0022-538X/89/041495-10$02.00/0
Copyright © 1989, American Society for
Microbiology
Genetic
Dissection
of the Transactivating Domain of the
Ela 289R
Protein of Adenovirus
Type
2
M. L. FAHNESTOCKt AND JAMES B. LEWISt*
Fred Hutchinson Cancer ResearchCenter, 1124 Columbia Street, Seattle, Washington 98104, andDepartmentof Pathology, University of Washington, Seattle, Washington 98195
Received 25 October1988/Accepted 8December 1988
A series of linker-scanning, deletion, and frameshift mutations were made in the pm975 variant of the adenovirus type 2 Elagene,whichexpressesonly the largerof thetwomajor Ela proteins. Most of thesewere within the46-amino-acidsegmentuniquetothe larger Ela protein product (the289R protein),whichconfers
onittheabilitytoactivate intranstheexpression of othergenes.The mutationswererecombined into virus and
assayed by in vitro transcription in nuclei isolated from infected cells for their ability to activate the transcription of other viral earlygenesand of the endogenous hsp7Ogene. Mutant Elaproteins from which the
289R-uniquesegmentwasremoved by deletionortruncation didnotcompletely lose theabilitytotransactivate bycomparisonwithavirus which makesnoElaatall, indicating thatsequencesoutside this domainareactive in the positive regulation of transcription. The Ela mutations tested fell into several classes: those that increasedtransactivation of virtually allgenes,thosethat severely depressed transactivation of allgenes, and those thatdepressed transactivation only moderately. Each mutation had similar effectsontheexpression of all
transcription units tested, indicatingacommon processin theirtransactivation. However,somemutantsin the third categorydecreasedtransactivation ofsomeinducedgenes moreseverely than of others. Suchgene-specific
defectssuggesttheexistence of subclasses ofEla-responsive transcription units, consistent with the involvement of diverse proteins in the transactivation of different genes. Two specific structural components of the transactivating domain, a putative metal-binding element and a region with high potential for n-sheet
formation at itscarboxy-terminus,appeartobe importanttothetransactivation function.
The289-amino-acid-residue(289R) protein product of the adenovirus Ela gene initiates the viral infectious cycle by activating in trans (transactivating) the expression of other viralgenesatthelevelof transcription(31, 32, 46, 51). The Ela 289R protein also specifically transactivates a few
cellulargenes during infection andnonspecifically activates the expression of mostgenes with which the Elagene is
cotransfected in transient expression systems (reviewed in reference 3). In addition, Ela-mediated activation of tran-scription has been observed in vitro with extracts of Ela-containing cells(20, 53) and uninfected cellextracts supple-mented with bacterially produced Ela protein (43) and synthetic peptides corresponding to the Ela amino acid sequence (13). The mechanism for thisEla-dependent acti-vation of transcription isnotclear, but a number of
impor-tant parameters have been defined. The enhancement of transcription does not require the continuous presence of Ela protein (53), nordoes itrequire protein synthesis (13, 43). Ela-mediated transactivation is accompanied by an
increasein thenumber oftranscribing complexes rather than by an increased rate ofinitiation, shown by template
com-mitment assays (53) and experiments with inhibitors of reinitiation in in vitrotranscription reactions (25). Nuclease protection assays demonstrate qualitative and quantitative alterations in the interaction of proteins with upstream promotersequences ofsome (but notall) susceptible genes
in the presence of Ela (reviewed in reference 21), but the protein itself does not bind DNA directly (6, 24). These
* Correspondingauthor.
tPresent address: Department of Microbiology, University of
California, LosAngeles,CA 90024.
tPresentaddress: ONCOGEN,3005 FirstAvenue, Seattle, WA
98121.
observations have led to the prediction that the Ela 289R protein indirectly affects transcription by catalyzing the formation of stable transcription complexes, by increasing the availability of or altering the activity of one or more
existing cellulartranscription factors (2).
On the basis of amino acid sequence homology and splicingpatterns among the Elaproteins of various adeno-virusserotypes, threefunctional domains have been identi-fiedinthe289R Elaprotein, termed conserved regions 1, 2, and 3(CR1, CR2, and CR3), each shown by geneticanalysis tobe important forone oranother ofthe multiple activities of Ela(reviewedinreference33).Thepositive effects ofEla
ontranscriptionareassociatedwithCR3,the46-amino-acid
segment unique to its 289R protein product. The smaller, 243-residue (243R) Ela protein, which lacks this segment, does not transactivate (3). The 289R-unique sequence has beenpostulatedtobeafunctionallydistinctdomaincarrying an independent transcriptional effector activity, a
supposi-tion supported by a recent demonstration that a
49-amino-acid synthetic peptide corresponding to the 46 residues unique to the 289R protein plus 3 adjacent amino acids encodedbythesecondexoniscapableofefficiently activat-ing transcription fromanEla-induciblegenefollowing coin-jection into mammalian cells (27) or addition to in vitro transcription reactions (13).
Described here are aseries ofsubstitution, deletion, and
truncation mutations in the transactivating domain ofthe Ela289R protein, constructed with the purpose of investi-gatingthe mechanism by which this viral effector manipu-latescellulartranscriptionsystems.Theeffect of eachonthe Ela-mediated transcriptional activation of five inducible
geneswasmeasured.Itwasfound that all mutationsaffected the transcription of all five target genes coordinately, al-though several affectedtheexpression ofsometargetgenes
1495
on November 10, 2019 by guest
http://jvi.asm.org/
more strongly than ofothers. These results are consistent with thecurrentmodel that transactivation isa
single
activ-ity effected through normal promoter sequences and the
multiple factorswhichbindthem,which differ for each gene
assayed (2, 8, 21). Inaddition, low-level activation ofsome geneswas seen even in the absence ofmost orall ofCR3,
and a protein carrying a mutation outside this segment
transactivated to higher levels than didthe wild-type
poly-peptide.
Thisobservationindicates that sequencesoutside ofthe Elatransactivating domainmayalsoaffect
positively
thetranscriptional regulation
ofsomeofallEla-inducible genes. The effects of these mutationsarediscussed intermsof thepossible
structuralorganization
ofthetransactivating
do-main.
MATERIALS AND METHODS
Cells and viruses. HeLa
monolayers
werepropagated
inDulbecco modified Eagle medium (DME)
containing
5% fetal bovine serum. HeLasuspension
cells were grown in Joklik modified minimal essential mediumcontaining
5% heat-inactivated calf serum, 75 UofpenicillinG perml, and 50 ,ug ofstreptomycin
perml. Human 293 cell monolayers(12)
werepropagated
in DMEcontaining
10% fetal bovine serum. Penicillin G andstreptomycin
were included to 100U/ml and 100
,ug/ml, respectively,
in mostexperiments.
Adenovirustype2(Ad2), dl309,andpm975werepropagated
on
suspension
HeLa cultures. All other viruses wereprop-agated
on 293 monolayers. Experiments were performedwith virus particles purified by banding in CsCl gradients.
Stocks were characterizedforPFU per milliliterby plaque
assayon293monolayersandforvirusparticlespermilliliter
by measuring
the A260 of sodium dodecyl sulfate(SDS)-disrupted
virions. All virus stocks used in theseexperiments showedparticle-to-PFU
ratios of 10 to 50. The valuesobtained for all mutant stocks fell in the range of 20 to 30
particles/PFU.
