0022-538X/09/$08.00⫹0 doi:10.1128/JVI.02115-08
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Epstein-Barr Virus Episome Stability Is Coupled to a Delay in
Replication Timing
䌤
†
Jing Zhou, Andrew R. Snyder, and Paul M. Lieberman*
The Wistar Institute, Philadelphia, Pennsylvania 19104
Received 7 October 2008/Accepted 3 December 2008
The temporal regulation of DNA replication is thought to be important for chromosome organization and genome stability. We show here that Epstein-Barr virus (EBV) genomes replicate in mid- to late S phase and that agents that accelerate replication timing of EBV reduce viral genome stability. Hydroxyurea (HU) treatment, which is known to eliminate EBV episomes, shifted EBV replication to earlier times in the cell cycle. HU treatment correlated with hyperacetylation of histone H3 and loss of telomere repeat factor 2 (TRF2)
binding at the EBV origin of plasmid replication (OriP). Deletion of TRF2 binding sites withinOriPor short
hairpin RNA depletion of TRF2 advanced the replication timing ofOriP-containing plasmids. Inhibitors of
class I histone deacetylases (HDACs) increased histone acetylation atOriP, advanced the replication timing of
EBV, and reduced EBV genome copy number. We also show that HDAC1 and -2 form a stable complex with TRF2 at OriP and that HU treatment inhibits HDAC activity. We propose that the TRF2-HDAC complex enhances EBV episome stability by providing a checkpoint that delays replication initiation at OriP.
Epstein-Barr virus (EBV) is a human gammaherpesvirus that has been implicated as a causal cofactor of several human malignancies, including Burkitt’s lymphoma, nasopharyngeal carcinoma, Hodgkin’s disease, and lymphoproliferative dis-eases during immunosuppression (19, 21, 32, 46). The virus typically establishes a latent infection in B lymphocytes, where it persists as a multicopy circular minichromosome that repli-cates in synchrony with the cellular genome (1, 45). The EBV origin of plasmid replication (OriP) binds the virus-encoded EBNA1 protein to form an efficient replicon that is essential for the plasmid stability of latent episomes (reviewed in refer-ences 25 and 41). EBNA1 binds to a minimal replicator se-quence withinOriP, referred to as the dyad symmetry (DS) region, which consists of two pairs of EBNA1 binding sites flanked by telomere repeat factor (TRF) binding sites (2, 8, 22, 44). This minimal replicator has been shown to function indis-tinguishably from cellular origins that are licensed to replicate once per cell cycle (5, 9, 33, 34). Epigenetic events are neces-sary for the establishment of a stable episomal origin, but the nature of these modifications has not been identified (24, 29). EBV episomes can be eliminated from some cell types by treatment with hydroxyurea (HU) (17, 40). HU has been used in clinical trials for treating EBV-positive central nervous sys-tem lymphomas (6, 38, 40). HU has also been used to eliminate the double minute satellite DNA that accumulates in many cancer cells and often carries amplified oncogenes or tumor suppressors, including c-myc and MDM2 (23, 39). The under-lying mechanism of action of HU has been attributed to its ability to inhibit ribonucleotide reductase and slow cell cycle progression (43). The mechanism of HU-induced loss of
dou-ble minutes and EBV episomes is not known but is thought to function through its effects on ribonucleotide reductase and DNA replication (18).
Chromatin organization and histone modifications have also been implicated in the regulation ofOriPreplication activity and plasmid stability. Previous studies have found that nucleo-somes flank DS elements and undergo cell cycle changes in histone modifications (48). In particular, histone H3 acetyla-tion was reduced in G1/S and then enriched in mid-S phase.
The histone H3 deacetylation in early S phase raised the ques-tion whetherOriP was subject to temporal control of DNA replication. The temporal control of DNA replication is thought to be important for gene expression and chromosome organization (11, 35, 49). There is strong evidence that chro-matin assembled early in S phase is hyperacetylated, and there-fore more permissive for transcription, while chromatin assem-bled in late S phase is hypoacetylated (47). In this study, we explore the possibility that replication timing plays a role in the genome stability of latent EBV episomes. We investigate the effect of HU treatment on the replication timing of EBV ge-nomes and explore the possibility that this is linked to the cell cycle deacetylation-acetylation of histone H3 at DS. Our find-ings suggest that histone deacetylation is linked to a delay in OriP replication timing and that this delay enhances the ge-nome stability of latent EBV.
MATERIALS AND METHODS
Cell culture.EBV-positive Burkitt lymphoma cell lines (Raji and MutuI) were maintained in RPMI medium supplemented with 10% fetal bovine serum, glu-tamine, penicillin, and streptomycin sulfate (Cellgro). D98/HR1 cells (EBV-positive adherent cells) and HeLa cells (EBV-negative adherent cells) were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate (Cellgro). The A39 mini-EBV lymphoblastoid cell line was cultured as described previously (33, 34). HU and valproic acid (VPA) (Sigma) were used at the concentrations indicated in each figure legend. Trichostatin A (TSA) was used at 100 ng/ml for 4 h, and sodium butyrate (NaB) was used at 1 mM for 4 h. HeLa cells were transfected with Lipofectamine 2000 (Invitrogen, Inc.) and selected with hygromycin (100
g/ml) for 10 days for generating stable pools containingOriPplasmids. * Corresponding author. Mailing address: The Wistar Institute,
Phila-delphia, PA 19104. Phone: (215) 898-9491. Fax: (215) 898-0663. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi .asm.org/.
䌤Published ahead of print on 10 December 2008.
