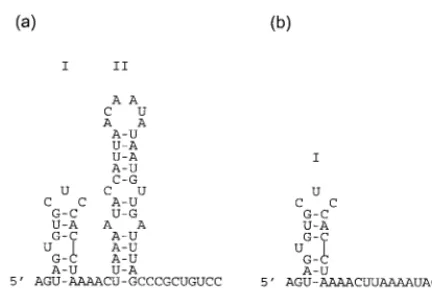
We were able to identify the first 32 bases of the 5⬘ negative-sense S segment as a region for which N possesses a preference over RNAs lacking this region. Analyses of the RNAs contain-ing 5⬘-terminal sequences by the Mfold program (18, 35) pre-dict the presence of a stem-loop structure, designated I, near the 5⬘ terminus (Fig. 9). It is possible that the observed pref-erence for binding is provided by this putative stem-loop,
FIG. 8. Competitive binding assays using a panel of RNAs. A 100-pg sample of32P-labeled BUNS5⬘(32)/3⬘(33) RNA and 100 ng of unlabeled competitor RNA were mixed, prior to the addition of 350 nM purified N, in a standard binding reaction. After incubation, the mixture was passed through a nitrocel-lulose filter and the retained labeled RNA was determined by scintillation count-ing and corrected by subtractcount-ing background counts of riboprobe in the absence of N. Results are expressed as percent competition. Thus, 100% competition would indicate that N bound the competitor RNA exclusively and 0% competi-tion would indicate that N bound the riboprobe exclusively. The competitor RNAs are described in Materials and Methods.
on November 9, 2019 by guest
http://jvi.asm.org/
which might act as a signal for N to begin binding the segment RNA. BUNS5⬘(65) is predicted to form a second stem-loop, II (Fig. 9a), and stem-loops similar to stem-loop II are predicted in the first 65 bases of the 5⬘ negative-sense termini of the other BUN segments (data not shown). However, RNA com-prising just stem-loop II (BUNS5⬘SL2 RNA) competed simi-larly to yeast RNA and is therefore not the sole contributor of the high degree of competition seen with BUNS5⬘(65). In addition, Mfold does not predict that BUNS5⬘(135) contains stem-loop II, although stem loop I is still predicted (data not shown). BUNS3⬘(33/22), an RNA possessing the 3⬘ terminus but lacking the 5⬘ terminus, did not demonstrate a greater degree of competition than yeast RNA, which suggests that the 3⬘terminus does not contribute significantly to the selectivity observed for BUNS5⬘(32)/3⬘(33). The preference of N for the 5⬘terminus is unlikely to be due to N binding dsRNA nonspe-cifically, since N did not possess a high preference for ORF dsRNA. Stem-loop I in the 5⬘terminus might also have impli-cations for the structure of the putative panhandle involving the ends of the RNA segment, particularly if this structure were retained after N binds, because loop I resembles the hook structures that have been proposed in the 5⬘termini of influ-enza virus and Thogoto virus RNAs (10, 17, 31).
Normally only full-length genome and antigenome segments are encapsidated in bunyavirus-infected cells, and hence a mechanism must exist to distinguish between these and viral mRNAs (or indeed cellular RNAs); it has been proposed that the presence of the additional primer-derived bases at the 5⬘ termini of viral mRNAs plays a role (25). We suggest that such additional bases may affect the secondary structure in the 5⬘ terminus, and experiments to test this hypothesis are under way. La Crosse bunyavirus N protein is reported to bind S segment-derived mRNA when N is present at high concentra-tions in infected cells, providing a feedback mechanism to regulate its own concentration (12), and this suggests that although authentic N is capable of binding RNAs other than genomes and antigenomes, it can only do so when present at high concentration. Competitive-binding experiments reported above suggest that BUN N is also capable of binding nonviral RNAs since yeast RNA could compete to a low but reproduc-ible degree (Fig. 8), which is presumably not sequence specific. Taken together, our observations may indicate two types of binding by N protein to RNA, selective or preferential binding to the 5⬘-terminal region and nonspecific binding. Other pro-teins have exhibited characteristics similar to those observed
with BUN N. Heterogeneous nuclear ribonucleoproteins are capable of both preferential binding to certain sequences and less sequence-specific binding at high concentrations, probably serving to hinder the formation of secondary structure in RNA (5, 6). Many positive-sense RNA virus core or coat proteins often possess the ability to selectively bind specific sequences (13, 15, 32, 34), sometimes binding hairpins in the 3⬘terminus, as well as binding RNA nonspecifically. Preferential binding to an encapsidation signal has also been observed for rabies virus (33) and human immunodeficiency virus (1).