Infectionswere
performed
asfollows.From 6x106
to8x106
HeLacellswereplatedper100-mm dish 18 hbeforeuse.Medium was removed, and the monolayers were washed once with DME without serum and inoculated with 0.5 to 0.75ml ofdiluted virus.After
adsorption
for1hat37°C,theinoculumwas removed, monolayerswere washed with
me-dium,
and 10 mlof freshmediumcontaining 5%fetalbovineserum (Hyclone) was added back to each. Cultures were returned to the incubator until harvest. A comparison of
multiple
Ad2preparations
showed that early-geneexpres-sion,
asmeasuredatthe levelofearly protein accumulation,is mostcomparable from infectiontoinfection if numberof
particles
rather than PFU per milliliter is used as the measure of virus concentration. Either 300 or 1,500 virusparticles
per cell wereused,
as indicated for each experi-ment.Generation ofmutations. Linker-scanning mutations (30)
were generated in plasmid pPFpm975, a derivative of
pEKpm975
(31)which encodesonlythe13S Ela mRNA and its 289-residue protein product. To generatepPFpm975, an1,100-base-pair
(bp)HpaI-EcoRI
fragment containingthe ad-enovirus Elb gene and M13 RF sequences was removed,leaving
adenovirus sequences from nucleotide positions1to 1569 intact in a pBR322 background, flanked by PstI and EcoRIsites. TwofragmentsofpPFpm975weresubclonedto useassubstratesfordeletion.The1,193-bp fragmentextend-ing
from the left end of the viral chromosome(PstI)
to anAhaIll sitein the Ela intronwasligated intovectorpUC19
(48)
togenerateplasmid
pEA. To prepareplasmid pCH,
theClaI-Hpal fragment encompassingnucleotides 916 to 1569 of theviral chromosomewas
ligated
intopUC18
(48).Nested deletion sets weregenerated by progressive exo-nuclease III digestion of adenovirus sequences from the BamHI sites in parentplasmids pEAandpCH.Theplasmids
wererestrictedwith BamHI and
digested
with exonuclease IIIfollowedby S1nuclease. Deleted endswererepaired
with Escherichia coli DNApolymerase
Ilarge fragment
(Kle-now). BamHI 10-mer linkers(BethesdaResearch
Laborato-ries)
wereligated
tothe deletedends,
followedby
restriction with BamHI and PstI (pEA) or EcoRI (pCH) to release deleted inserts. These were then religated intoBamHI-PstI-cut
pUC19
DNAforpEA
derivativesorBamHI-EcoRI-digested
pUC18
DNAforpCH derivatives,
and theresulting
plasmids
wereusedtotransform E. coliHB101toampicillin
resistance. Deletionendpoints
of viral DNA in the recombi-nants were determinedby
sequenceanalysis
on double-strandedplasmid
DNA. Mutant Ela genes were recon-structed from thecomplementary
deletion sets by ligatingappropriate
gel-purified
inserts fromBamHI-EcoRI-re-stricted
pCH
derivatives intocomplementary
BamHI-EcoRI-digested
pEA derivatives. Theposition
of the linker within each mutantgenewas verifiedby
sequenceanalysis
onplasmid
DNA.Mutant EIA sequenceswererecombined into
H5dl309,
a variantofadenovirus type 5(AdS),by
the methodsofStow(45)andof Montelletal.
(31).
Resulting
viruseswereplaque
purified
and screenedby
restrictiondigestion
of viral chro-mosome DNA for the appearance ofa novel BamHI frag-mentandfor the lossof thewild-type
Hindlll Gfragment
on doubledigestion
with the twoenzymes.Immunoblotanalysisof viralproteins. Viral
proteins
were detectedintotal celllysates by
immunoblotby
the methods of Palmer et al. (35). To preparelysates,
infected andmock-infected monolayers were washed twice with cold
phosphate-buffered
saline(PBS)
and thenscraped
intoelec-trophoresis sample
buffer(24a)at aconcentration ofapprox-imately 107
cells per ml. Eachlysate
was sonicated with three 15-spulses
at 30 W and boiled for 3 minprior
to fractionation by electrophoresis in 10% (for the72,000-Mr
DNA-binding protein
[72K DBP]) or 15%(ElA)polyacryl-amide-SDS
gels
andelectrophoretic
transferofproteins
tonitrocellulose
(BA;
Schleicher andSchuell),
followedby
reactionwith
antibody
and1251I-labeled Staphylococcus
au-reusprotein
A. For detection of Elaproteins,
a 500-folddilution ofa
polyclonal
antiserum raised in rabbitsagainst
abacterially produced
Trp-E:ElA fusionprotein
was used(44). A
polyclonal
rabbit antiserum to the Ad2 72K DBP(supplied by
CarlAnderson),
diluted 1:1,000, was used fordetection ofthatviral
product.
Results werequantitated by
densitometry
ofautoradiograms.
Tofacilitatecomparison
ofdata among
experiments,
numerical values forprotein
accu-mulation obtainedby densitometryin eachexperimentwere normalizedtothatforpm975.Transcriptioninisolated nuclei.
Transcriptional
activation of all viralearly
genes and several cellular genes wereassayed by run-on
transcription
in nuclei isolated from infected HeLa cells. HeLa monolayers were infected as described above. Infections with all viruseswerebegun
and cultures were harvested atroughly
the same time on the sameday,
andsamples
wereprocessed
andanalyzed
as a grouponthesameday.
Atthe timesindicatedpostinfection,
cultures were harvested as follows.
Monolayers
were washed twice with cold PBS and scraped into 5 ml of PBS perplate.
Nucleiwere isolatedby lysis
in 10 mMTris(pH
7.4)-10mMNaCl-3 mM
MgCl2-3
mMdithiothreitol-20 U ofon November 10, 2019 by guest
http://jvi.asm.org/
RNasin (Promega Biotec) per ml-0.5% Nonidet P-40 for 3
min on ice with occasional agitation. Nuclei were pelleted for 10minat 1,500 rpm(IEC model DPR-6000), washed once with buffer without Nonidet P-40, and finally resuspended in 40 lI of storage buffer (5 mM MgCl2, 10 mM Tris [pH 7.5], 0.5 M sorbitol, 2.5% Ficoll, 0.3 mM spermidine, 1 mM dithiothreitol, 50% glycerol) per plate. All samples were stored at
-70°C
until use.Transcription reactions were carried out for 20 min at
30°C,
essentially as described by Weinheimer et al. (49). The purified, radiolabeled transcription products were collected by centrifugation, washed once with 70% ethanol, dried under vacuum, and suspended in 1 ml of hybridization buffer (0.3 M NaCl, 100 mM Tris [pH 8], 10 mM EDTA, 100,ugof sheared salmon sperm DNA per ml, 100 ,ug of yeast tRNA per ml, 1x Denhardt solution (29a), 50% formamide [49]). A107
cpm amount of each sample was diluted to 1.5 ml with hybridization buffer and applied to filters and prehybridized for 6 to 24 h at 42°C in the same buffer, on which DNAs corresponding to genes to be assayed were immobilized. After hybridization for 24 h at42°C with mixing, filters were washed with 2x SET (0.3 M NaCl, 100 mM Tris [pH 8], 5 mM EDTA-0.1% SDS for 0.5 h at room temperature and then for 1 h at60°Cin0.lx SET-0.1% SDS. Following this stringent wash, filters were incubated in RNase A (5,ug/ml)in 2 x SET for 20min, washed again for 1 h in two changes ofO.lx SET-0.1% SDS, and exposed to preflashed X-ray film at -70°C with an intensifying screen.