2154
on November 8, 2019 by guest
http://jvi.asm.org/
Antibodies.The following rabbit polyclonal antibodies were used: rabbit poly-clonal anti-EBNA1 and TRF2 were raised against a recombinant full-length EBNA1 and TRF2, and rabbit immunoglobulin G (IgG) (Santa Cruz), polyclonal ORC2 (BD Pharmingen), polyclonal acetyl H3 (AcH3) (Upstate), polyclonal MCM3 (Abcam), mSin3 (Santa Cruz Biotech), MTA (Santa Cruz Biotech), and monoclonal RRM2 (Abnova) were used according to the manufacturers’ sug-gestions.
Plasmids. OriPwild-type (wt) and mutant (nm⫺) plasmids have been de-scribed previously and consist ofOriPsequences, EBNA1, enhanced green flu-orescent protein, and hygromycin genes as a pREP10 (Invitrogen) derivative (8). Short hairpin TRF2 (shTRF2) has been described previously (7).
Replication Timing Assays.Cells were incubated with 50M bromodeoxyuri-dine (BrdU) for 30 min. Labeled cells were then fixed in 70% ethanol and resuspended in propidium iodide (PI) staining buffer for 30 min. Stained cells were sorted at 50,000 cells per fraction for six fractions (G1, S1 to S4, and G2/M)
by fluorescence-activated cell sorting (FACS). Three hundred microliters of lysis buffer I (50 mM Tris-HCl [pH 8.0], 1 M NaCl, 10 mM EDTA, 0.5% sodium dodecyl sulfate [SDS]; 0.2 mg/ml proteinase K, 10l heat-denatured sonicated single-stranded DNA [10 mg/ml]) was added to the collected cells and incubated at 50°C for 2 h. DNA was extracted with phenol-chloroform, precipitated with alcohol, and then dissolved in 500l 1⫻Tris-EDTA buffer. Sonication was used to generate fragments of⬃0.25 to 2 kb (average, 700 bp). DNA was then heat denatured at 95°C for 5 min and cooled down on ice. Fifty microliters was kept as input. Fifty microliters of 10⫻immunoprecipitation (IP) buffer (100 mM NaPO4[pH 7.0], 1.4 M NaCl, 0.5% Triton X-100) was added to the DNA
solution to make a final 1⫻solution; 4l anti-BrdU (stock, 25g/ml; BD Pharmingen) was then incubated with the solution for 60 min at room temper-ature with rotation, and then 3.5g (10l) of rabbit anti-mouse IgG antibody (Sigma) was added to each tube and rotated for 30 min at room temperature. The precipitated DNA was centrifuged for 5 min at 14,000 rpm and washed twice in 750l 1⫻IP buffer, and the pellet was resuspended in 200l lysis buffer II (10 mM EDTA, 50 mM Tris-HCl [pH 8.8], 0.5% SDS, 0.25 mg/ml proteinase K). DNA was incubated at 37°C overnight, and then another 100l lysis buffer II was added and left for 1 h at 50°C. DNA again was phenol-chloroform extracted twice and precipitated with ethanol in the presence of glycogen. Real-time PCR was used to compare the BrdU incorporation on different regions with specific primers. Real-time PCR analysis of DNA was quantified using the standard curve method on an ABI 7000 thermocycler and normalized to the input of each fraction. The data were then normalized to the total signal (from G1, to S1 to S4,
to G2/M, and set as 100%). At least three independent chromatin IPs (ChIPs)
were performed for each data point. The error bars in the figures represent standard deviations from three real-time PCRs from the three ChIP experi-ments.
Primers used for real-timer PCR were as follows: for DS, ATGTAAATAAA ACCGTGACAGCTCAT (forward) and TTACCCAACGGGAAGCATATG (reverse); for actin, GCCATGGTTGTGCCATTACA (forward) and GGCCAG GTTCTCTTTTTATTTCTG (reverse); for lamin B2, GTGCACAGCGCCAG GTTA (forward) and GTGCACAGCGCCAGGTTA (reverse); and for beta-globin, AGGAGAAGACTGCTGTCAATGC (forward) and TCCTTGAGCCT CTCTTATAACCTTGA (reverse).
Genome maintenance assay.Cells (approximately 1⫻106
cells per sample) were collected and resuspended in 100l SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.0.). After brief sonication, IP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris [pH 8.0], 167 mM NaCl) was added to 1 ml and then incubated with proteinase K for 2 to 3 h at 50°C. Three hundred microliters was removed and subjected to phenol-chloroform extraction and ethanol precipitation. Precipitated DNA was then assayed by real-time PCR using primers for the DS region of EBV and normalized by the cellular DNA signal at the actin gene locus.
HDAC activity assay.A histone deacetylase (HDAC) activity assay kit (Bio-Vision, CA) was used to measure the HDAC activity. Briefly, the HDAC flu-orometric substrate and assay buffer were added to cell extract or IP samples in a 96-well format and incubated at 37°C for 30 min. The reaction was stopped by adding lysine developer, and the mixture was incubated for another 30 min at 37°C. A fluorescence plate reader with excitation at 355 nm and emission at 460 nm was used to quantify HDAC activity. The Bradford protein assay (Bio-Rad, CA) was used to measure the concentration of protein from input. The HDAC activity is presented as the relative fluorescence units per microgram input protein.
Additional methods.ChIP assays were performed as described previously (48). Centrifugal elutriation was performed as described previously (10, 48). Cell cycle progression and length of S phase were measured by BrdU pulse-labeling com-bined with PI staining as described previously (3). RRM2 small interfering RNA
was obtained from Dharmacon as a Smartpool and was transfected with Dhar-macon transfection reagent according the manufacturer’s protocol. TRF2 shRNA has been described previously (7).