The prediction of a binding site near the 5⬘ terminus for which N is highly selective is in agreement with the suggestion of Kolakofsky and colleagues (12, 25), who envisaged a sce-nario in which N binds a site in the 5⬘terminus of the segment RNA as it is being transcribed. Addition of the next N mono-mer could occur through its combined affinity for the RNA and the bound protein, and in this regard, homotypic interactions between tomato spotted wilt virus N proteins have already been reported (30). Encapsidation of the RNA as it is being transcribed removes the necessity for highly cooperative bind-ing unless the transcription elongation event is progressbind-ing more quickly than the binding of N to the nascent chain.
ACKNOWLEDGMENTS
We thank Carol Noonan for assisting with protein purification, Ewan Dunn for supplying some of the transcription plasmids, and Paul Yeo for helpful discussion.
J.C.O. was supported by a CASE studentship from BBSRC and Roche Products Ltd., and work in the laboratory of R.M.E. is sup-ported by grants from the Wellcome Trust and the MRC.
REFERENCES
1.Allen, P., B. Collins, D. Brown, Z. Hostomsky, and L. Gold.1996. A specific RNA structural motif mediates high affinity binding by the HIV-1 nucleo-capsid protein (NCp7). Virology225:306–315.
2.Bishop, D. H. L., M. E. Gay, and Y. Matsuoko.1983. Non-viral heteroge-neous sequences are present at the 5⬘ends of one species of snowshoe hare bunyavirus S complementary RNA. Nucleic Acids Res.11:6409–6418. 3.Black, D. L., R. Chan, H. Min, J. Wang, and L. Bell.1998. The
electro-phoretic mobility shift assay for RNA binding proteins, p. 109–136. In C. W. J. Smith (ed.), RNA:protein interactions, Oxford University Press, Oxford, United Kingdom.
4.Bouloy, M., N. Pardigon, P. Vialet, S. Gerbaud, and M. Girad.1990. Char-acterisation of the 5⬘and 3⬘ends of viral messenger RNAs isolated from BHK21 cells infected with Germiston virus (Bunyavirus). Virology75:50–58. 5.Burd, C. G., and G. Dreyfuss.1994. Conserved structures and diversity of
functions of RNA-binding proteins. Science265:615–621.
6.Burd, C. G., and G. Dreyfuss.1994. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J.13:1197–1204.
7.Dunn, E. F., D. C. Pritlove, H. Jin, and R. M. Elliott.1995. Transcription of a recombinant Bunyavirus RNA template by transiently expressed Bunyavi-rus proteins. Virology211:133–143.
8.Elliott, R. M. (ed.).1996. The Bunyaviridae. Plenum Press, New York, N.Y. 9.Eshita, Y., B. Ericson, V. Romanowski, and D. H. L. Bishop.1985. Analyses of the mRNA transcription processes of snowshoe hare bunyavirus S and M RNA species. J. Virol.55:681–689.
10. Flick, R., G. Neumann, E. Hoffmann, E. Neumeier, and G. Hobom.1996. Promoter elements in the influenza vRNA terminal structure. RNA2:1046– 1057.
11. Go¨tt, P., R. Stohwasser, P. Schnitzler, G. Darai, and E. K. F. Bautz.1993. RNA binding of recombinant nucleocapsid proteins of hantaviruses. Virol-ogy194:332–337.
12. Hacker, D., R. Raju, and D. Kolakofsky.1989. La Crosse virus nucleocapsid protein controls its own synthesis in mosquito cells by encapsidating its mRNA. J. Virol.63:5166–5174.
13. Hacker, D. L.1995. Identification of a coat protein binding site on southern bean mosaic virus RNA. Virology207:562–565.
14. Jin, H., and R. M. Elliott.1993. Characterization of Bunyamwera virus S RNA that is transcribed and replicated by the L protein expressed from recombinant vaccinia virus. J. Virol.67:1396–1404.
15. Khromykh, A. A., and E. G. Westaway.1996. RNA binding properties of core protein of the flavivirus Kunjin. Arch. Virol.141:685–699.