DNAs used as probes included the following. For Ela, single-stranded DNA from an M13 derivative containing the right strand of Ad2 DNA from nucleotides 267 to 1530 was used (pHEB4-R). For E1B, phage DNA from an M13 derivative containing the right strand of Ad2 (nucleotides 1767 to 2045) was used (mpSK-ElB). The probe for the E2 gene was phage DNA from an M13 derivative containing the left strand of the Ad2 chromosome fromnucleotides 21338 to 23924 (mpEcoB-EX). The E3 probe was phage DNA from an M13 derivative carrying Ad2 nucleotides 27000 to 28653, right strand only (mpHinH-EH). The probe used for E4 was single-stranded DNA "rescued" by M13 infection from a pBS+ vector (Vector Cloning Systems) into which nucleo-tides 33594 to 34933 had been subcloned(pHinF-KH). M13 DNA carrying nucleotides 6575 to 7262 of the Ad2 right strand was used as probe for the major late promoter (R1131 [10]). VA RNAs were detected with adouble-stranded probe (pVA [7]) containing Ad2 DNA between nucleotides 9831 and 11555. Plasmid pUR-HS70 (40), obtained from Joseph Nevins, was used to detect transcripts from thehsp70 genes. A cDNA to the human,-actin gene (pHF,BA-1 [14]) was used as a control in all experiments.
Densitometry was used to quantitate the intensity of hybridization to the various probes. The image from each slot was scanned three times, at a different position each time. Data for Ela, Elb, and E4 wereobtained from a 36- to 38-h exposure. All other measurements were derived from a 16- to 18-h exposure. M13mpl9single-stranded phage DNA, pBS+ single-stranded DNA, and pBR322 were included in all experiments as controls. No background signal was seen with phage or single-stranded pBS+ probes. pBR322 (and single-stranded DNA obtained by M13 rescue of the pBS-plasmid; not shown) did hybridize to some RNA product from infected cells, especially at later times postinfection. This background noise was subtracted from values obtained by densitometry before the data wereplotted.
Computer analysis of nucleic acid and protein structure. The GenePro program (Riverside Scientific) was used for
nucleotide sequence analysis and prediction ofprotein se-quence. Prediction ofprotein secondary structure was per-formed with both GenePro and PROTLYZE
(Scientific
and Educational Software).RESULTS
Generationandcharacterization ofviruses mutated in Ela. Avariant Ela gene thatmakesonlythe 13S mRNA
(pm975
[31])wasmutagenizedsothatmutantsof the289-residue Ela protein could be studied in the absence of the 243-residue protein encoded by the 12S mRNA.
Eight
linker-scanning
mutants were generated which
replaced
in combination all but 14 aminoacidsofthe46 thatarepresentin 289R butnot 243R. In addition, a truncation mutantwas madeby
intro-ducingaframeshift in the
protein-coding
sequenceafter the first codonof the 289R-unique sequence, and twoin-frame deletion mutations weremade,
oneremoving
42 residues fromCR3 andoneremovingCR2,aconservedsequence that is required forEla-mediatedrepression
ofenhancer-driventranscription (27, 37). The
position
of the linker in each mutant gene was verifiedby
DNA sequenceanalysis
onplasmid DNA. Figure 1 shows the
predicted
amino acidreplacementsin the mutantEla
proteins
andtheirpositions
withinthe 289R sequence. Also included are twoadditional
Elavariants, pm1098and
pmlll2
(27),
forcomparison
withpreviously described Ela mutants.
Infection was chosen over transfection as the means for
manipulatingmutantgenesfor severalreasons.
First,
viruses are veryreliable vehicles forintroducing
foreign
DNAinto cells. Second, infectionis the best wayto ensure consistent production of sufficient Elaprotein
foranalysis,
as insur-ance that differences inprotein
production
from mutant genes are notresponsible
for thephenotypes
observed. Third, infection is theonly
practical
meansforassaying
the effects of multiple Ela mutations on thetranscription
of multiple target genes in the sameexperiment.
Each mutant Ela genewastherefore recombined intothe chromosome ofvirus d1309 (38). This virus is
essentially
isogenic
for Ela with Ad2 but isavariant ofaclosely
relatedvirus,
AdS.The mutant viruses used for theexperiments
describedhere are thus derived from Ad2 for nucleotides 1 to 1339 and fromAdS forthe remainder ofthe viral chromosome. d1309 was
included as a control in all
experiments,
as were Ad2/5pm975,
the dl309derivativecarrying
thepm975
gene from which the mutants described here weregenerated,
andd1312,
adl309derivative which does notexpress Ela dueto the deletionofmost of the genefrom the viral chromosome(38).
Assay ofmutants for activation of viral andcellular genes. Two assay systems wereused to assess theeffects of these mutations on Ela-mediated transactivation. To measure
their effects on the
transcriptional
activation of a series of target genes, the extent oftranscription
of all viralearly
genes and the
endogenous
hsp70
gene, whoseexpression
is increased in the presence of Ela(22),
wasmeasureddirectly
by
transcription
in nuclei isolated from cells infected withwild-type andmutant viruses. In
addition,
the accumulation of the stableproteinproduct
of theE2agene, the 72KDBP,
was measured by immunoblot on
lysates
of infected cells from cultures infected inparallel
to and harvested at the same timeas those usedfor assays oftranscription.
Results from one setofexperiments
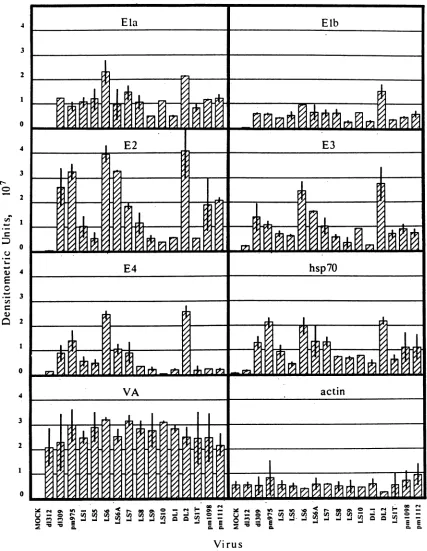
are shown inFig.
2. Datafrom quantitation of theautoradiographic
images
are shown inFig. 3 and4.
Direct measurement of
transcriptional
activity
in nuclei isolated from infected cells showed considerablevariability
on November 10, 2019 by guest
http://jvi.asm.org/
CR I CR 2 CR 3
F
.1l
I
108 19
GPV SMPNLVPEVI DLTCHEAGFP PSDDEDEEGE EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTCG MFVYSPVSEP w.t. GP .GS DEEGE EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTCG MFVYS DL2
E..I GSGIVYS DLI
E AGGYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTCG MFVYS LS1
E EFVLDYVEHP GHGCRSSGSG RRNTGDPDIM CSLCYMRTCG MFVYS LS5 E EFVLDYVEHP GHGCRSCHYH jj3 PDIM CSLCYMRTCG MFVYS LS6 E EFVLDYVEHP GHGCRSCHYH RRNT RIRM CSLCYMRTCG MFVYS LS6A E EFVLDYVEHP GHGCRSCHYH RRNTGDPI CSLCYMRTCG MFVYS LS7
E EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM ISGSGMRTCGMFVYS LS8 E EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCSG GMFVYS LS9
E EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTCTIS LS10 ER GkS&CGAPR ARLQVLSLSP EEYGGPRYYV FALLYEDLWH VCLQ* LSlT
E EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTC| MFVYS p.1098 E EFVLDYVEHP GHGCRSCHYH RRNTGDPDIM CSLCYMRTCG MFVY P.1112
FIG. 1. Positions of and predicted amino acid replacements in mutations generated in the 289Rprotein oftheadenovirustype 2 Ela gene. Domain structure, defined by sequence and functional analysis (32), is indicated schematically at the top,withtheamino acidsequenceof domains inwhichmutations were made indicated below. Amino acids substituted by aBamHI linkerareboxed.Dotsindicateresidues that havebeen lost indeletionmutants. Unrelated sequence at thecarboxy terminus of truncation mutantLS1T resulting from translationin an alternatereadingframeafterframe shift is underlined. The nameassigned to each mutant Ela gene and the virus thatcarriesit isindicated totheright of its sequence (w.t., wild type). The positions of the two point mutations in mutantspm1098andpmll2areindicatedbyabox aroundthe amino acid change that each produces.
