Crystal Structure of the Dimerized N Terminus of Porcine
Circovirus Type 2 Replicase Protein Reveals a Novel Antiviral
Interface
Gao Luo,a,bXiongfei Zhu,a,bYang Lv,bBomin Lv,bJin’e Fang,a,bShengbo Cao,a,bHuanchun Chen,a,bGuiqing Peng,a,b Yunfeng Songa,b
aState Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan, China bCollege of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
ABSTRACT Two replicase (Rep) proteins, Rep and Rep=, are encoded by porcine
circovirus (PCV) ORF1; Rep is a full ORF1 transcript, and Rep=is a truncated transcript generated by splicing. These two proteins are crucial for the rolling-circle replication (RCR) of PCV. The N-terminal sequences of Rep and Rep= are identical and interact to form homo- or heterodimers. The three types of dimers perform different func-tions during replication. A structural examination of the interfacing termini has not been performed. In this study, a crystal structure of dimerized Rep protein N termini was resolved at 2.7 Å. The dimerized protein was maintained by nine intermolecular hydrogen bonds and 15 pairs of hydrophobic interactions. The amino acid residue Ile37 participates in 11 of the hydrophobic interactions, mostly with its side chain. To find the predominant sites for protein dimerization and virus replication, a series of mutant proteins and virus replicons were generated by alanine substitution. Of all the single amino acid substitutions, the mutation at Ile37 showed the greatest effect on protein dimerization and virus replication. A double mutation at Leu35 and Ile37 almost eliminated protein dimerization and had the greatest negative effect on virus replication. These studies demonstrate that Leu35 and Ile37 are the most important residues for protein dimerization and are crucial for virus replication. Our results also show that PCV replication can be decreased by disrupting the dimerization of Rep or Rep= at the N terminus, suggesting that the structural interface responsible for dimerization offers a promising antiviral target.
IMPORTANCE Porcine circovirus type 2 (PCV2) is one of the most economically
damag-ing pathogens affectdamag-ing the swine industry. Although vaccines have been available for more than 10 years, the virus still remains prevalent. More effective strategies for disease prevention are clearly required. The Rep and Rep= proteins of the virus have identical N-terminal regions that interact with each other, allowing the formation of homo- or heterodimers. The heterodimer has crucial functions during different stages of viral repli-cation. Here, we resolved the crystal structure of the Rep (Rep=) dimerization domain. The individual residues involved in the intermolecular interaction were visualized in the protein structure, and several interactions were verified by mutant analysis. Our studies show that disrupting the interaction decreases viral replication, thus revealing a new tar-get for the design of antiviral agents.
KEYWORDS porcine circovirus type 2 (PCV2), replicase (Rep), dimerization, crystal
structure, antiviral interface
P
orcine circovirus (PCV) is a member of the genusCircovirusof the familyCircoviridae (1). Two types of PCV have been identified. Porcine circovirus type 1 (PCV1) is a nonpathogenic virus isolated from PK-15 cells (2, 3), while porcine circovirus type 2Received25 April 2018Accepted20 June 2018
Accepted manuscript posted online5 July 2018
CitationLuo G, Zhu X, Lv Y, Lv B, Fang J, Cao S, Chen H, Peng G, Song Y. 2018. Crystal structure of the dimerized N terminus of porcine circovirus type 2 replicase protein reveals a novel antiviral interface. J Virol 92:e00724-18. https://doi.org/10.1128/JVI.00724-18.
EditorRozanne M. Sandri-Goldin, University of California, Irvine
Copyright© 2018 American Society for Microbiology.All Rights Reserved.
Address correspondence to Yunfeng Song, [email protected].
crossm
on November 6, 2019 by guest
http://jvi.asm.org/
acids [aa], respectively) and in sequence (⬎80% amino acid identity) (13). The ORF1 gene can also generate a Rep= protein (168 aa in PCV1 and 178 aa in PCV2) by alternative transcript splicing (14). Rep and Rep=have a common N-terminal domain (118 aa) that contains three essential motifs for the initiation of virus replication. While the function of the rolling circle I (RC-I) motif is unknown, RC-II is involved in divalent ion coordination, and RC-III has nicking/joining activities (15). The C-terminal portion of Rep contains a P-loop motif with NTPase and a helicase domain, both of which are absent in Rep=(16, 17). Rep and Rep=can form homo- or heterodimers by association of their N termini to generate Rep-Rep, Rep-Rep=, and Rep=-Rep=(13, 18). The relative amounts of Rep and Rep= RNAs vary during replication, suggesting that the three complexes may play different roles at different stages of replication (19).
The PCV genome is copied by rolling-circle replication (RCR) (20). The replication origin contains a putative stem-loop structure in which a conserved nonamer is positioned at the apex (19). In contrast to the role played by Rep during RCR in Escherichia coli plasmids and geminiviruses, the PCV Rep protein cannot by itself promote virus replication. Instead, a Rep-Rep= complex is required (21). To initiate replication, the Rep-Rep=complex cleaves the DNA in the loop region and covalently binds to the 5=end. The cellular DNA polymerase then commences DNA synthesis at the free 3=OH extremity, displacing the strand bound to the Rep-Rep=complex. At the conclusion of replication, the newly synthesized DNA replaces the displaced strand to regenerate a double-stranded genome that can support additional rounds of replica-tion. The displaced strand is joined by the Rep-Rep= complex to generate a circular single-stranded molecule (17).
The two components of the Rep-Rep= complex have distinct roles during the process of replication. Rep is responsible for the initial steps that permit DNA synthesis to proceed, whereas Rep=joins the ends of the displaced strand to produce a closed circular genome (20). However, the interactions that allow Rep and Rep=to form homo-or heterodimers have not been explhomo-ored in detail. In this study, the crystal structure of the Rep (Rep=) dimerization domain was resolved to identify residues involved in the intermolecular association.
RESULTS
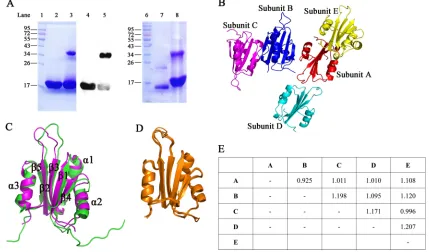
Expression and purification of the Rep N-terminal fragment.A fragment con-taining N-terminal amino acid residues 1 to 150 of the Rep protein (here designated RepN) was expressed in soluble form inE. coliBL21(DE3). The protein was purified by Ni2⫹affinity column chromatography, followed by gel filtration chromatography. As predicted from the sequence, the purified protein yielded a single band with a molecular weight (MW) of 18 kDa when subjected to SDS-PAGE (Fig. 1A, lane 2), but only if the protein sample was prepared by incubation at 100°C in the presence of dithiothreitol (DTT). Without this treatment, an additional band at 36 kDa was also observed (Fig. 1A, lane 3). In a Western blot assay, both bands were stained by His tag antibody (Fig. 1A, lanes 4 and 5). These results suggest that the Rep protein N-terminal fragment can form dimers and that an intermolecular disulfide bond can be formed between some paired monomers.