RESULTS
Delay in replication timing of EBV.Previous studies of the
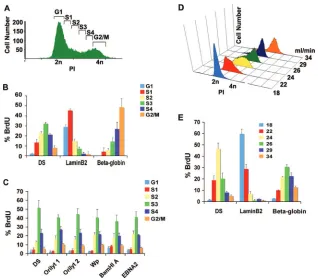
replication timing of EBV have been controversial. An earlier study reported that EBV replicates in early S phase (28), while a subsequent study using a different method found that EBV episomes replicate in late S phase (4). To help resolve this question, we assayed EBV genome replication timing by two different methods using Raji cells, an EBV-positive Burkitt lymphoma cell line that lacks functional viral DNA polymerase and replicates exclusively by episomal replication (Fig. 1). For the first method, Raji cells were pulse-labeled with BrdU for 30 min and then subjected to flow cytometry using FACS to frac-tionate cells based on their stage in the cell cycle. DNA syn-thesis (BrdU incorporation) was measured by BrdU-specific IP and real-time PCR analysis. Using this method, we compared EBV DNA replication to that of two cellular replicons with known temporal regulation of replication. The lamin B2 origin has been characterized as one of the earliest-firing origins, while the beta-globin locus origin is known to replicate late in the cell cycle. As expected, we found that lamin B2 replicates in the early fractions (S1), while beta-globin replicates in the late fractions (S4 and G2/M). In contrast, the EBV DS region
was found to replicate after lamin B2 (primarily in fraction S3) but not as late as beta-globin (Fig. 1B). To determine if other regions of the EBV genome replicated at the same time as DS, we compared six different EBV genome locations spanning essentially the entire 170 kb of EBV. We found that all regions tested replicated in the same S phase fraction as DS (Fig. 1C). This suggests that the EBV genome replicates within a single stage of the S phase, which is consistent with other reports that estimate that EBV latent genome replication is completed within a 20- to 40-min window in S phase (29).
The S phase timing of replication was further evaluated by a second method using centrifugal elutriation, which separates cells based on their morphology. Centrifugal elutriation suc-cessfully separated Raji cells according to their stage of the cell cycle as determined by FACS analysis after PI staining (Fig. 1D). Cells were pulse-labeled with BrdU, fractionated by cen-trifugal elutriation, and then analyzed by BrdU IP and real-time PCR analysis for EBV DS region or cellular lamin B2 and beta-globin loci. As with the FACS method, centrifugal elu-triation revealed that DS replicates in mid to late S phase, while lamin B2 replicates in the G1and early S fractions and
beta-globin replicates in late S and G2fractions. The S phase
delay in replication timing of EBV genomes was also observed in several different latently infected B-cell lines, including the MutuI Burkitt lymphoma cell line and the lymphoblastoid cell line containing the mini-EBV genome (A39) (see Fig. S1 in the supplemental material). The S phase delay in replication tim-ing profile was further confirmed ustim-ing three-color TaqMan probes to simultaneously measure DS and reference lamin B2 or beta-globin origins in the same PCRs (see Fig. S2 in the supplemental material). While the precise stage of S phase may vary among cell types and experimental designs, the delay
on November 8, 2019 by guest
http://jvi.asm.org/
in replication timing of EBV relative to lamin B remains in-variant in all cell types and experimental protocols.
HU treatment advances the replication timing of EBV.HU
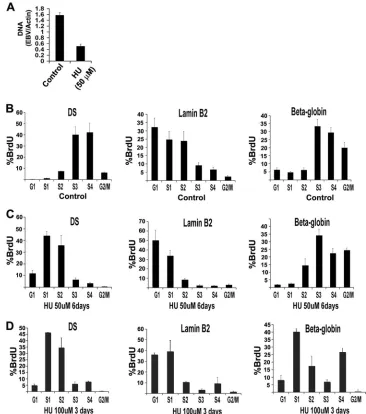
is the only pharmacological agent that has been reported to reduce and eliminate EBV genomes from latently infected cells. The mechanism of HU-induced episome loss is not known. We therefore explored whether HU treatment had any effect on the replication timing of EBV episomes (Fig. 2). First, we demonstrated that treatment of Raji cells with HU reduced EBV episome copy number using real-time PCR analysis of EBV DNA relative to a cellular DNA region in the actin open reading frame. We found that treatment with 50M HU for 6 days led to a⬃3-fold reduction in EBV genome copy number (Fig. 2A). Higher concentrations of HU (⬎200 M) caused cell cycle arrest, and continuous growth in 50M HU reduced Raji cell viability (data not shown). We therefore assayed the replication timing of EBV relative to lamin B2 and beta-globin in untreated Raji cells (control) (Fig. 2B) or in cells treated with 50M HU for 6 days (Fig. 2C) or 100M HU for 3 days (Fig. 2D). We found that treatment with 50M and 100M HU caused a dramatic shift in EBV replication from the S3-S4 fraction in controls to the S1-S2 fraction in treated samples. Lamin B2 replication remained early in HU treatment, while beta-globin replication was slightly advanced in S phase with 50
M and substantially advanced by 100 M HU treatment. Similar effects of HU on OriP replication timing were observed in latently infected Mutu I cells (see Fig. S3 in the
supplemen-tal material). These findings indicate that HU treatment accel-erates the replication timing of EBV, as well as a late-firing cellular replicon at the beta-globin locus.