in the transactivating potential of the various mutant Ela proteins. As expected, little or no expression of viral early genes ortheendogenoushsp7Ogene was seenin the absence of Ela products, represented by infection with dl312, while
wild-type controls dl309 and pm975 appeared to be roughly
equivalentin theirability to stimulate transcription from the genestested. LS9 and DL1consistently stimulated the least transcription from all genes, indicating a severe defect for transactivation in these mutant Ela proteins. Oddly, how-ever,neither of these mutations appeared to be as defective for transactivation as dl312. At the other end of the scale,
LS6and DL2 activated higher levels of transcription from all viral genes than did the wild-type controls. Enhancement of
transactivation of the hsp7Ogene was not seen at thistime
point with either of these mutants, but was noted with mutantDL2 at 6hpostinfection in other experiments (M. L. Fahnestock, Ph.Dthesis, University of Washington, Seattle, 1988).
Allothermutationsreduced Ela-mediated transactivation of the transcription units assayed to various degrees. LS1,
LS7,and LS8 were moderately defective fortransactivation of all target genes, while LS6A generally appeared to be nondefective for transactivation or only slightly so compared with pm975. Other mutations showed gene-specific defects. LS1T appeared to be almost equivalent in defect to LS9 and DL1 but showed a greater ability to activate the E3
tran-scriptionunit. LS5 was somewhat more effective at eliciting transcription from E3 and E4 than were LS9 and DL1, but wasequivalently defective for activation of the E2 andhsp70
genes. LS1O, pm1098, andpmlll2 were exceptionally de-fective for transactivation of E4. The specificity of this defect is underscored by the fact thatpm1098 and pmlll2
were minimally defective for the transactivation of all other genes assayed.
A DNA encoding the two adenovirus virus-associated
A.
2
A R!39
El A 2-43RL.
Lr fl C13C 2 CT)
A Cl. J WJJ_LCJ CQC:L
Te m bb
0s_
E2 DSP- - -_
B.
.L.i 9)
Lr
E2
F 3
E4 p!
,.1
;1.^. v,A
0So
-r
P1
o fl <I: T ) CY)c I, U t
')n E E fn O
co 'IO COn _z
2 ::6 ca. __j j
rl- 0cocoP2- C\JH
0) 0 0 0 ~j -j 0 -J -J -J -J 0 0 .-J
E0
co
F--C.
EL0
Go _
[image:4.612.86.537.75.303.2]- m . mm m - m - - . . a. m mm m FIG. 2. Comparisonoftranscriptionalactivityof viral and cellu-lar genes and accumulation of Ela and the E2a DBPs 8 h after infection of HeLa cells with 300 virus particles ofwild-type and mutantviruses per cell.(A) Exposures of immunoblots for Elaand E2aproteins. Thepositionsof both are marked atleft. (B)A 36-h exposureof filters fromanassayoftranscriptioninisolatednuclei. Eachcolumn represents a singlefilter hybridized with RNAfrom cells infected by a single virus, indicated at the top. The genes represented byeach rowofbands areindicatedatleft.MLP,Major late promoter.
4 e%90
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.325.561.415.629.2].8
3
2
0
4
3
0
I-c)
._
*-C
E
z._
:#
:
ou
2
1
0
4
3
2
1
0
4
3
2
1
0
q ,, V0EE-- - 0E E ° ,, x, EE 0 0 1 - E
Virus
FIG. 3. Graph of numerical data from densitometry of autoradiograms of transcriptionassaysinisolated nuclei shown in Fig. 2. Bar height indicates theintensityof hybridization to the gene indicated during infection by a given virus, identified below. Numbers are the average of data fromtwoexperiments. theline within each barindicatesthe range of values that wereaveraged.
(VA) RNAs was included as a measure of efficiency of
infection bythedifferentviruses. Thetranscriptionunitsfor
these RNAsareactiveatearlytimes(41), and their expres-sion is Elaindependent at early times postinfection in our hands. Hybridization to this probe was quite strong in all
infectedcultures,indicating
successful
infectioninall cases,and was insensitive to a-amanitin (Fig. 2), consistent with
transcription byRNApolymerase III(50). Hybridizationto all other probes was abolished when a-amanitin was in-cluded in the transcription reaction mix, consistent with theirbeingtranscribed byRNA polymerase II.
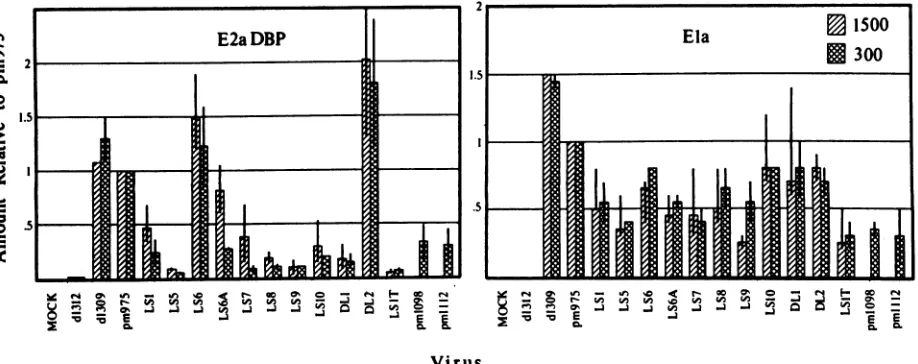
As shown in Fig. 4, the amount of E2a DBP detected
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.94.521.69.620.2]E2a
DBP
Ela . 1500S.l
E 2 1.5IM
300CL 0
1.5
0 10
...w I
19
a
I a4.4 I
c .5
I
I0 -5
E
.< r-m0
&I & to 0
t w w 0 - 00&
.Jjj J J
a--a 0. ..
jj-g=a An ;;an % < g-- 00 V, 0 e4 (-0 "
m =C7, 1 .. -w, ~j-i-_a 0
C6 - E E
Virus
FIG. 4. Comparison of accumulationofElaproteinsand E2a DBP inwild-type-and mutant-infected cellsat8hpostinfectionwith 300 particlespercell and6 hpostinfectionwith1,500particlespercell.Datafromdensitometricscansof immunoblotswerenormalizedtovalues forpm975. Valuesaretheaverageof results fromtwoexperiments with300particlespercell and threeexperimentswith1,500particlesper cell. The line within eachbarindicates the range of values that wereaveraged.
during these infections varied markedly, mirroring defects
seen in transactivation of the E2 gene at the level of transcription. Additional data from experiments performed
at higher multiplicity with harvest at 6 h postinfection are
included for comparison. LS5, LS9, LS10,DL1, and LS1T all showed very littleaccumulation ofDBPby8 h postinfec-tion. Slightly greater accumulation was detected in cells
infected with LS1, LS7, LS6A, pm1098, andpmlll2. LS6
andDL2showedincreased accumulation ofthe DBPrelative
to pm975. The relative protein accumulation found 8 h after
infection with several mutants (LS6A, LS7, LS8, pm1098,
andpmlll2) at 300 particles per cell was depressed
com-pared with the relatively moderate defect seen above in
transcription rates. This difference might reflect a lag be-tween transcription and processing of an mRNA and its subsequent translation into protein. DBPaccumulationwas
relatively greaterwith mutants LS6A, LS7, and LS8 after
infection with 1,500 particles per cell and harvest at the
earlier time point. Together, these observations confirm a
lesserdefectin mutantsLS6A, LS7, and LS8 such that early
deficits in transcription canbeovercomeby increased time
orgenedosage.pm1098andpmlll2were not included in the assays athigh multiplicity.