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 2
on November 6, 2019 by guest
Crystal structure of RepN protein. Although RepN contains 150 amino acid residues, electron density was not visible after Ser112 in the structural determination. A RepN protein crystal was dissolved in phosphate-buffered saline (PBS) and subjected to SDS-PAGE on a 12% gel. The molecular weight of the crystallized protein (Fig. 1A, lane 7) was significantly less than that of the purified RepN protein (Fig. 1A, lane 8). The molecular weight of the crystallized protein, as determined by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), was 12.8 kDa for the monomer and 25.5 kDa for the dimer. We therefore predicted that a cleavage at Ser112 had occurred during crystallization. A dimerized protein band was also present in the crystallized protein (Fig. 1A, lane 7), suggesting that the crystallized protein could still form a dimer with disulfide bond.
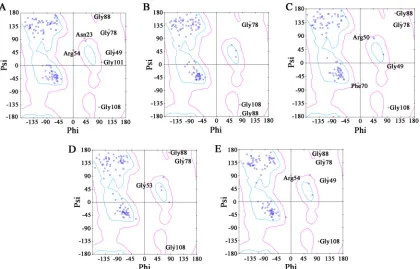
The structure of RepN was resolved by molecular replacement, and the resolution was refined to 2.7 Å. The RepN crystal belongs to space group C2 and contains five subunits (A to E) (Fig. 1B). The overall structure of the RepN monomer is defined by three␣-helices and five-strands. Ramachandran plot analysis of the five subunits was performed using Discovery Studio Visualizer (Biovia, San Diego, CA, USA). In the five subunits, there are a total of 25 amino acid residues in the disallowed region, and 20 of these are glycines; 7 residues are in the generously allowed region, and the rest (95.99%) are in the core region (Fig. 2 and Table 1). There are no obvious structural variations among the five subunits, and the root mean square deviations (RMSDs) between different subunits range from 0.925 to 1.207 Å (Fig. 1D and E). While the crystal structure has greater variability than the nuclear magnetic resonance (NMR)-resolved structure (PDB accession number2HWO) (22), the overall RMSD between the crystal structure and NMR structure is 1.970 Å. Large variations appear at the␣-helix 2 (Q68 to L77) and the lineage between-strands 2 and 3 (E48 to H57) (Fig. 1C); the
FIG 1Protein expression and crystal structure. (A) Purified RepN protein was mixed with 2⫻SDS loading buffer containing 0.1 M DTT and then heated to 100°C for 10 min (lane 2). Unheated protein without DTT was loaded in lane 3. The purified protein was identified by Western blotting with a His tag antibody. The DTT-treated protein was loaded in lane 4 while the sample without DTT was loaded in lane 5. The protein crystal was dissolved in PBS, mixed with 2⫻SDS loading buffer (without DTT and heat treatment), and then analyzed using SDS-PAGE (lane 7). Purified protein was loaded as a control (lane 8). (B) Five subunits (shown in different colors) were resolved in the crystallized protein. (C) The crystal structure was aligned with the NMR-resolved structure illustrating the structural similarity. The NMR structure is shown in green, and the crystal structure is shown in purple. (D) The five subunits in the resolved crystal structure were superimposed and showed high structural conservation. (E) Root mean square deviations (RMSDs; in angstroms) between each subunit are listed in the table.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.36.465.75.327.2]RMSDs of these two fragments are 2.154 and 2.144 Å, respectively. The␣-helices 1 and 3 and-strands 1 to 5 have relatively lower structural variation (Fig. 1C), and the RMSDs range from 0.624 to 1.522 Å.
There are four protein interaction patterns predicted by PISA (23) among the five subunits (Table 2), i.e., the dimers A⫹E or B⫹C, A⫹B, A⫹D, and B⫹D. The dimeric structures A⫹E and B⫹C share identical interaction patterns. They have the highest buried surface area and lowest solvation free energy upon formation of the interface (ΔiG) (Table 2). In addition, only this interaction pattern has an opportunity to form an
intermolecular disulfide bond. Thus, we conclude that the dimeric A⫹E or B⫹C is the actual pattern for protein interaction and use the B⫹C dimer as the basis for our subsequent protein interaction analyses.
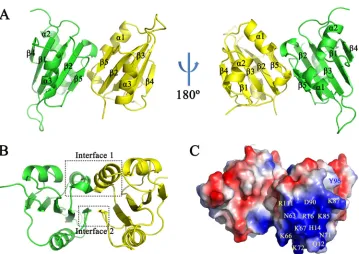
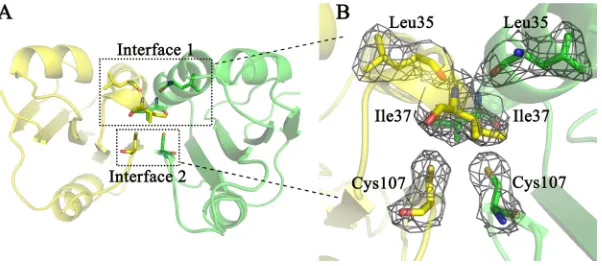
Interactions between dimeric subunits.Contacts between the two subunits of the RepN dimer occur at amino acids in␣-helix 1 and-sheet 5 (Fig. 3A). The buried surface area is 488.4 Å2in subunit B and 490.5 Å2in subunit C. The functional sites, such as
double-stranded DNA (dsDNA) binding and single-stranded DNA nicking, are on the solvent-accessible surface of the dimers (Fig. 3C). The intermolecular interaction is com-posed by hydrogen bonds (H bonds) and hydrophobic interactions. Two independent interfaces are observed in the dimeric structure (Fig. 3B). Interface 1 is composed of residues Arg33 to Ser38 and contains seven H bonds (Fig. 4A and B), while interface 2 includes six residues from Leu103 to Gly108 and contains two H bonds (Fig. 4D and E).
The intermolecular H bonds are predicted by PISA (23). Nine intermolecular hydro-gen bonds across the interface are identified between residues Arg33 and Ser38, Asp34 and Ser38, Leu35 and Ile37, and Glu106 and Glu108 (Table 3). Among the H bonds, six are identified as main chain-main chain H bonds, two are side chain-side chain H bonds, and one is main chain-side chain (Table 3). The intermolecular hydrophobic interactions are predicted by PSAIA (24), and as defined by PSAIA, hydrophobic interaction is an interaction between the hydrophobic amino acid residues with their side chains in 4.5-Å distance (24). Amino acid residues Leu35, Pro36, Ile37, Phe40, Phe43, and Ile105
FIG 2Ramachandran plot of the crystal structure. Panels A to E represent the individual Ramachandran plots of subunits A to E. Glycines are represented as triangles, prolines are shown as squares, and all other amino acid residues appear as circles. The areas within cyan lines are core regions, areas within pink lines are generously allowed regions, and the areas outside the pink lines are disallowed regions. Most of the amino acid residues in the disallowed area are glycines (20/25).