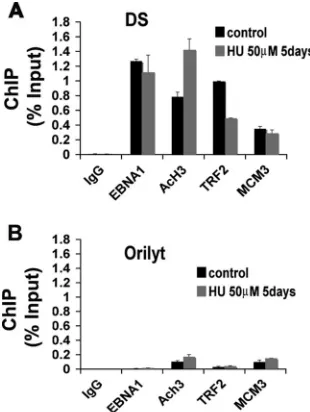
HU alters histone modification and TRF2 binding at OriP. Earlier studies ofOriPrevealed that TRF2 and histones were closely positioned to the EBNA1 binding sites in DS (8, 48). Furthermore, histone H3 positioned at DS was deacetylated in the early S phase. We therefore examined whether HU treat-ment altered the TRF2 binding or histone H3 acetylation at DS (Fig. 3). Raji cells were treated with 50M HU for 5 days and then assayed by ChIP with antibodies specific for EBNA1, AcH3, TRF2, or the cellular replication factor MCM3. We found that HU treatment had a weak effect on EBNA1 bind-ing, but increased AcH3, and decreased TRF2 binding by⬃ 2-fold (Fig. 3A). MCM3 binding did not change, but its cell cycle profile was not examined in these studies. The specificity for DS was demonstrated by comparing the binding at control region OriLyt, which is not an active origin during EBV latent infection in Raji cells. These experiments indicate that HU treatment leads to a remodeling of the DS region of OriP. Specifically, HU induced H3 acetylation and reduced TRF2 binding.
TRF2 site deletion or shRNA depletion of TRF2 advances
replication timing of OriP.Previous studies have shown that
[image:3.585.133.451.66.345.2]shRNA depletion of TRF2 or substitution mutations in TRF2 binding sites in OriPinhibit replication and plasmid mainte-nance (7, 8). We next tested whether shRNA depletion of
FIG. 1. EBV genomes replicate in mid to late S phase in Raji cells. (A) FACS analysis of PI-stained Raji cells was used to sort cells based on their position in the cell cycle. (B) The replication timing of DS, lamin B2, and beta-globin loci was determined by real-time PCR quantification of BrdU-containing DNA after pulse-labeling. Percent BrdU represents the relative distribution of DNA in each fraction of the cell cycle. (C) Different regions of EBV were amplified by real-time PCR, and results are presented as percent BrdU for each locus. (D) Raji cells were fractionated by centrifugal elutriation and then analyzed by FACS after PI staining. (E) Raji cells fractionated by centrifugal elutriation as shown in panel D were then assayed for replication timing of DS, lamin B2, and beta-globin loci. Error bars indicate standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
TRF2 altered the replication timing ofOriP(Fig. 4A and B). For these experiments, we utilized the EBV-positive adherent cell line D98/HR1 because of its high transfection efficiency relative to Raji cells. Transfection of shTRF2 effectively re-duced TRF2 protein without affecting the control protein SNF2h, as indicated by Western blotting (Fig. 4C). As in Raji cells, DS replicated in the later S phase fractions (S4) relative to lamin B2 (S1 to S2) in control transfected D98/HR1 cells. When TRF2 was depleted by TRF2-specific shRNA, DS rep-lication advanced to the S3 fraction, while no change was observed in lamin B2 (Fig. 4A and B). To better evaluate the role of TRF binding sites in regulatingOriPreplication timing, we assayed the replication timing of plasmids containing wt
OriPor a mutantOriPwith substitution mutations in all three nanomer binding sites for TRF2 (OriPnm⫺) (Fig. 4D and E). Plasmids were transfected into HeLa cells and selected with
hygromycin for 10 days. Replication timing was assayed with BrdU pulse-labeling and cell sorting, as in Fig. 1A. We found that plasmids in HeLa cells containingOriPwt replicated in S3 to S4, while plasmids withOriPnm⫺replicated primarily in the S2 fraction. These results suggest that TRF2 binding sites in DS delay the replication timing ofOriP-containing plasmids. Furthermore, these results suggest that the delay in replication timing correlates with the enhanced plasmid stability ofOriP
wt relative toOriPnm⫺.
[image:4.585.106.472.68.482.2]VPA alters OriP protein binding and histone modification. The other major effect of HU was an increase in histone H3 acetylation. We therefore tested the effect of several HDACs on the replication timing of EBV genomes in Raji cells. We found that TSA and NaB had a potent effect on the replication timing but were highly toxic to Raji cells (see Fig. S4 in the supplemental material). As an alternative, we used VPA,
FIG. 2. HU advances replication timing of EBV. (A) Raji cells were treated with 50M HU or control for 6 days and then assayed for EBV genome copy number. EBV copy number was assayed by real-time PCR analysis of DS relative to cellular actin. (B to D) Replication timing of Raji cell DS, lamin B2, or beta-globin was analyzed by FACS under control conditions (B) or with 50M HU for 6 days (C) or 100M HU for 3 days. Error bars indicate standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
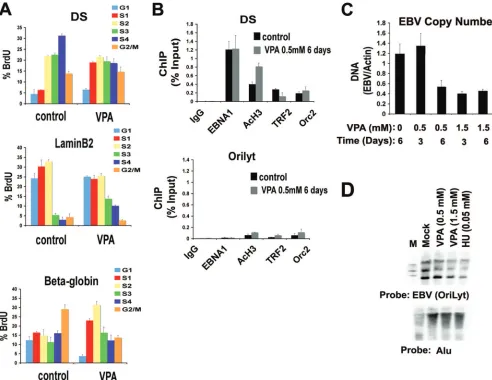
which inhibits class I HDAC and is less toxic to Raji cells. VPA was assayed for its effect on replication timing in Raji cells (Fig. 5A). VPA treatment (0.5 mM for 6 days) advanced the EBV replication from S4 to mostly S1 and S2. VPA had little effect on lamin B2 replication but caused an acceleration of the late-firing beta-globin from the G2/M fraction to S2. This
in-dicates that VPA treatment advances replication timing of EBV and a cellular late-firing replicon to early stages of the cell cycle.