The same lysates in which the E2 DBP was quantitated
were also assayed for Ela products to verify that defects in
transactivation couldnotbeattributedtodecreasedlevelsof
theeffectorprotein inmutant-infected cells. Figure4shows
that theamountofElaprotein productspresent atthis time during
infection
differed by no more than threefold amongvirusescarrying in-frame mutations whatever the conditions used for infection and that the variation observed did not correlate with the effect of the mutation on transactivation.
Osborne et al. reported that a decrease of 10-fold in wild-type Ela mRNA levels after infection at low multiplicity has no apparent effect on the progress of infection or on focus
formation in assays of transformation (34). Also, other
studieshave shown that inclusion of protein synthesis
inhib-itors during the early phase of infection with wild-type viruses decreases ElaRNA production (and thus
presum-ablythe amountof Ela protein) 10- to 20-fold but does not
decreasethe amounts of RNA transcribed from other early genes (26). These experiments were performed before
anti-sera to the Ela protein products were available, so
corre-sponding information on Ela protein accumulation is lack-ing. However, they support the idea that the level of
expressionofearlygenes is relativelyinsensitive to moder-ate fluctuations in the amount of Ela protein present and thus that the differences in transactivation of viral and cellular genes noted for the mutant Elaproteins assayed in theseexperiments cannotbeattributed to defects in
expres-sion ofthe mutant Elagenes themselves.
DISCUSSION
The simultaneous assay of expression of several
Ela-inducible target genes during the early phase of infection
with wild-type adenovirus and with variants carrying
muta-tions in the transactivating domain ofthe major289R Ela
protein showed a variety of phenotypes with respect to
Ela-mediated transactivation.Allofthemutations described
hereaffectedtheactivity ofthe Elaprotein,shownby either increasesordecreasesin thetranscriptionalactivity of early
viralgenes and the cellularhsp70gene.Removal of42ofthe 46 amino acids ofthe 289R-unique domain by deletion or
truncation produced a severe deficit in transcription ofall genes. This reaffirms the importance of that region to the
activationoftranscription by Ela.
However, no mutant was completelydefective for trans-activationofallgenesbycomparisonwithdl312,suggesting
that sequencesoutsidethetransactivating domain, and
prob-ablyamino-terminal toit, cancontribute topositive
regula-tion of transcription by the Ela protein, at least during
infection. Precedent for such a conclusion exists in a few reports(forexample, reference 6) that sometransactivation activity is associated withthe 243R as well as with the 289R
protein. Somegeneralproperty of the Elapolypeptide may be responsiblefor this phenomenon, such asits acidity (pl
4.5
[16]).
The activating domains of several yeast and mammalian transcriptional regulatory proteins have beenshown to beacidic (39), and acidic translation products of randomly cloned fragments of E. coli DNA can serve as
transcriptional activatorsinSaccharomyces cerevisiae (29). The transactivating domain, on the other hand, is distin-guished by numerous basicamino acidsand twoconserved
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.82.541.72.254.2]sequencemotifs(see below)thatdiffer
considerably
from the restof the Ela 289Rprotein.
Ittherefore seemsunlikely
that sequencesoutside this domain could influencetranscription
by
thesame mechanism.In
general,
each of the Ela-inducible genes tested wasaffected
similarly by
agiven
mutation withinCR3, aswould beexpected
ifCR3-dependent transcriptional
activation of all genes involvedacommonprocess. Forexample,
linker-scanning
mutantLS9 showed the leastability
toactivate all genestested,
while LS6 enhanced the induction oftranscrip-tion of all viralgenes. Ini
addition,
thepatternof response of each gene tothe full spectrum ofmutationswassimilar,
asjudged by
visualinspection
ofFig.
3. However, in a few cases an Ela mutation affected theexpression
ofspecific
genes
preferentially.
Mutant LS5 wasrelatively
more defi-cient for activation of E2 andhsp7O
thanfor activation of the E3 and E4 genes. Alterations near thecarboxy
terminus of thetransactivat'ing
domain(LS10,
pm1098,
andpm1112)
substantially
decreased induction of E4 but had little effect on the other genesassayed.
These observations-could
be rationalized if Ela-mediated activation oftranscription
in-volved interactions amongmultiple
proteins,
with differentsUibsets used for different genes. A
single transcription
"factor" common to all
Ela-susceptible
genes is thus notrequired
toexplain
the effect of the viralprotein
on theactivity
ofdisparate
promoters transcribedby
distinct poly-merases,only susceptibility
to a common Ela function which is eliminated in theseverely
defective mutantsLS9,
DL1,andLSlTand enhanced inLS6. It is
impossible
tosay from this information whether direct interaction between Ela andmultiple
factors is reflected in these results or the interaction ofsome other constituent of the transactivationpathway
withmultiple
otherproteins.
The mutants de-scribed here may be useful inanswering
suchquestions
in the future.Two genes, Ela and Elb, conformed to the
general
pattern of increased
expression
withLS6 and DL2 butwerelargely
insensitive to the mutations that decreased transac-tivation of the othertargetgenes.Furthermore,in thecaseofEla, the amount of Ela
protein
observed in infected cellswas not
proportional
totranscription
levels. Forexample,
the increased
transcription
seenwithLS6and DL2 wasnot reflected inhigher
levels of Elaprotein.
This suggeststhat the Ela gene isregulated
in acomplex
fashion at severallevels,
possibly including
posttranscriptional
control at the level oftranslationorprotein stability.
TheBibgene,
although
essentially
inactive in the absence of Elaexpression (during
infection wilth d1312), was little affected in anegative
senseby
any of the mutations in the transactivatorprotein. Insensitivity
of the Bib gene to mutations in Ela has beenreported
previously
(4, 42). The cause of thisphenomenon
is unknown. It isinteresting
that the structureof itspromoterissimpler
thanthatof the other viralearly
genes,consisting
ofonly
abinding
site fortranscription
factorSpl
andaTATAbox(52). Itcontainsno inducible elements, which are associated with all other adenovirusearly
transcription
units(8, 21). Thepeculiarities
of its
regulation by
Elamight
be duetothisunique
promoterstructure or
perhaps
to cis effects from itsproximity
to the Elatranscription
unit andupstreamenhancer elements (17, 19). The latterpossibility
issupported by
theespecially
dramatic
(threefold)
effect on Bibtranscription
of mutantDL2, which is defective for enhancer
suppression
(see below).An additional
layer
ofcomplexity
in activation oftran-scription by
Ela isindicatedby
theeffects ofmutantDL2onthat process. Enhanced
transcription,
above levels inducedby
thewild-type
289R Elaprotein,
from all geneswas seenduring
infection witha viruscarrying
this mutation. CR2, adomain which is involved in the
negative
regulation
ofmetal
binding
beta sheet140 85
6EEFVLDYVEHPGH
HYHRRNTGDPDINNMRTCG.MFVYS
R E L A T V
E
T R A N S
R
p T 0 N
2.50_
2.25_
2.00_
1.75_
1
.50_
1.25_
1.00~
0.