[image:6.612.66.282.69.222.2]16. Kolakofsky, D., and D. Hacker.1991. Bunyavirus RNA synthesis: genome FIG. 9. Predicted secondary structure of the 5⬘terminus of BUN S segment
RNA. Secondary structures of BUNS5⬘(65) (a) and BUNS5⬘(32) RNA (b) were predicted using Mfold (18, 35). The most energetically favorable structures are shown, and stem-loops I and II are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
transcription and replication. Curr. Top. Microbiol. Immunol.169:143–159. 17. Leahy, M. B., J. T. Dessens, and P. A. Nuttall.1997. Striking conformational similarities between the transcription promoters of Thogoto and influenza A viruses: evidence for intrastrand base pairing in the 5⬘promoter arm. J. Vi-rol.71:8352–8356.
18. Mathews, D. H., J. Sabina, M. Zuker, and D. H. Turner.1999. Expanded sequence dependence of thermodynamic parameters provides robust predic-tion of RNA secondary structure. J. Mol. Biol.288:911–940.
19. Obijeski, J. F., D. H. L. Bishop, E. L. Palmer, and F. A. Murphy.1976. Segmented genome and nucleocapsid of La Crosse virus. J. Virol.20:664– 675.
20. Pardigon, N., P. Vialat, M. Girard, and M. Bouloy.1982. Panhandles and hairpin structures at the termini of Germiston virus (Bunyavirus). Virology 122:191–197.
21. Patterson, J. L., and D. Kolakofsky.1984. Characterization of La Crosse virus small genome transcripts. J. Virol.49:680–685.
22. Pettersson, R. F., and C. H. von Bonsdorf.1975. Ribonucleoproteins of Uukuniemi virus are circular. J. Virol.15:386–392.
23. Qiagen.1997. The QIAexpressionist, 3rd ed. Qiagen, Hilden, Germany. 24. Raju, R., and D. Kolakofsky.1986. Translational requirement of La Crosse
virus S-mRNA synthesis: in vivo studies. J. Virol.61:96–103.
25. Raju, R., and D. Kolakofsky.1987. Unusual transcripts in La Crosse virus-infected cells and the site for nucleocapsid assembly. J. Virol.61:667–672. 26. Raju, R., and D. Kolakofsky.1989. The ends of La Crosse virus genome and
antigenome RNAs within nucleocapsids are base paired. J. Virol.63:122– 128.
27. Richmond, K. E., K. Chenault, J. L. Sherwood, and T. L. German.1998.
Characterization of the nucleic acid binding properties of tomato spotted wilt virus nucleocapsid protein. Virology248:6–11.
28. Samso, A., M. Bouloy, and C. Hannoun.1975. Presence de ribonucleopro-teins circulaires dans le virus Lumbo (Bunyavirus). C. R. Acad. Sci. Ser. D 280:779–782.
29. Severson, W., L. Partin, C. S. Schmaljohn, and C. B. Jonsson.1999. Char-acterization of the Hantaan nucleocapsid protein-ribonucleic acid interac-tion. J. Biol. Chem.274:33732–33739.
30. Uhrig, J. F., T.-R. Soellick, C. J. Minke, C. Philipp, J.-W. Kellmann, and P. H. Schreier.1999. Homotypic interaction and multimerization of nucleo-capsid protein of tomato spotted wilt tospovirus: identification and charac-terization of two interacting domains. Proc. Natl. Acad. Sci. USA96:55–60. 31. Weber, F., O. Haller, and G. Kochs.1997. Conserved vRNA end sequences of Thogoto-orthomyxovirus suggest a new panhandle structure. Arch. Virol. 142:1029–1033.
32. Wei, N., and T. J. Morris.1991. Interactions between viral coat protein and a specific binding region on turnip crinkle virus RNA. J. Mol. Biol.222:437– 443.
33. Yang, J., D. C. Hooper, W. H. Wunner, H. Koprowski, B. Dietzschold, and Z. F. Fu.1998. The specificity of rabies virus RNA encapsidation by nucleo-protein. Virology242:107–117.
34. Zhou, M., A. K. Williams, S.-I. Chung, L. Wang, and E. W. Collisson.1996. The infectious bronchitis virus nucleocapsid protein binds RNA sequences in the 3⬘terminus of the genome. Virology217:191–199.
35. Zuker, M., D. H. Mathews, and D. H. Turner.1999. Algorithms and ther-modynamics for RNA secondary structure prediction: a practical guide. NATO ASI Ser.70:11–43.