The effect of VPA treatment ofOriPbinding proteins was examined using ChIP assay. We found that treatment with 0.5 mM VPA for 6 days led to a⬃2-fold increase in AcH3 and a corresponding⬃2-fold decrease in TRF2 binding to the DS region of OriP. No significant changes were observed in EBNA1 or ORC2 binding, and no significant binding of any of these factors were observed at control region OriLyt (Fig. 5B, lower panel). These findings indicate that VPA affects protein binding and histone modification atOriPsimilarly to HU treat-ment (Fig. 3) and TRF2 depletion (Fig. 4).
VPA causes EBV episome loss.Since VPA produced effects
similar to those of HU on EBV replication timing and protein interactions, we tested whether VPA caused the loss of EBV genome copy number. Raji cells were incubated with 0.5 or 1.5 mM VPA for 3 or 6 days and then assayed by real-time PCR
[image:5.585.84.239.69.275.2]FIG. 3. HU induces H3 acetylation and TRF2 dissociation from DS. Raji cells were treated with 0M (control) or 50M HU for 5 days and then assayed by ChIP with antibodies to EBNA1, AcH3, TRF2, MCM3, or control IgG. ChIP DNA was assayed by real-time PCR with primers specific for DS (A) or for OriLyt (B). Error bars indicate standard deviations.
FIG. 4. TRF2 binding delays replication timing ofOriP. (A and B) Replication timing was analyzed by the FACS method in EBV-positive D98/HR1 cells after transfection with control or shTRF2 expression plasmids. Replication timing was determined for DS (A) or lamin B2 (B) loci. (C) Western blot of D98/HR1 cells transfected with the control or shTRF2 plasmids used for the experiments shown in panels A and B. Immunoblots with anti-TRF2 (top panel) or anti-SNF2h (lower panel) are shown. (D) Replication timing of DS region onOriPwt orOriPnm⫺ plasmids in HeLa cells after 10 day of hygromycin (100g/ml) selection. (E) Same as in panel D, but replication timing was determined for the lamin B2 locus. Error bars indicate standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
for EBV DNA relative to cellular actin DNA (Fig. 5C). We found that 1.5 mM VPA caused a⬃2- to 3-fold reduction in EBV copy number at 3 and 6 days. Similarly, we found that 0.5 mM VPA caused⬃3-fold reduction at 6 days, although there was little effect detected after 3 days. The EBV genome copy number reduction caused by VPA and HU treatment of Raji cells was also observed by Southern blot analysis (Fig. 5D). An OriLyt probe was used to detect the EBV genome (upper panel) and an Alu probe was used as a loading control for total cellular DNA (lower panel). These findings indicate that VPA eliminates EBV genome copy number in a dose- and time-dependent manner.
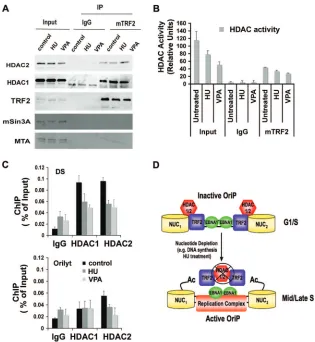
HDAC1 and -2 associate with TRF2 and are inhibited by HU.HU treatment caused a significant increase in histone H3 acetylation atOriP(Fig. 3A). To determine if H3 acetylation at
OriPis partially regulated by TRF2 interaction with HDAC complexes, we assayed TRF2 IPs for the presence of class I
HDACs (Fig. 6A). We found that HDAC2 associates with TRF2 as measured by IP from Raji extracts (Fig. 6A, top panel). However, treatment with HU or VPA did not signifi-cantly reduce HDAC2 interaction with TRF2. TRF2 did not associate with mSin2 or MTA, two abundant components of well-characterized class I HDAC-containing complexes (Fig. 6A, lower panels). The effect of HU and VPA on HDAC activity was measured by a fluorescence-based HDAC assay (Fig. 6B). We found that both HU and VPA inhibited total cellular HDAC activity by⬃20 and⬃50% and inhibited to a lesser extent the HDAC activity associated TRF2 IPs (Fig. 6B). The association of HDAC1 and HDAC2 withOriPwas mea-sured by ChIP assay in Raji cells after HU, VPA, or mock treatment (Fig. 6C). Both HU and VPA caused HDAC1 and HDAC2 dissociation from OriP. This dissociation correlates with the partial loss of TRF2 binding atOriP(Fig. 2A). Taken together, these results suggest that HU, as well as VPA,
dis-FIG. 5. VPA advances EBV replication timing and reduces viral genome copy number. (A) Raji cells treated with 0.5 mM VPA or control for 6 days and then assayed for replication timing at DS, lamin B2, and beta-globin loci. (B) ChIP assays with antibodies to EBNA1, AcH3, TRF2, ORC2, or control IgG by real-time PCR for binding at DS (top panel) or control OriLyt (lower panel) regions of EBV. (C) EBV genome copy number was assayed in Raji cells treated with 0.5 mM VPA for 3 or 6 days or with 1.5 mM VPA for 3 or 6 days or in control untreated cells, as indicated. (D) Southern blot of Raji cell DNA after mock treatment (lane 1) or treatment with 0.5 mM VPA (lane 2), 1.5 mM VPA (lane 3), or 0.05 mM HU (lane 4) for 6 days. DNA was cut with BamHI and hybridized with a probe to EBV OriLyt (top panel) or Alu repeats (lower panel). M, 1-kb molecular weight ladder. Error bars indicate standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.44.536.68.448.2]rupts TRF2 and HDAC association with OriP and that this dissociation correlates with advanced replication timing (Fig. 6D).