75_
0.
50_
0.25_
0.00
I
I I
975 312 LS1
I I Ia a --- I I a I
LS5 LS6 LS6A LS7 LS8 LS9 LS1O01098 1112
VIRUS
0E3
ME4
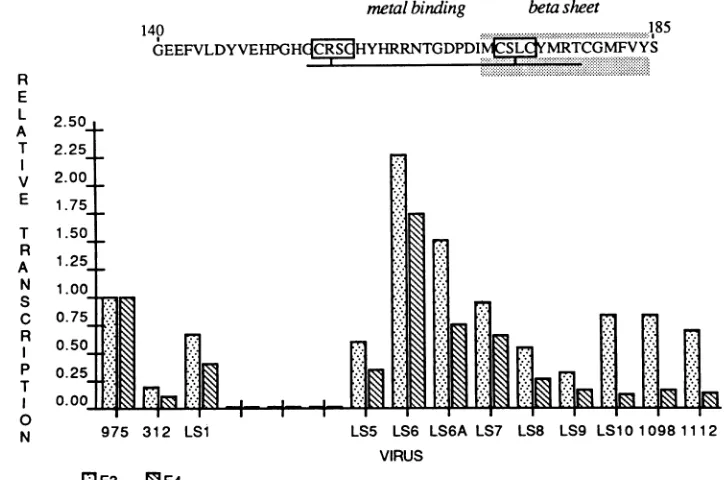
FIG. 5. Positions of structural elements within thetransactivating domainasdefinedby sequence andcomputer analysisand assays of transactivation potentialofmutant Elaproteins. The46-amino-acid sequence of the289R-unique segmentisalignedabove agraphof the relativetransactivatingabilities ofmutantscarryinglinker-scanningandpointmutations, sothat the effect of each mutationontranscription is shown below eachposition.Datafortwoof the sixEla-responsivegenesassayedareshown.975,pm975;312, d1312; 1098,pm1098;1112,
pm1112. Transcriptionis shown relativetothat inpm975.
.1.
I I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.127.490.434.674.2]Pk A
A
LIAIAMAIi
AVAd 7
m3
*Y
C
_. NA-VI-vX
&IAi-A
v
iAd
AA.
AALMiAdl12
o J1H'I'. 0
O3
00
0 000OCDt L
AnA
A A #cd Ad2
2
8'9
RAA
nfA
.,. A | Ad4~~4~ Ad 2
t1 / ~243R| ~~~~ 1
50 100 150 200 250
Amino acid position
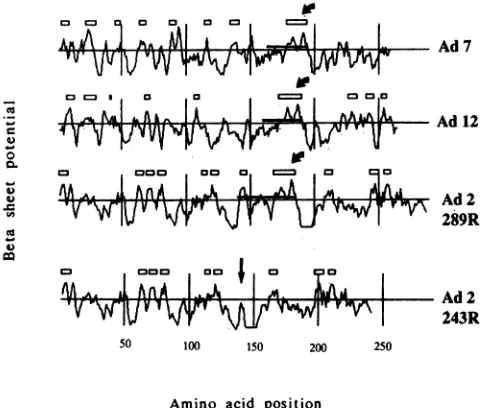
FIG. 6. Computer analysisofp-sheetpotentialinthelargeEla proteinsofdivergentadenovirusserotypesand the 243Rproteinof
Ad2. Boxes above each graphindicate sequences with calculated potentialsufficient topredictp-sheetformation. Ahorizontal bar in
eachgraphindicates thepositionof thesegmentuniquetothelarger
Elaproteinineachserotype.Curvedarrowsindicate thepositions
ofthe conservedP-sheetstructure. Astraightarrow indicates the
positionof thesplice junctioninthe Ad2 243Rproteinwhichdeletes
CR3.
enhancer-driven transcription by Ela proteins (27, 37), is deleted inDL2. In transient expression assays, cotransfec-tion with the DL2
Ela
mutant results in greatly enhanced transcriptionfroma reportercatgene drivenby the simian virus 40 enhancer relative to that seen aftercotransfection with thepm975 or LS6variant, consistent with the loss of enhancer repression capabilities with this mutation (Bill Demers,personal communication). Since Elaproteinlevels in cells infected with the DL2 virus were no higher than those seenduringinfectionwithpm975ord1309,theeffect of DL2ontransactivationcannotbeexplained as the resultof hyperexpressionof Ela from reliefofnegative autoregula-tionbyEla geneproducts.Amorelikely explanationfor this phenomenonisthat theexpressionofallgenesassayed here, andtherefore possibly all Ela-sensitive genes, isunder the influence ofan enhancerelement(s). This isconsistent witha recent report that an enhancer at the left end of the adenovirusgenome positivelyaffects thetranscriptionof all viral genes(18).
Comparison of sequences among the Ela proteins of severaladenovirusserotypesdefinestwostructuralmotifs in CR3 thatare also implicated in transactivation by the
mu-tantsdescribed here.Theirpositionsareindicated inFig.5. The first isaputative metal-bindingdomaincomposedoftwo cysteine pairs separated by13amino acids, many ofwhich
are basic or hydrophobic in nature (1). Mutants carrying
mutations which affect the cysteine pairs and amino acids
immediately carboxy-terminaltothem(LS5, LS9, and LS8)
are highly defective for transactivation. Interestingly, the
Elaproteins produced by LS5 and LS9, shown in Fig. 2, migrate aberrantlyinpolyacrylamide gels, indicating that the sequencessubstituted inthesemutationsmaybeinvolved in formation ormaintenance ofconformation orare sites ofa
modification(s)of the Elaproteinwhichenhancesfunction. Mutations betweenthecysteine pairs (LS6, LS6A,andLS7)
had a negligible effect on transactivation. In fact, one of these mutations (LS6) is an "up" mutation for thatactivity.
Inthismutant, two amino acids (Thr-164 and Gly-165)which may bepresent at the same position in the Ela proteins of all
sIerotypes
(23, 47) are replaced. Its phenotype suggests that these residues may function to down regulate transactiva-tion.Overlapping this metal-binding segment, extending ap-proximately between amino acids 170 and 184 in Ad2, is a sequence predicted by computer analysis with two programs
bythe rules of Chou and Fasman (5) to have highpotential for
p-sheet
formation. Output from one of these programs (PROTLYZE), showing the position of this structure and its occurrence in the Elaproteins of multiple adenovirus sero-types, isreproducedinFig. 6. Mutationslying solely in this region (LS10, pm1098, and pmlll2) have an especially deleterious effect on transcription of the viral E4 gene.Although prediction of secondary structure in proteins is
currently an inaccurate process, the presence of a similar structurein multiple divergent proteins is morelikely to be
significant. Theimportance of this sequence to transactiva-tion is underscored by the observation that all defective
in-frame mutations in Ela generated by random methods
whichhave beendescribed in theliteraturefallwithin it(hr3,
hr4, and hr5[11, 15]andpm1098andpmlll2 [27]).