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 4
on November 6, 2019 by guest
[image:4.585.43.463.70.339.2]participate in hydrophobic interaction (Table 4). In total, 15 pairs of hydrophobic interactions are found in the interface (Table 4), and the Ile37 residues in both subunits participate in 11 of these interactions (Table 4). To get a more detailed understanding of these intermolecular interactions, a two-dimensional interface image was generated by Ligplot⫹ (25). Ligplot⫹ recognizes the sulfur atom as hydrophobic; hence, the Cys107 residues are shown to participate in the hydrophobic interactions (Fig. 5) whereas they do not according to PSAIA (Table 4). As shown in Fig. 5, the Ile37 residues participate in hydrophobic interactions mostly via their side chain atoms (CD1, CG1, and CG2). In the SDS-PAGE analysis, some crystallized protein is found to form disulfide bonds (Fig. 1A, lane 7). We did not detect a dimeric state with an intermolecular disulfide bond; however, the Cys107 residues in the two chains are probably close enough to form a disulfide bond if rotation of the C-S bond occurs (Fig. 6B).
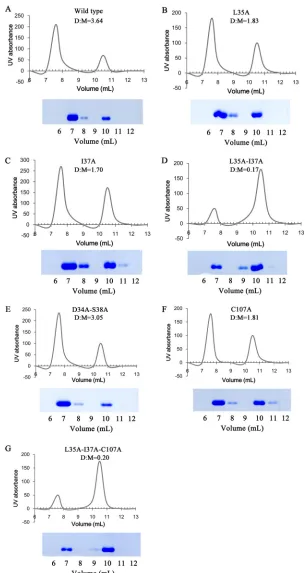
[image:5.585.40.379.83.363.2]A series of mutant proteins, single mutants L35A, I37A, and C107A, double mutants L35A I37A and D34A S38A, and the triple mutant L35A I37A C107A, were constructed to investigate the predominant factors for protein dimerization. All mutant proteins were expressed inE. coliBL21(DE3) and purified by Ni2⫹affinity column chromatog-raphy and then concentrated to 4 mg/ml. Five hundred microliters of these proteins was loaded onto a Superdex 75 gel filtration column (GE Life Sciences) and eluted under a flow rate of 0.5 ml/min. The amounts of protein monomer and dimer were
TABLE 1Crystallographic data and refinement statistics
Parameter Valuea
Data collection
Space group C2
Cell parameter (Å)
a, b, c 84.93, 71.21, 131.72
␣,,␥ 90.00, 104.34, 90.00
Wavelength (Å) 1.0
Resolution range (Å) 50–2.7
No. of reflections 46,536
Completeness (%) 97.9 (99.0)
Rmerge(%) 8.1 (59.7)
I/ 12.2 (2.0)
Redundancy 4.4 (4.3)
Refinement statistics
Resolution (Å) 50–2.7
Rwork/Rfree(%) 22.4/28.9
No. of atoms 4,107
Protein 4,043
Solvent 64
RMSD
Bond length (Å) 0.009
Bond angle (°) 1.515
Ramachandran plot (%)
Core 95.99
Allowed 2.91
Disallowed 1.10
aValues in parentheses are for the last shell.
TABLE 2Overall analysis of the protein interface
Interfacing
structures Buried area (Å2) ⌬iG (kcal/mol)a
No. of H bonds
No. of salt bridges
No. of disulfide bonds
A⫹E 493.4 ⫺3.2 9 0 0
B⫹C 489.4 ⫺3.3 9 0 0
A⫹B 381.5 ⫺1.0 6 5 0
A⫹D 367.4 2.7 4 1 0
B⫹D 72.4 1.3 2 2 0
aΔiG, solvation free energy gain upon formation of the interface.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.41.370.657.729.2]monitored by UV absorbance. The eluted proteins were then subjected to SDS-PAGE after incubation at 100°C for 10 min in the presence of DTT. The amount of dimer or monomer was estimated by calculating the area under each peak in the chromato-grams; the ratios of dimer to monomer (D/M) are indicated in Fig. 7. In the wild-type protein, the ratio between the dimer and monomer was 3.64 (Fig. 7A). Of the single FIG 3Structural overview of the dimerized protein. (A) Secondary structure of the dimerized protein. The monomers contact each other at␣-helix 1 and-sheet 5. (B) The protein dimer has two interfaces; interface 1 is composed of residues from Arg33 to Ser38, and interface 2 is composed of residues from Leu103 to Gly108. (C) The electrostatic surface was generated by PyMOL. Blue represents positive-charged regions, and red indicates negative-charged regions. The dsDNA binding-related residues are indicated in yellow text. Tyr96 (Y96), which is responsible for single-stranded DNA nicking and covalent binding, is indicated in blue text.
FIG 4Stereoscopic view of interfaces 1 and 2. The residues participating in intermolecular H bonds in interface 1 (A and B) and interface 2 (D and E) are shown as sticks. Carbon atoms are distinguished between the subunits using green and yellow. Nitrogen atoms are shown in blue, oxygen atoms are in red, and sulfur atoms are in yellow. Intermolecular H bonds are indicated by black dashes (B and E). Electron density maps of the two interfaces are shown in panels C and F. The contour level at 1.0 sigma was generated for a 2Fo⫺Fcmap.
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 6
on November 6, 2019 by guest
[image:6.585.43.402.66.320.2] [image:6.585.41.401.491.687.2]amino acid substitutions, I37A had the greatest impact on protein dimerization, with a D/M of 1.7 (Fig. 7C), although L35A and C107A also had some effect (Fig. 7B and F). Monomer levels observed for the double mutation D34A L38A were similar to those of the wild-type protein (D/M of 3.05) (Fig. 7F). Notably, the double mutation L35A I37A resulted in a significant change in the dimer-monomer ratio (D/M of 0.17), with most of the protein migrating as a monomer (Fig. 7D). A similar result was obtained with the triple mutation L35A I37A C107A (Fig. 7G). These results indicate that Ile37 is a critical residue for Rep dimerization and that the effect will be enhanced when Ile37 interacts with Leu35.
Enzyme activities in wild-type and mutant proteins.The full-length Rep protein exhibits ATPase and helicase activities. To determine whether these activities were present in the mutant proteins, the full-length wild-type Rep protein and three mutants (D34A L38A, C107A, and L35A I37A C107A) fused with a SUMO tag, were expressed in E. coli and purified by Ni2⫹affinity column chromatography. Enzyme activities were then compared in ATP hydrolysis and DNA unwinding assays. As shown in Fig. 8, ATPase and helicase activities were unaffected by any of the mutations tested. Since the triple mutant (L35A I37A C107A) showed no change in enzyme activities, single or double mutations at Leu35 and Ile37 would probably not affect helicase or ATPase activities of the full-length protein.