DISCUSSION
EBV latent infection has been estimated to contribute to
⬃1% of all human cancers (31). The mechanisms that control latent cycle replication and episome stability are therefore of great interest for potential targets of therapeutic intervention. HU treatment has been used in a clinical setting to eradicate latent viral genomes, but its mechanism of action has remained poorly understood. In this work, we found that HU can alter a programmed delay in the replication timing of EBV genomes and OriP-containing episomes (Fig. 1 and 2). We found that HU treatment caused the dissociation of TRF2 from the DS region of OriPand altered the normal cell cycle pattern of histone deacetylation that occurs in early S phase at DS (Fig. 3). The importance of histone deacetylation was further sub-stantiated by experiments with HDAC inhibitors (e.g., VPA),
which prevented the early S phase histone deacetylation at OriP, advanced replication timing, and destabilized the latent EBV episome (Fig. 5). HU inhibited HDAC activity and elim-inated TRF2-HDAC binding atOriP(Fig. 6). These findings suggest that a TRF2-HDAC complex mediates a replication timing delay important for EBV genome stability.
[image:7.585.134.449.66.408.2]The latent episome of EBV can replicate for many genera-tions without significant loss of copy number. Metaphase chro-mosome tethering correlates well with plasmid stability for EBV, Kaposi’s sarcoma-associated herpesvirus, and human papillomavirus, but other factors must also contribute to this process (25). Efficient plasmid maintenance depends on un-known epigenetic changes in the viral chromosome and the coupling of DNA synthesis to plasmid segregation (24, 27). Therefore, it is likely that DNA and chromatin structures formed atOriPcontribute to the plasmid maintenance process. Our previous studies of OriP found that DS is flanked by positioned nucleosomes that are subject to cell cycle changes in histone modifications (48). Genetic studies have demonstrated
FIG. 6. HU and VPA alter HDAC activity and association withOriP. (A) Raji cells treated with HU (0.05 mM) or VPA (0.5 mM) or mock treated for 6 days were subject to IP with control IgG or TRF2 antibody. Immunoprecipitates were then assayed by Western blotting with antibodies specific for HDAC2, HDAC1, TRF2, mSIN3, and MTA, as indicated. (B) HDAC activity in Raji cell extracts and IPs was measured by fluorescence assay (BioVision, Inc). Raji cells were treated with HU or VPA or mock treated as described for panel A. Input extracts were normalized by Bradford assay using bovine serum albumin as a standard. (C) ChIP assays with antibodies to HDAC1, HDAC2, or control IgG were used to measure binding toOriP(top panel) or control OriLyt (lower panel) in Raji cells after 6 days of HU, VPA, or mock treatment, as described for panel A. (D) Schematic model of TRF2-HDAC regulation ofOriPreplication in early G1/S and its subsequent derepression of replication in
mid/late S phase.
on November 8, 2019 by guest
http://jvi.asm.org/
that the TRF2 binding sites within DS also contribute signifi-cantly to episome stability (7, 8, 26). More recent studies found that DNA recombination proteins and replication pausing at DS is also important for episome stability (10). The new find-ings from this study, which indicated that replication timing is coupled to EBV episome stability, further support a model that complex epigenetic regulation and checkpoint mechanisms contribute to EBV genome maintenance during latent infec-tion.
The effect of HU on EBV episome maintenance has been well established, but its mechanism of action has not been elucidated (6, 18, 40). In this work, we found that HU treat-ment altered the replication timing and chromatin organiza-tion ofOriP. Several mechanisms have been reported to reg-ulate replication timing. InSaccharomyces cerevisiae, deletion of the HDACRpd3caused late-firing origins to fire synchro-nously with early replication origins (42). Control of replica-tion timing has been linked to the S phase checkpoint protein RAD53, indicating that replication timing was important for genome stability during S phase (12, 37). In higher eukaryotes, early replication timing correlates with histone acetylation, and tethering of HDAC2 delays replication timing (16). Replica-tion timing is also controlled by the intra-S phase checkpoint kinases ATM and ATR, which regulate origin firing in re-sponse to replication stress (36). Moreover, dormant replica-tion origins can be activated during replicareplica-tion stress induced by low levels of HU (15). This suggests that replication timing is regulated by mechanisms that link S phase checkpoints with histone acetylation. In concordance with these models, we found that HU treatment prevented the normal histone H3 deacetylation that occurs at the DS region ofOriPin early S phase (Fig. 2 and 3). We propose that OriP is subject to a TRF2-HDAC checkpoint that regulates replication initiation at DS by histone modification. Histone deacetylation at DS may also explain why replication does not always initiate from
OriP(29, 30).
HU is a known inhibitor of ribonucleotide reductase, which is required to generate deoxynucleoside triphosphates for DNA replication during S phase. At high concentrations (⬃1 mM), HU can provoke significant replication defects and cause cell cycle arrest. However, at the low concentrations used in our experiments (⬃50 M), HU did not cause a cell cycle arrest (see Fig. S3 in the supplemental material) and did not elicit an S phase checkpoint response (data not shown). Nev-ertheless, low-level HU treatment alters the replication timing ofOriPand destabilizes EBV genome maintenance. Our stud-ies indicate that HU treatment operates through ribonucle-otide reductase inhibition, since small interfering RNA deple-tion of the RRM2 subunit phenocopies HU effects on EBV replication timing, histone deacetylation at DS, and genome stability (see Fig. S7 in the supplemental material). HU treat-ment may affect EBV replication timing by altering the rate at which S phase progresses. However, low levels of HU had only modest effects on S phase duration and the percentage of cells in S phase (see Fig. S5 in the supplemental material). In con-trast, HU treatment caused a significant loss of TRF2 binding and a corresponding increase in histone H3 acetylation at OriP (Fig. 3). We suggest that low-level HU mimics the nucleotide depletion observed in mid-S phase and that nucleotide deple-tion evokes a signal that reduces TRF2 binding, inhibits
HDAC activity, and promotes replication initiation at OriP
(Fig. 6D). Under normal conditions (no HU treatment), the TRF2-HDAC complex may prevent OriP from initiating among the first wave of cellular replicons. This would protect EBV genomes from replicating in cellular conditions that are mutagenic or suboptimal for the completion of DNA replica-tion. The delay in replication timing may also favor the asso-ciation of EBV genomes with chromatin and segregation ma-chinery necessary for episome stability.