The splicing event which removes the Ela 289R-unique
coding sequence in Ad2 completely deleted the two sub-structuresdescribed above from the Elaproteins of diver-gent serotypes(Adl2, Ad7, and Ad4) (23, 47),shown for the Ad2 243R ElaproteininFig. 6.However, the more
amino-terminalresidues oftheuniquesegment of the Ad2 289R Ela
protein areactually retainedin thesmallerproteinsof other serotypes due toutilization of a splice donor at adifferent
position
in the common Ela precursor RNA (36). If exon structurereflects domain structure, assuggested byGilbert(9), the probable metal-binding sequence and'
p-sheet
to-gethermayconstituteasingle functional subunit ofthe Ela
protein. However, the example of LS1 indicates that the upstream residues do influence Ela-mediated
transactiva-tionandmightbe a secondfunctionalclusterin the
transac-tivating domain. No conserved functional groups in this
sectionofthe Elaprotein sequence areevide'nt from com-puteranalysis of secondarystructure andchargein the Ela
proteins of multiple serotypes. Furthermutagenesis within thissegmentofthe Ad2transactivating domainisnecessary todefine its contributiontothemechanism oftransactivation
by Ela.
The results
presented
heredemonstrate thattransactiva-tion of viral and cellulargenes by the Ad2 289R Elagene
product
during
adenovirus infection isacomplexfunctionofseveralactivities. Maximal activationrequiresthepresence
of the 46-amino-acid segment
unique
to the larger Elaprotein. However, since mutants which retain
little
orno sequencefromthis segmentoftheprotein retainsomeabilityto activate at least some genes, a second, much weaker
positive
regulatoryactivityhasbeenpostulated, mediated bysequences outside the transactivating domain. The
discov-ery of this activity, which affects the transcription of both viral and cellulargenes, coupled with the information that
Ela-inducible transcription units can be differentially
af-fectedby specific mutations inElaincreases thedifficulty in separating any process inwhich the Elaprotein(s)
partici-patesfrom its
positive
effectsontranscription. Thispointis atodds with theconclusions ofLillieetal. intheir analysis ofthephenotypes ofmutantspm1098andpmlll2(27). From assays ofsteady-state RNAlevels 16 h postinfection, withon November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.60.300.75.279.2]cytosine arabinoside used to prevent replication, thus ex-tending the early phase and amplifying early-gene expres-sion, these authors reported that pm1098 andpmlll2 are extremely (50-fold) defective for transactivation of viral early genes relative to the wild-type Ela 289R protein and yet are equivalent in transforming ability. In the assays reported here, in which transcription was measured directly during the early phase of infection in the absence of drugs, these two mutants were no more than twofold defective for the activation of any gene but E4. They were by no means as defective as the Ela- mutant d1312 or many of the other Ela variants assayed.
The mutations in the Ela 289R protein described here include several phenotypes with respect to transactivation which had not previously been reported or studied in depth. The results of their analysis in assays of transcription point toward more than one mechanism by which Ela can affect gene expression positively and to the possible definition of subclasses of Ela-responsive genes by the gene-specific effects of modification of the Ela 289R protein. Until the underlying mechanisms behind Ela activities can be eluci-dated, direct assays can be developed for them, and the spectrumof genes that they affect can be examined in some detail, correlations drawn or denied between the positive regulation of transcription by Ela and its other functions may represent oversimplifications.
ACKNOWLEDGMENTS
We thank R. Morgan Wain and Kathleen Critchett for expert technical assistance, Carl Anderson for antiserum to theDBP, and Arnold Berk for providing plasmids pEKpm975 andpKS103.
This investigation was supported by Public Health Service grants CA29600 and CA39636 from the National Institutes of Health.
LITERATURE CITED
1. Berg, J. M. 1986. Potential metal-binding domains in nucleic acid binding proteins. Science 232:485-487.
2. Berk, A. J. 1986. Adenovirus promoters and Ela transactiva-tion. Annu. Rev. Genet. 20:45-79.
3. Berk, A. J. 1986. Functions of adenovirusElA.CancerSurveys 5:367-387.
4. Carlock, L. R., and N. C. Jones. 1981.Transformation-defective mutant of adenovirus type 5 containing a single altered ElA mRNA species. J. Virol. 40:657-664.
5. Chou, P. Y., and G. D. Fasman. 1978. Empirical predictions of protein conformation. Annu. Rev. Biochem. 47:251-276. 6. Ferguson, B., B. Krippl, 0. Andrisani, N. Jones, H. Westphal,
and M. Rosenberg. 1985. ElA 13S and 12SmRNA products made in Escherichia coli both function as nucleus-localized transcription activators but do not directly bind DNA. Mol. Cell. Biol. 5:2653-2661.
7. Fowlkes, D. M., and T. Shenk. 1980. Transcriptional control regions of the adenovirus VAl RNA gene. Cell22:405-413. 8. Garcia, J., F. Wu, and R. Gaynor. 1987. Upstream regulatory
regions required to stabilizebinding to the TATAsequencein an adenovirus early promoter. NucleicAcids Res. 15:8367-8385. 9. Gilbert, W. 1985. Genes in pieces revisited. Science 228:
823-824.
10. Gingeras, T. R., D. Sciaky, R. E. Gelinas, J. Bing-Dong, C. E. Yen, M. M. Kelly, P. A. Bullock,B. L. Parsons, K. E. O'Neill, and R. J. Roberts. 1982.Nucleotide sequencesfrom the adeno-virus-2 genome. J. Biol. Chem. 257:13475-13491.
11. Glenn, G. M., and R. P. Ricciardi. 1985. Adenovirus 5 early region1A host range mutants hr3, hr4, and hr5 contain point mutations which generate single amino acid substitutions. J. Virol. 56:66-74.
12. Graham, F. L., J. Smiley, W. C. Russell, and R. Naiva. 1977. Characteristics of ahuman cell linetransformedby DNA from humanadenovirus 5. J. Gen. Virol. 36:59-74.
13. Green, M.,P.Loewenstein, R. Pusztal, and J. Symington. 1988. An adenovirus ElA protein domain activatestranscription in vivo and in vitro in the absence of protein synthesis. Cell 53:921-926.
14. Gunning, P., P.Ponte, H.Okayama,J. Engel, H. Blau, and L. Kedes. 1983.Isolation andcharacterizationoffull-lengthcDNA clonesfor human a-, 3-, and -y-actin mRNAs: skeletal butnot
cytoplasmic actins have an amino-terminal cysteine that is subsequentlyremoved. Mol. Cell. Biol.3:787-795.
15. Harrison, T., F. Graham, and J. Williams. 1977. Host-range mutants ofadenovirus type 5defectiveforgrowth in HeLa cells. Virology77:319-329.
16. Harter, M., and J. Lewis.1978. Adenovirus type 2earlyproteins synthesized invitro andinvivo:identification in infected cells of the38,000- to50,000-molecular-weight protein encodedbythe left endof theadenovirus type 2 genome. J.Virol.26:736-749. 17. Hearing, P., and T. Shenk. 1983. The adenovirus type 5ElA transcriptional control region contains a duplicated enhancer element. Cell33:695-703.
18. Hearing, P., and T. Shenk. 1986. The adenovirus type 5 Ela enhancer contains two functionally distinct domains: one is specific forElAand the other modulatesallearly units incis. Cell 45:229-236.
19. Hen, R., E. Borrelli, P. Sassone-Corsi, and P. Chambon. 1983. Anenhancer element is located 340base pairs upstreamfrom the adenovirus-2 ElA capsite. Nucleic Acids Res. 11:8747-8760.