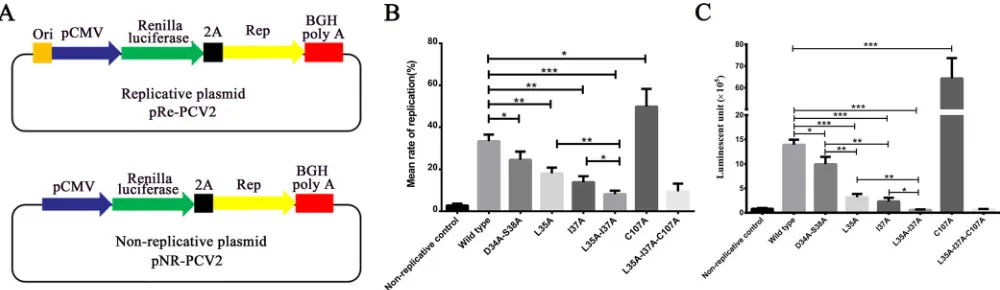
[image:7.585.42.371.82.199.2]Replication assay.To assess the role of Rep dimerization in virus replication, a PCV2 replicon carrying a luciferase reporter gene was constructed (pRe-PCV2) (Fig. 9A).
TABLE 3Residues and atoms participating in intermolecular hydrogen bonds
Subunit C Subunit B
Distance (Å) Type Positiona Residue Atomb Position Residue Atom
33 Arg O 38 Ser N 2.75 Main chain-main chain 34 Asp OD1 38 Ser OG 2.90 Side chain-side chain 35 Leu O 37 Ile N 2.95 Main chain-main chain 37 Ile N 35 Leu O 2.80 Main chain-main chain 38 Ser N 33 Arg O 2.80 Main chain-main chain 38 Ser OG 33 Arg O 3.34 Side chain-main chain 38 Ser OG 34 Asp OD1 2.45 Side chain-side chain 106 Glu O 108 Gly N 2.78 Main chain-main chain 108 Gly N 106 Glu O 2.78 Main chain-main chain
aPosition is the amino acid number in the Rep protein, starting from the methionine.
bAtom designations conform to standard International Union of Pure and Applied Chemistry (IUPAC)
nomenclature for amino acids. The first letter represents the name of the atom, and the second letter and number indicate the position of the atom in the amino acid.
TABLE 4Residues participating in hydrophobic interaction in the protein interfacea
Subunit B Subunit C
Positionb Amino acid Position Amino acid
35 Leu 35 Leu
35 Leu 36 Pro
35 Leu 37 Ile
36 Pro 35 Leu
36 Pro 36 Pro
36 Pro 37 Ile
37 Ile 35 Leu
37 Ile 36 Pro
37 Ile 37 Ile
37 Ile 40 Phe
37 Ile 43 Phe
37 Ile 105 Ile
40 Phe 37 Ile
43 Phe 37 Ile
105 Ile 37 Ile
aHydrophobic interactions involving Ile37 are in boldface.
bPosition is the amino acid number in the Rep protein, starting from the methionine.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.41.370.547.721.2]Mutations were introduced by overlap extension PCR to yield six mutant replicons (L35A, I37A, L35A I37A, D34A S38A, C107A, and L35A I37A C107A). The replicon plasmids, as well as a nonreplicative control, were transfected into PK-15 cells. At 72 h posttransfection, the cells were collected for real-time quantitative PCR (qPCR) and luciferase assays. The wild-type replicon generated 12-fold more viral DNA and had 17-fold more luciferase activity than the nonreplicative plasmid (P⬍0.001), demon-strating that the replicon plasmid replicates effectively in the cells (Fig. 9B and C). Compared to levels with the wild-type replicon, the mutation I37A resulted in a 2.4-fold decrease in DNA (P⬍0.01) and a 6-fold decrease in luciferase activity (P⬍0.001), while L35A resulted in 1.8-fold (P⬍ 0.01) and 4.3-fold (P⬍ 0.001) decreases in DNA and luciferase activity, respectively (Fig. 9B and C). The double mutation L35A I37A resulted in a significant decrease in both DNA replication (4.1-fold reduction;P⬍0.001) and luciferase activity (23-fold reduction;P⬍0.0001) (Fig. 9B and C). The D34A S38A double mutation had some negative effect on virus replication but not as much as the L35A or I37A mutation. In addition, the mutation C107A resulted in significantly enhanced virus DNA replication and luciferase activity; this enhancement was not observed in the L35A I37A C107A mutant (Fig. 9B and C).
DISCUSSION
In the last 20 years, PCV2 has spread rapidly across the world and caused consid-erable economic losses to the pork industry. Although commercial vaccines have been
FIG 5Intermolecular H bonds and hydrophobic interactions in a two-dimensional view. Residues participating in H bonds are modeled in a ball-and-stick style. The green dashes represent intermolecular hydrogen bonds, and distances (in angstroms) between atoms are superimposed on the dashed lines. Residues involved in hydrophobic contacts are indicated by arcs with spokes. Atom designations conform to standard International Union of Pure and Applied Chemistry (IUPAC) nomenclature for amino acids. The first letter represents the name of the atom, and the second letter and number indicate the position of the atom in the amino acid.
FIG 6Spatial location of Cys107. (A) Cys107 is located in interface 2, while Leu35 and Ile37 are located in interface 1. However, Cys107 is near Leu35 and Ile37 in the secondary structure. (B) The cysteine residues in the subunits are close to each other in the dimer. The distance between the sulfur atoms is 4.9 Å. Disulfide bond formation should be possible with rotation of the C-S bond.
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 8
on November 6, 2019 by guest
[image:8.585.42.542.77.217.2] [image:8.585.57.358.566.699.2]FIG 7Analysis of protein interaction by mutagenesis. A series of mutations was introduced into the protein. The expressed proteins were purified by Ni2⫹affinity column chromatography, concentrated to 4 mg/ml, and loaded onto a Superdex 75 column. The eluted protein in each fraction was monitored by UV absorbance (yaxis). Thexaxis represents the elution volume. The eluted proteins were mixed with SDS loading buffer with DTT, heated to 100°C for 10 min, and identified by SDS-PAGE. The numbers under the SDS-PAGE images correspond to the elution volume (xaxis). The amount of monomer or dimer protein was estimated by calculating the area under each peak in the chromatograms, and the ratio between dimer and monomer (D/M) is indicated in each panel.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:9.585.51.356.69.642.2]deployed for more than 10 years, the virus is still prevalent in swine herds (26) as new variants with increased virulence have emerged and spread (27). Improved antiviral strategies are required to stanch the spread and infection of PCV2.