Epigenetic events have been implicated in the establishment of a stable EBV episome. Our previous studies indicated that nucleosome positioning and histone deacetylation at the DS region of OriP were important for plasmid stability. We now show that disruption of the normal cell cycle histone deacety-lation pattern can cause genome instability. HDAC inhibitors (e.g., VPA, TSA, and NaB) promoted the loss of EBV ge-nomes, similar to HU. Remarkably, HDAC inhibitors are well-established initiators of the EBV lytic cycle and induce apop-tosis in cells where lytic replication is blocked (14, 20). In cells where lytic cycle replication is blocked, like for the replication defective genomes of Raji cells, HDAC inhibitors lead to a loss of EBV episomes. Our data indicates that HDAC inhibitors have a primary effect on chromatin organization atOriPand a fundamental role in maintaining the stable viral episome. This raises the possibility that lytic replication is a programmed response to cellular conditions that destabilize episome main-tenance. If so, lytic induction therapies, which combine reac-tivation signals with inhibitors of viral lytic replication, may have the added benefit of eliminating nonreactivated latent virus genomes (13).
In conclusion, we have implicated the TRF2-HDAC com-plex in the regulated delay of replication timing ofOriP. We provide evidence that TRF2-HDAC is an important target of HU, which is the only clinically tested pharmacological agent that eliminates EBV episomes from latently infected cells. We found that HU treatment causes a dissociation of TRF2-HDAC and an inhibition of TRF2-HDAC activity. This corresponds to the loss of the programmed delay in replication timing and a decrease in episome stability. We propose that TRF2-HDAC provides OriP with a replication checkpoint that enhances viral genome stability. These finding provide new insights into the mechanism through which HU eliminates EBV genomes from infected cells, and they may provide new opportunities to dis-rupt latent EBV infection in human tumors.
ACKNOWLEDGMENTS
We thank Andreas Wiedmer for technical support and the Wistar Cancer Center Core Facilities for their assistance.
This work was funded by NCI grant CA93606 to P.M.L, an NIH NRSA grant to A.S., and a Lymphoma Research Foundation Fellow-ship to J.Z.
REFERENCES
1.Adams, A.1987. Replication of latent Epstein-Barr virus genomes in Raji cells. J. Virol.61:1743–1746.
2.Bashaw, J. M., and J. L. Yates.2001. Replication fromoriPof Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J. Virol.75:10603–10611.
3.Bhaskara, S., B. J. Chyla, J. M. Amann, S. K. Knutson, D. Cortez, Z. W. Sun, and S. W. Hiebert.2008. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell30:61–72. 4.Carroll, S. M., J. Trotter, and G. M. Wahl.1991. Replication timing control can be maintained in extrachromosomally amplified genes. Mol. Cell. Biol. 11:4779–4785.
on November 8, 2019 by guest
http://jvi.asm.org/
5.Chaudhuri, B., H. Xu, I. Todorov, A. Dutta, and J. L. Yates.2001. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc. Natl. Acad. Sci. USA98:10085–10089.
6.Chodosh, J., V. P. Holder, Y. J. Gan, A. Belgaumi, J. Sample, and J. W. Sixbey.1998. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. J. Infect. Dis.177:1194–1201. 7.Deng, Z., C. Atanasiu, J. S. Burg, D. Broccoli, and P. M. Lieberman.2003.
Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate repli-cation of Epstein-Barr virus OriP. J. Virol.77:11992–12001.
8.Deng, Z., L. Lezina, C. J. Chen, S. Shtivelband, W. So, and P. M. Lieberman. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell9:493–503.
9.Dhar, S. K., K. Yoshida, Y. Machida, P. Khaira, B. Chaudhuri, J. A. Wohlschlegel, M. Leffak, J. Yates, and A. Dutta.2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287–296.
10.Dheekollu, J., Z. Deng, A. Wiedmer, M. D. Weitzman, and P. M. Lieberman. 2007. A role for MRE11, NBS1, and recombination junctions in replication and stable maintenance of EBV episomes. PLoS One2:e1257.
11.Donaldson, A. D.2005. Shaping time: chromatin structure and the DNA replication programme. Trends Genet.21:444–449.
12.Early, A., L. S. Drury, and J. F. Diffley.2004. Mechanisms involved in regulating DNA replication origins during the cell cycle and in response to DNA damage. Philos. Trans. R. Soc. London B359:31–38.
13.Feng, W. H., G. Hong, H. J. Delecluse, and S. C. Kenney.2004. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J. Virol. 78:1893–1902.
14.Feng, W. H., and S. C. Kenney.2006. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expres-sion. Cancer Res.66:8762–8769.
15.Ge, X. Q., D. A. Jackson, and J. J. Blow.2007. Dormant origins licensed by excess Mcm2 7 are required for human cells to survive replicative stress. Genes Dev.21:3331–3341.
16.Goren, A., A. Tabib, M. Hecht, and H. Cedar.2008. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev.22:1319–1324.