20. Hoeffier, W. K., and R. G. Roeder. 1985. EnhancementofRNA polymerase IIItranscription bythe ElA gene product of ade-novirus. Cell41:955-963.
21. Jones, N. C., P. W. Rigby, and E. B. Ziff. 1988. trans-Acting protein factors and the regulation ofeukaryotic transcription: lessons from studies on DNA tumor viruses. Genes Dev. 2:267-281.
22. Kao, H.-T., and J. R. Nevins. 1983. Transcriptional activation and subsequent control of the human heat shock gene during adenovirusinfection. Mol. Cell. Biol. 3:2058-2065.
23. Kimelman, D., J.S.Miller, D.Porter, and B. E. Roberts. 1985. Ela regions of the human adenoviruses and of the highly oncogenic simian adenovirus 7 are closely related. J. Virol. 53:399-409.
24. Ko, J.-L., B. Dalie, E. Goldman, and M. Harter. 1986. Adeno-virus-2 earlyregion IA protein synthesized in Escherichia coli extractsindirectlyassociates withDNA.EMBOJ.5:1645-1651. 24a.Laemmli, U. K. 1970. Cleavage of structuralproteinsduringthe assembly of the head ofbacteriophage T4. Nature (London) 227:680-686.
25. Leong, K., and 4. Berk. 1986. Adenovirus early region 1A protein increasesthe numberoftemplate molecules transcribed in cell-free extracts. Proc. Natl.Acad. Sci. USA 83:5844-5848. 26. Lewis, J. B., and M. B. Mathews. 1980. Control of adenovirus early geneexpression: a classof immediateearlyproducts.Cell 21:303-313.
27. Lillie, J. W., M.Green, and M. R. Green. 1986. Anadenovirus Ela protein region required for transformation and transcrip-tionalrepression. Cell 46:1043-1051.
28. Lillie, J. W., P. M. Loewenstein, M. R. Green, and M. Green. 1987. Functional domains of adenovirus type 5 Ela proteins.
Cell 50:1091-1100.
29. Ma, J., and M. Ptashne. 1987. A new class ofyeast transcrip-tionalactivators. Cell 51:113-119.
29a.Maniatis, T., E. F. Fritsch, andJ. Sambrook. 1982. Molecular cloning: alaboratory manual. Cold Spring HarborLaboratory,
ColdSpring Harbor, N.Y.
30. McKnight, S. L., and R. Kingsbury. 1982. Transcriptional
control signals of a eukaryotic protein-coding gene. Science 217:316-324.
31. Montell, C., E. F. Fisher,M. H.Caruthers,andA.J.Berk.1982. Resolving the functions of overlapping viral genes by site-specific mutagenesis at amRNA splice site. Nature (London)
295:380-384.
32. Moran, E., T.Grodzicker,R.J.Robert, M.B.Mathews, andB. Zerler. 1986. Lytic and transforming functions of individual
on November 10, 2019 by guest
http://jvi.asm.org/
products of the adenovirus ElA gene. J. Virol. 57:765-775. 33. Moran, E., and M. B. Mathews. 1987. Multiple functional
domainsin theadenovirusElAgene.Cell 48:177-178. 34. Osborne, R. F., R. B. Gaynor, and A. J. Berk. 1982. TheTATA
homology and the mRNA 5' untranslated sequence are not required for expression of essential adenovirus ElAfunctions. Cell29:139-148.
35. Palmer, D. K., K. O'Day, M. H. Wener, B. S. Andrews, and R.L. Margolis. 1987. A 17-kD protein (CENP-A) copurifies with nucleosome core particles and with histones. J. Cell Biol. 104:805-815.
36. Perricaudet, M., J.-M.le Moullec, P. Tiollais, and U. Pettersson. 1980. Structure of two adenovirus type 12 transforming poly-peptides and their evolutionary implications. Nature (London) 288:174-176.
37. Schneider, J. F., F. Fisher, C. R. Goding, and N. C. Jones. 1987. Mutational analysis of the adenovirus Ela gene: the role of transcriptional regulation in transformation. EMBO J. 6:2053-2060.
38. Shenk, T., N. Jones, W. Colby, and D. Fowlkes. 1980.Functional analysis ofadenovirus-5 host-range deletion mutants defective for transformation of rat embryo cells. Cold Spring Harbor Symp. Quant. Biol.44:367-375.
39. Sigler, P. 1988. Acid blobs and negative noodles. Nature (London) 333:210-212.
40. Simons, M. C., K. Kitchener, H.-T. Kao, E. Hickey, L. Weber, R. Voellmy, M. Heintz, and J. R. Nevins. 1987. Selective induction of human heat shock gene transcription by the ade-novirus ElA gene products, including the 12S ElA product. Mol. Cell. Biol. 7:2884-2890.
41. Soderlund, H., U. Petterson, B. Vennstrom, L. Philipson, and M. B.Mathews. 1976. A newspecies of virus-coded low molec-ularweight RNA fromcells infected with adenovirus type 2. Cell7:585-593.
42. Solnick, D., and M. A. Anderson. 1982. Transformation-defi-cient adenovirus mutant defective in expression of region 1A butnotregion 1B.J. Virol. 42:106-113.
43. Spangler, R., M. Bruner, B. Dalie, and M. Harter. 1987. Activation of adenovirus promoters by the adenovirus Ela protein in cell-freeextracts. Science237:1044-1046.
44. Spindler, K. R., D.S.E.Rosser,andA.J. Berk. 1984. Analysis of adenovirustransforming proteins from early regions 1A and 1B with antisera to inducible fusion antigens produced in Escherichiacoli. J.Virol. 49:132-141.
45. Stow, N. D. 1981.Cloning ofaDNAfragment from the left-hand terminus of the adenovirus type 2 genome and its use in site-directedmutagenesis. J. Virol. 37:171-180.
46. Svensson, C., and G.Akusjarvi. 1984.Adenovirus2early region ElA stimulates expression of both viral and cellular genes. EMBOJ. 3:789-794.
47. Tokunaga,O., T. Yaegashi, J. Lowe, L. Dobbs, and R. Padman-abhan. 1986. Sequence analysis in the El region of adenovirus type 4 DNA. Virology. 155:418-433.
48. Vieira, J., and J. Messing. 1982. The pUC plasmids, an M13mp7-derivedsystemfor insertion mutagenesis and sequenc-ing withsynthetic universal primers. Gene 19:259-268. 49. Weinheimer, S. P., andS. L. McKnight. 1987. Transcriptional
andpost-transcriptional controls establish the cascade ofherpes simplex virus protein synthesis.J. Mol. Biol. 195:819-834. 50. Weinmann, R., T. G. Brendler, H. J. Rashkas, and R. G.
Roeder. 1976. Lowmolecular weight viral RNAs transcribed by RNA polymerase III during adenovirus 2 infection. Cell 7: 557-566.
51. Winberg, G., and T. Shenk. 1984. Dissection of overlapping functions within the adenovirus type 5 ElA gene. EMBO J. 3:1907-1912.
52. Wu, L., D. Rosser, M.Schmidt, and A. Berk. 1987. A TATA box implicatedin ElAtranscriptional activation of a simple adeno-virus 2 promoter. Nature (London) 326:512-515.
53. Yoshinaga, S.,N.Dean, M. Han, and A. J. Berk. 1986. Adeno-virus stimulation of transcription by RNA polymerase III: evidence foranElA-dependent increaseintranscriptionfactor
IIIC concentration. EMBOJ. 5:343-354.