Because Rep and Rep=are essential for virus replication, they are potential targets for virus-specific antiviral drug design. Rep and Rep=interact at their N termini to form homo- or heterodimers. Although the roles of the Rep-Rep and Rep=-Rep=homodimers remain unclear, the Rep-Rep=heterodimer has proved to be crucial for several steps in virus replication (28). The interface between interacting proteins has been proposed as promising targets for antiviral chemotherapy (29, 30). Many drugs directed to the interfaces of various proteins have been approved or are at different phases of clinical trials (31–33). In this study, we demonstrated a correspondence between protein dimerization and virus replication. Mutations that decreased the ability of Rep to dimerize also reduce virus replication. Our results suggest that the Rep dimer interface is a promising antiviral target. Our study also provides detailed structural information concerning the interface that will be valuable for designing antiviral inhibitors.
The N terminus of the Rep protein has two DNA binding sites that are important for initiating replication. Tyr96 cleaves a single-stranded DNA target by nucleophilic attack with its hydroxyl group and then forms a phosphotyrosine diester to bind at the 5=end of the DNA (16). Meanwhile, the N terminus of Rep also binds noncovalently to the FIG 8Effect of mutations on the helicase and ATPase activities of full-length Rep protein. (A) Helicase activities of the wild-type (wt) and mutant proteins. (B) ATPase activities of the wild-type and mutant proteins. The bars represent the standard deviations of three assays. The mutations had no significant (ns) effects on the ATPase or helicase activities of the full-length Rep protein.
FIG 9The effect of mutations on virus replication. (A) Diagram of the PCV2 replicon. A fragment containing the PCV2 origin of replication, the CMV promoter, and sequences encoding theRenillaluciferase gene, FMDV 2A, PCV2 Rep, and the BGH polyadenylation signal were inserted into the pMD18-T vector to generate a replicative plasmid, pRe-PCV2. A nonreplicative plasmid, pNR-PCV2, was constructed as a control by excluding the origin of replication. (B) Determination of replicon replication level by real-time qPCR. (C) Determination of replication level byRenillaluciferase assay. The bars represent the standard deviations of three assays.*,P⬍0.05;**,P⬍0.01;***,P⬍0.001.
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 10
on November 6, 2019 by guest
[image:10.585.55.356.70.224.2] [image:10.585.43.543.542.687.2]double-stranded hexanucleotide H1/H2 direct repeats on the right arm of the stem-loop (14). Numerous residues in the N terminus of Rep, Gly10, Gln12, His14, Arg16, Asn63, Phe64, Lys66, Lys67, Asn71, Lys72, Val73, Asp90, Arg111, Gln115, and Arg116, are involved in the binding of dsDNA (22). We localized the positions of these residues in the dimeric structure. Tyr96 was found on both sides of the dimer surface. The dsDNA binding-related residues together constitute a region of strong positive charge at the top and bottom of the protein (Fig. 3C). All the functional sites within the N terminus of Rep are located in a solvent-accessible region of the dimeric protein, indicating that the dimerization of the protein offers no steric hindrance to the nicking and binding of substrate DNA.
When we analyzed the purified RepN protein by SDS-PAGE, two protein bands (18 kDa and 36 kDa) were observed (Fig. 1A), and both were recognized by the His tag antibody in a Western blot assay (Fig. 1A). Heating the sample with DTT caused the 36-kDa protein band to disappear (Fig. 1A, lanes 2 and 4). A protein sample derived from a crystal also generated dimeric and monomeric bands when subjected to SDS-PAGE (Fig. 1A, lane 7). In the crystal structure, the Cys107 residues of both monomer subunits are in close proximity to each other in the dimer interface. The intermolecular disulfide bond was completely eliminated in the C107A mutant, indi-cating that the native Cys107 residue in the Rep N terminus can form a disulfide bond in the protein dimer. Usually, the cytoplasm of a eukaryotic cell is stated to be a reducing environment, and the disulfide bonds in cytoplasm tend to be broken rather than formed (34). Interestingly, the infection of PCV2 can caused oxidative stress in cells, and the reactive oxygen species (ROS) increased in a time-dependent manner in PCV2-infected cells (35, 36). The ROS in cells was reported to increase the level of disulfide-bonded proteins (DSBPs) in a mammalian cell cytoplasm (37). Thus, we consider that some protein dimers can still form a disulfide bond in cells during virus replication. But this bond does not promote virus replication. As reported previously, the homo- and heterocomplexes are not equally abundant and may have distinct roles at specific stages during virus replication (19). If the different complexes are in dynamic equilibrium, the formation of disulfide bonds between subunits may block dissociation and prevent the monomer subunits from participating in the formation of other dimers. This may be why the C107A mutation enhances the replication of PCV. These results suggest that eliminating the potential disulfide bond between Cys107 residues might be a possible approach to increase PCV2 titers in cells.
The intermolecular interaction analysis showed that the dimeric protein is main-tained by hydrophobic interactions and H bonds. Ile37 is located in the center of the interface and is involved in more hydrophobic interactions than other residues on the interface, with most of the hydrophobic forces imposed on its side chain (Fig. 5). When Ile37 was replaced with Ala, effectively eliminating the side chain, dimerization was significantly impaired (Fig. 7C). Leu35 interacts with Ile37 hydrophobically and by H bonding, and it should be an important factor in the interaction between monomers. The double mutation at Leu35 Ile37 affects protein dimerization and virus replication more than either I37A or L35A alone. We also tested the double mutation D34A S38A; these two sites have less hydrophobic interaction but participate in five intermolecular H bonds (Fig. 5 and Table 4). The D34A S38A mutation did not decrease protein dimerization or virus replication as much as the single mutation I37A or double mutation L35A I37A. These results suggest that the primary factor driving Rep protein dimerization is hydrophobic interaction, while the intermolecular H bonds stabilize the dimer.
We present a model of the Rep-Rep= complex binding to the PCV replicating genome that incorporates the structural data from this study as well as current information concerning Rep and Rep=function (Fig. 10). In this model, Rep=nicks and covalently binds the positive DNA strand via Tyr96, and the Rep C-terminal domain binds noncovalently to the negative strand. Rep= is responsible for the nicking and joining of the viral DNA, while Rep unwinds the dsDNA in a 3=-to-5=direction. The
on November 6, 2019 by guest
http://jvi.asm.org/
cellular DNA polymerase binds to the free 3=end of the positive strand and conducts leading strand synthesis.
In summary, we have resolved the structure of the dimerized N terminus of the PCV Rep protein and identified the amino acid residues that participate in the interaction between monomer subunits. Modifications that affected protein dimerization also decreased virus replication. The interface between monomers is therefore a potential target for the development of anti-PCV agents.