17.Jiang, R., M. Kanamori, Y. Satoh, M. Fukuda, K. Ikuta, M. Murakami, and T. Sairenji.2003. Contrasting effects of hydroxyurea on cell growth and reduction in Epstein-Barr virus genomes in EBV-infected epithelioid cell lines vs Burkitt’s lymphoma cell lines. J. Med. Virol.70:244–252. 18.Jiang, R., J. L. Zhang, Y. Satoh, and T. Sairenji.2004. Mechanism for
induction of hydroxyurea resistance and loss of latent EBV genome in hydroxyurea-treated Burkitt’s lymphoma cell line Raji. J. Med. Virol.73: 589–595.
19.Kieff, E.2007. Epstein-Barr virus and its replication, 5th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA.
20.Klass, C. M., L. T. Krug, V. P. Pozharskaya, and M. K. Offermann.2005. The targeting of primary effusion lymphoma cells for apoptosis by inducing lytic replication of human herpesvirus 8 while blocking virus production. Blood105:4028–4034.
21.Klein, E., L. L. Kis, and G. Klein.2007. Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene26:1297–1305.
22.Koons, M. D., S. V. Scoy, and J. Hearing. 2001. The replicator of the Epstein-Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J. Virol.75:10582–10592.
23.Kuttler, F., and S. Mai.2007. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin. Can-cer Biol.17:56–64.
24.Leight, E. R., and B. Sugden.2001. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell. Biol.21:4149– 4161.
25.Lindner, S. E., and B. Sugden.2007. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal repli-cation in human cells. Plasmid58:1–12.
26.Lindner, S. E., K. Zeller, A. Schepers, and B. Sugden.2008. The affinity of EBNA1 for its origin of DNA synthesis is a determinant of the origin’s replicative efficiency. J. Virol.82:5693–5702.
27.Nanbo, A., A. Sugden, and B. Sugden.2007. The coupling of synthesis and partitioning of EBV’s plasmid replicon is revealed in live cells. EMBO J. 26:4252–4262.
28.Nonoyama, M., and A. Tanaka.1975. Plasmid DNA as a possible state of Epstein-Barr virus genomes in nonproductive cells. Cold Spring Harbor Symp. Quant. Biol.39:807–810.
29.Norio, P., and C. L. Schildkraut.2004. Plasticity of DNA replication initia-tion in Epstein-Barr virus episomes. PLoS Biol.2:e152.
30.Norio, P., and C. L. Schildkraut.2001. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science294:2361–2364.
31.Parkin, D. M.2006. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer118:3030–3044.
32.Rickinson, A. B., and E. Kieff.2007. Epstein-Barr virus, 5th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. 33.Ritzi, M., K. Tillack, J. Gerhardt, E. Ott, S. Humme, E. Kremmer, W.
Hammerschmidt, and A. Schepers.2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell Sci. 116:3971–3984.
34.Schepers, A., M. Ritzi, K. Bousset, E. Kremmer, J. L. Yates, J. Harwood, J. F. Diffley, and W. Hammerschmidt.2001. Human origin recognition com-plex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J.20:4588–4602.
35.Schwaiger, M., and D. Schubeler.2006. A question of timing: emerging links between transcription and replication. Curr. Opin. Genet. Dev.16:177–183. 36.Shechter, D., V. Costanzo, and J. Gautier.2004. ATR and ATM regulate the
timing of DNA replication origin firing. Nat. Cell Biol.6:648–655. 37.Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi, C. Obuse,
T. Tsurimoto, and H. Yoshikawa.1998. Regulation of DNA-replication origins during cell-cycle progression. Nature395:618–621.
38.Slobod, K. S., G. H. Taylor, J. T. Sandlund, P. Furth, K. J. Helton, and J. W. Sixbey.2000. Epstein-Barr virus-targeted therapy for AIDS-related primary lymphoma of the central nervous system. Lancet356:1493–1494. 39.Snapka, R. M., and A. Varshavsky.1983. Loss of unstably amplified
didrofolate reductase genes from mouse cells is greatly accelerated by hy-droxyurea. Proc. Natl. Acad. Sci. USA80:7533–7537.
40.Srinivas, S. K., J. T. Sample, and J. W. Sixbey.1998. Spontaneous loss of viral episomes accompanying Epstein-Barr virus reactivation in a Burkitt’s lymphoma cell line. J. Infect. Dis.177:1705–1709.
41.Sugden, B., and E. R. Leight.2001. Molecular mechanisms of maintenance and disruption of virus latency. Curr. Top. Microbiol. Immunol.258:3–11. 42.Vogelauer, M., L. Rubbi, I. Lucas, B. J. Brewer, and M. Grunstein.2002.
Histone acetylation regulates the time of replication origin firing. Mol. Cell 10:1223–1233.
43.Yarbro, J. W.1992. Mechanism of action of hydroxyurea. Semin. Oncol. 19:1–10.
44.Yates, J. L., S. M. Camiolo, and J. M. Bashaw.2000. The minimal replicator of Epstein-Barr virus oriP. J. Virol.74:4512–4522.
45.Yates, J. L., and N. Guan.1991. Epstein-Barr virus-derived plasmids repli-cate only once per cell cycle and are not amplified after entry into cells. J. Virol.65:483–488.
46.Young, L. S., and A. B. Rickinson.2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer4:757–768.
47.Zhang, J., F. Xu, T. Hashimshony, I. Keshet, and H. Cedar.2002. Estab-lishment of transcriptional competence in early and late S phase. Nature 420:198–202.
48.Zhou, J., C. M. Chau, Z. Deng, R. Shiekhattar, M. P. Spindler, A. Schepers, and P. M. Lieberman.2005. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J.24:1406–1417.
49.Zink, D.2006. The temporal program of DNA replication: new insights into old questions. Chromosoma115:273–287.