MATERIALS AND METHODS
Expression and purification of the N terminus of Rep.The DNA fragment encoding amino acids 1 to 150 at the N terminus of the Rep protein (RepN) was amplified by PCR using two specific primers (Table 5, primers 1 and 2). The PCR product was then inserted into the NdeI and XhoI sites of the prokaryotic expression vector pET-30a (Novagen, Darmstadt, Germany), yielding pET30a-RepN. The resulting construct encodes RepN with a 6⫻His tag at its C terminus without a flexible linker.E. coli BL21(DE3) competent cells were transformed with this plasmid, incubated until the culture reached an optical density at 600 nm (OD600) of 0.8, and then induced with 0.5 mM isopropyl -D -1-thiogalactopyranoside (IPTG) at 37°C for 4 h. The expressed protein was purified by Ni2⫹affinity column chromatography, followed by gel filtration chromatography. The purified protein was identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
[image:12.585.102.306.68.240.2]FIG 10A model of Rep-Rep=complex binding to the PCV replicating genome. The Rep and Rep=proteins interact with each other by their N-terminal domains (dark cyan). Rep=covalently binds to the 5=end of the positive strand of the viral genome by Tyr96. The C-terminal domain of Rep (purple) noncovalently binds to the negative strand of the viral genome and unwinds the parental double-stranded genome. The cellular DNA polymerase (yellow) initiates the new progeny DNA strand (blue) synthesis from the free 3=OH extremity.
TABLE 5Primers for the PCR, site-directed mutagenesis, and real-time qPCR
No. Primer name Primer sequence (5=–3=)a
1 RepN-s ATCATATGCCGAGCAAGAAGAATGGA 2 RepN-a ATCTCGAGCAGCCCGCGGAAATTTCT 3 Rep-OE-s CGCGGATCCATGCCCAGCAAGAAG 4 Rep-OE-a CCGCTCGAGTCAGTAATTAATTTCAT
5 L35A-s GCAAGAAAATACGGGATGCTCCAATATCCCTATTTGAT 6 L35A-a ATCAAATAGGGATATTGGAGCATCCCGTATTTTCTTGC 7 I37A-s ATACGGGATCTTCCAGCATCCCTATTTGATTATTTT 8 I37A-a AAAATAATCAAATAGGGATGCTGGAAGATCCCGTAT 9 L35A-I37A-s GGGATGCTCCAGCATCCCTATTTGATTATT 10 L35A-I37A-a AGGGATGCTGGAGCATCCCGTATTTTCTTG
11 D34A-S38A-s CGGGCTCTTCCAATAGCTCTATTTGATTATTTTATTGTTGGCG 12 D34A-S38A-a GAGCTATTGGAAGAGCCCGTATTTTCTTGCGCTCG 13 C107-s ACTGATCGAGGCGGGAGCTC
14 C107-a GAGCTCCCGCCTCGATCAGT 15 qPCR-Rluc-s TGGGATGAATGGCCTGATAT 16 qPCR-Rluc-a ATGCTGCAAATTCTTCTGGTTC
aDigestion sites are underlined.
September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 12
on November 6, 2019 by guest
[image:12.585.43.373.560.729.2]Western blotting. The protein was transferred from an SDS-PAGE gel to a nitrocellulose filter membrane (Millipore, Darmstadt, Germany). Western blotting was performed with 6⫻His tag monoclonal antibody (Invitrogen, Grand Island, NY, USA), followed by horseradish peroxidase-conjugated goat anti-mouse IgG (Boster, Wuhan, China) as the secondary antibody. Binding was detected using Super-Signal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA) and visualized using an MF-ChemiBIS, version 3.2, chemiluminescence imaging system (DNR Bio-Imaging Systems, Mahale Hahamisha, Israel).
Crystallization, data collection, and structure determination.Purified RepN protein was concen-trated to 16 mg/ml using a centrifugal filter unit (Millipore, Billerica, MA, USA). Three commercial crystallization kits (HR2-110, HR2-114, and HR2-144; Hampton, Aliso Viejo, CA, USA) were screened by 1:1 mixing with the protein via the hanging-drop vapor diffusion method at 4°C. Crystals were acquired under three different conditions: 0.1 M Bis-Tris, pH 6.5, 45% polypropylene glycol P 400; 0.1 M HEPES sodium, pH 7.5, 0.2 M sodium citrate tribasic dihydrate, 30% (⫹/⫺)-2-methyl-2,4-pentanediol; 0.1 M sodium cacodylate trihydrate pH 6.5, 0.2 M magnesium acetate tetrahydrate, 30% (⫹/⫺ )-2-methyl-2,4-pentanediol. Conditions were then optimized by changing the concentration of salt and precipitant and by varying the pH. The best crystal was obtained using 0.1 M HEPES sodium pH 8.0, 0.45 M sodium citrate tribasic dihydrate, and 50% (⫹/⫺)-2-methyl-2,4-pentanediol.
Liquid nitrogen was used to flash-cool crystals in a cryoprotectant solution containing 30% ethylene glycol and 70% reservoir solution. Data collection was performed on a 3W1A beamline (wavelength, 1.0 Å; temperature, 100 K) at the Beijing Synchrotron Radiation Facility (BSRF) using a mar345 detector. During data collection, the distance from the crystal to the detector was 150 mm, and the oscillation angle was from 0 to 209° with a frame width 1.0. The exposure time was set to 30 s. All diffraction images were integrated, merged, and scaled using HKL-3000 software (38). Initial phases were obtained by molecular replacement with an NMR-resolved structure of the PCV2 Rep nuclease domain (PBD accession number2HWO) (22) using Phaser. Model building was initially performed automatically with ARP/wARP (39). Manual model rebuilding was accomplished using COOT (40), and subsequently the structure was refined using the PHENIX program (41). Structural figures were drawn using PyMOL (42).
Site-directed mutagenesis by overlap extension PCR. The mutations were designated using standard nomenclature (e.g., L35A indicates that leucine has been replaced by alanine at amino acid residue 35). Six mutants (L35A, I37A, C107A, D34A S38A, L35A I37A, and L35A I37A C107A) were generated by overlap extension PCR as previously described (43). Using the L35A mutation as an example, two separate PCRs were performed using primers 3 and 6 and primers 4 and 5 (Table 5). The two PCR products were isolated by agarose gel electrophoresis and recovered using by a gel extraction kit (Omega Bio-Tek, Norcross, GA, USA). A third PCR was conducted using equimolar amounts of the two products as templates with primers 3 and 4 (Table 5) to generate the L35A mutant gene fragment. All mutations were verified by sequence analysis.
Helicase activity assay.The helicase activity of the wild-type and mutant Rep proteins was analyzed using a fluorescence resonance energy transfer (FRET) assay as described previously (44). Briefly, two cDNA molecules were synthesized. One was a 3=BHQ-2-labeled 22-mer (5=-GGTTCTGAGGGTGGCGGTA CTA-3=), and the other was a 5=Cy5-labeled 37-mer (5=-TAGTACCGCCACCCTCAGAACCTTTTTTTTTTTTTT T-3=). A 15-nt overhang is generated when the two strands anneal. A capture strand (5=-TAGTACCGCC ACCCTCAGAACC-3=) was employed to prevent reannealing of the two separated substrate DNA strands generated during the unwinding assay. The substrate DNAs for the FRET assay were combined at a 1.2:1 (BHQ-2/Cy5) molar ratio. The mixture was heated briefly to 95°C and then cooled slowly in a 37°C incubator. Annealing efficiency was evaluated by fluorescence (excitation, 635 nm; emission, 670 nm). In the unwinding activity assay, 3M purified Rep or mutant protein in reaction buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, and 10 mM MgCl2) was added to a 96-well black plate and combined with 240 nM annealed DNA substrate and 1.2M capture strand. The reaction was initiated by adding 2.5 mM ATP per well. After incubation at 37°C for 60 min, fluorescence was measured for each well.
ATPase activity analysis.The ATPase reaction was conducted using 3M full-length protein and 100M ATP in reaction buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2). After incubation at 37°C for 30 min, an equal volume of Kinase-Glo Plus reagent (Promega, Madison, WI, USA) was added to each well to quantitate the remaining ATP. Luminescence was measured for each well using an Infinite 200 Pro multimode reader (Tecan, Männedorf, Switzerland). ATPase activity was calculated as follows: 1⫺(Lnp⫺Lex)/Lnp, whereLnpis the luminescence generated by the nonprotein control, andLexis the luminescence generated by the experimental sample.
Construction of PCV2 replicon.A PCV2 rolling-circle replibased plasmid (Fig. 9A) was con-structed to test the influence of mutations on virus replication. TheRenillaluciferase gene was inserted into the HindIII and BamHI sites of the pcDNA3.1(⫹) plasmid to generate pcDNA-Rluc. The complete PCV2repgene, fused with a foot-and-mouth disease virus (FMDV) 2A fragment at its 5=end, was inserted into the BamHI and XhoI sites of pcDNA-Rluc to generate pcDNA-Rluc-Rep. Using the pcDNA-Rluc-Rep plasmid as the template, a PCR amplicon was generated that included a human cytomegalovirus (CMV) promoter (pCMV), genes encoding Renillaluciferase, FMDV 2A, PCV2 Rep, and the bovine growth hormone (BGH) polyadenylation signal. The region containing the PCV2 replication origin was added upstream of the promoter. The PCR amplicon was then inserted into the pMD18-T vector by TA cloning to generate the PCV2 replicon, pRe-PCV2. A nonreplicative plasmid (pNR-PCV2) was also constructed by excluding the origin region. The mutant replicon was generated by replacing the wild-type ORF1 gene with the corresponding mutant genes.
Real-time qPCR assay for replication.Replication was quantitated using real-time qPCR as previ-ously described (45). Briefly, 72 h after transfection, cells were harvested, and replicon DNA was extracted
on November 6, 2019 by guest
http://jvi.asm.org/
measured using a 1450 MicroBeta TriLux microplate multidetector (PerkinElmer, Waltham, MA, USA).
Statistical analysis.In the enzyme activity and virus replication analyses, three independent assays were conducted per group, and the difference in results between groups was analyzed by Student’st test. A difference with aPvalue of⬍0.05 was considered significant, aPvalue of⬍0.01 was considered very significant, and aPvalue of⬍0.001 was considered extremely significant.
Accession number(s).Coordinates and structure factors were deposited in the RCSB Protein Data Bank under accession number5XOR.
ACKNOWLEDGMENTS
This study was supported by the National Program on Key Research Project of China (2016YFD0500400 and 2018YFD0501004), the National Natural Science Foundation of China (31472221 and 31772711), and the Hubei Major Scientific and Technological Innovation Plan (2018ABA107).
We thank Ke Shi (Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota) for his contributions in the data analyses.
REFERENCES
1. Finsterbusch T, Mankertz A. 2009. Porcine circoviruses—small but pow-erful. Virus Res 143:177–183.https://doi.org/10.1016/j.virusres.2009.02 .009.
2. Tischer I, Rasch R, Tochtermann G. 1974. Characterization of papovavirus-and picornavirus-like particles in permanent pig kidney cell lines. Zentralbl Bakteriol Orig A 226:153–167.
3. Dulac GC, Afshar A. 1989. Porcine circovirus antigens in PK-15 cell line (ATCC CCL-33) and evidence of antibodies to circovirus in Canadian pigs. Can J Vet Res 53:431– 433.
4. Harding JC. 2004. The clinical expression and emergence of porcine circovirus 2. Vet Microbiol 98:131–135.https://doi.org/10.1016/j.vetmic .2003.10.013.
5. Chae C. 2005. A review of porcine circovirus 2-associated syndromes and diseases. Vet J 169:326 –336.https://doi.org/10.1016/j.tvjl.2004.01.012. 6. Opriessnig T, McKeown NE, Harmon KL, Meng XJ, Halbur PG. 2006.
Porcine circovirus type 2 infection decreases the efficacy of a modified live porcine reproductive and respiratory syndrome virus vaccine. Clin Vaccine Immunol 13:923–929.https://doi.org/10.1128/CVI.00074-06. 7. Meng XJ. 2013. Porcine circovirus type 2 (PCV2): pathogenesis and
interaction with the immune system. Annu Rev Anim Biosci 1:43– 64.
https://doi.org/10.1146/annurev-animal-031412-103720.
8. Kixmoller M, Ritzmann M, Eddicks M, Saalmuller A, Elbers K, Fachinger V. 2008. Reduction of PMWS-associated clinical signs and co-infections by vaccination against PCV2. Vaccine 26:3443–3451. https://doi.org/10 .1016/j.vaccine.2008.04.032.
9. Ramamoorthy S, Meng XJ. 2009. Porcine circoviruses: a minuscule yet mammoth paradox. Anim Health Res Rev 10:1–20.https://doi.org/10 .1017/S1466252308001461.
10. Cheung AK. 2003. The essential and nonessential transcription units for viral protein synthesis and DNA replication of porcine circovirus type 2. Virology 313:452– 459.https://doi.org/10.1016/S0042-6822(03)00373-8. 11. Lv QZ, Guo KK, Zhang YM. 2014. Current understanding of genomic DNA
of porcine circovirus type 2. Virus Genes 49:1–10. https://doi.org/10 .1007/s11262-014-1099-z.
12. Yang X, Chen F, Cao Y, Pang D, Ouyang H, Ren L. 2012. Complete
genome sequence of porcine circovirus 2b strain CC1. J Virol 86:9536.
https://doi.org/10.1128/JVI.01406-12.
13. Mankertz A, Caliskan R, Hattermann K, Hillenbrand B, Kurzendoerfer P, Mueller B, Schmitt C, Steinfeldt T, Finsterbusch T. 2004. Molecular biol-ogy of porcine circovirus: analyses of gene expression and viral replica-tion. Vet Microbiol 98:81– 88.https://doi.org/10.1016/j.vetmic.2003.10 .014.
14. Steinfeldt T, Finsterbusch T, Mankertz A. 2001. Rep and Rep=protein of porcine circovirus type 1 bind to the origin of replication in vitro. Virology 291:152–160.https://doi.org/10.1006/viro.2001.1203. 15. Ilyina TV, Koonin EV. 1992. Conserved sequence motifs in the initiator
proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res 20: 3279 –3285.https://doi.org/10.1093/nar/20.13.3279.
16. Steinfeldt T, Finsterbusch T, Mankertz A. 2007. Functional analysis of cis-and trans-acting replication factors of porcine circovirus type 1. J Virol 81:5696 –5704.https://doi.org/10.1128/JVI.02420-06.
17. Steinfeldt T, Finsterbusch T, Mankertz A. 2006. Demonstration of nicking/joining activity at the origin of DNA replication associated with the rep and rep’ proteins of porcine circovirus type 1. J Virol 80: 6225– 6234.https://doi.org/10.1128/JVI.02506-05.
18. Mankertz A, Hillenbrand B. 2002. Analysis of transcription of porcine circovirus type 1. J Gen Virol 83:2743–2751.https://doi.org/10.1099/0022 -1317-83-11-2743.
19. Cheung AK. 2012. Porcine circovirus: transcription and DNA replication. Virus Res 164:46 –53.https://doi.org/10.1016/j.virusres.2011.10.012. 20. Cheung AK. 2015. Specific functions of the Rep and Rep proteins of
porcine circovirus during copy-release and rolling-circle DNA replication. Virology 481:43–50.https://doi.org/10.1016/j.virol.2015.01.004. 21. Mankertz A, Hillenbrand B. 2001. Replication of porcine circovirus type 1
requires two proteins encoded by the viral rep gene. Virology 279: 429 – 438.https://doi.org/10.1006/viro.2000.0730.
22. Vega-Rocha S, Byeon IJ, Gronenborn B, Gronenborn AM, Campos-Olivas R. 2007. Solution structure, divalent metal and DNA binding of the endonuclease domain from the replication initiation protein from
por-September 2018 Volume 92 Issue 18 e00724-18 jvi.asm.org 14
on November 6, 2019 by guest
cine circovirus 2. J Mol Biol 367:473– 487.https://doi.org/10.1016/j.jmb .2007.01.002.
23. Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774 –797.https://doi.org/10.1016/j .jmb.2007.05.022.
24. Mihel J, Sikic M, Tomic S, Jeren B, Vlahovicek K. 2008. PSAIA—protein structure and interaction analyzer. BMC Struct Biol 8:21.https://doi.org/ 10.1186/1472-6807-8-21.
25. Laskowski RA, Swindells MB. 2011. LigPlot⫹: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51: 2778 –2786.https://doi.org/10.1021/ci200227u.
26. Afghah Z, Webb B, Meng XJ, Ramamoorthy S. 2017. Ten years of PCV2 vaccines and vaccination: Is eradication a possibility? Vet Microbiol 206:21–28.https://doi.org/10.1016/j.vetmic.2016.10.002.
27. Ssemadaali MA, Ilha M, Ramamoorthy S. 2015. Genetic diversity of porcine circovirus type 2 and implications for detection and control. Res Vet Sci 103:179 –186.https://doi.org/10.1016/j.rvsc.2015.10.006. 28. Faurez F, Dory D, Grasland B, Jestin A. 2009. Replication of porcine
circoviruses. Virol J 6:60.https://doi.org/10.1186/1743-422X-6-60. 29. Loregian A, Marsden HS, Palu G. 2002. Protein-protein interactions as
targets for antiviral chemotherapy. Rev Med Virol 12:239 –262.https:// doi.org/10.1002/rmv.356.
30. Zhan P, Li W, Chen H, Liu X. 2010. Targeting protein-protein interactions: a promising avenue of anti-HIV drug discovery. Curr Med Chem 17: 3393–3409.https://doi.org/10.2174/092986710793176357.
31. Corbi-Verge C, Kim PM. 2016. Motif mediated protein-protein interac-tions as drug targets. Cell Commun Signal 14:8.https://doi.org/10.1186/ s12964-016-0131-4.
32. Zhao Y, Yu S, Sun W, Liu L, Lu J, McEachern D, Shargary S, Bernard D, Li X, Zhao T, Zou P, Sun D, Wang S. 2013. A potent small-molecule inhibitor of the MDM2-p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J Med Chem 56:5553–5561.https://doi.org/ 10.1021/jm4005708.
33. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosen-berg SH, Elmore SW. 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19:202–208.https://doi.org/10.1038/nm.3048.
34. Saaranen MJ, Ruddock LW. 2013. Disulfide bond formation in the
cyto-plasm. Antioxid Redox Signal 19:46 –53.https://doi.org/10.1089/ars.2012 .4868.
35. Chen X, Ren F, Hesketh J, Shi X, Li J, Gan F, Hu Z, Huang K. 2013. Interaction of porcine circovirus type 2 replication with intracellular redox status in vitro. Redox Rep 18:186 –192.https://doi.org/10.1179/ 1351000213Y.0000000058.
36. Chen X, Ren F, Hesketh J, Shi X, Li J, Gan F, Huang K. 2012. Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-B pathway. Virology 426:66 –72.https://doi.org/10.1016/j.virol.2012 .01.023.
37. Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. 2004. Protein disulfide bond formation in the cytoplasm during oxida-tive stress. J Biol Chem 279:21749 –21758.https://doi.org/10.1074/jbc .M312267200.
38. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. 2006. HKL-3000: the integration of data reduction and structure solution—from diffrac-tion images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859 – 866.https://doi.org/10.1107/S0907444906019949. 39. Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated
macromo-lecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179.https://doi.org/10.1038/nprot.2008.91. 40. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular
graphics. Acta Crystallogr D Biol Crystallogr 60:2126 –2132.https://doi .org/10.1107/S0907444904019158.
41. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948 –1954.https:// doi.org/10.1107/S0907444902016657.
42. Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1. Schrodinger LLC, New York, NY.https://pymol.org/2/.
43. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59.https://doi.org/10.1016/0378-1119(89)90358-2. 44. Fang J, Li H, Peng G, Cao S, Zhen FF, Chen H, Song Y. 2013. Methods for
detecting ATP hydrolysis and Nucleic acid unwinding of Japanese en-cephalitis virus NS3 helicase. J Virol Methods 194:33–38.https://doi.org/ 10.1016/j.jviromet.2013.08.005.
45. Faurez F, Dory D, Henry A, Bougeard S, Jestin A. 2010. Replication efficiency of rolling-circle replicon-based plasmids derived from porcine circovirus 2 in eukaryotic cells. J Virol Methods 165:27–35.https://doi .org/10.1016/j.jviromet.2009.12.013.