0022-538X/10/$12.00 doi:10.1128/JVI.00342-09
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
The Potent Anti-HIV Activity of CXCL12
␥
Correlates with Efficient
CXCR4 Binding and Internalization
䌤
Jeffrey D. Altenburg,
1Qingwen Jin,
1Bashar Alkhatib,
2and Ghalib Alkhatib
1*
Department of Microbiology and Immunology, Indiana University School of Medicine,1and Department of Biology,
Indiana University Purdue University School of Science,2Indianapolis, Indiana
Received 16 February 2009/Accepted 3 December 2009
We previously demonstrated that the naturally occurring splice variant stromal cell-derived factor 1␥/ CXCL12␥ is the most potent CXCL12 isoform in blocking X4 HIV-1, with weak chemotactic activity. A conserved BBXB domain (B for basic and X for any residue) located in the N terminus (24KHLK27) is found in all six isoforms of CXCL12. To determine whether the potent antiviral activity of CXCL12␥is due to the presence of the extra C-terminal BBXB domains, we mutated each domain individually as well as in combi-nation. Although binding of CXCL12␥to heparan sulfate proteoglycan (HSPG) was 10-fold higher than that observed with CXCL12␣, the results did not demonstrate a direct correlation between HSPG binding and the potent antiviral activity. CXCL12␥ mutants lacking the conserved BBXB domain (designated␥B1) showed increased binding to HSPG but reduced anti-HIV activity. In contrast, the mutants lacking the C-terminal second and/or third BBXB domain but retaining the conserved domain (designated B2, B3, and B23) showed decreased binding to HSPG but increased anti-HIV activity. The B2, B3, and B23 mutants were associated with enhanced CXCR4 binding, receptor internalization, and restored chemotaxis. Internalization of CXCR4 was more potent with CXCL12␥ than with CXCL12␣ and was significantly reduced when the conserved BBXB domain was mutated. We concluded that the observed potent anti-HIV-1 activity of CXCL12␥ is due to increased affinity for CXCR4 and to efficient receptor internalization.
Chemokines are small, structurally related chemoattractant cytokines characterized by conserved cysteine residues. Based on the positions of the first N-terminal cysteines, chemokines fall into four subfamilies. The CC and CXC subfamilies have been well characterized. The CC subfamily includes the fol-lowing: regulated on activation, normal T-cell expressed and secreted (RANTES), monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory peptides 1 (MIP-1). The prototype of the CXC subfamily is interleukin-8 (IL-8)/ CXCL8. The C chemokine (lymphotactine) and CX3C chemo-kine (fractalchemo-kine) subfamilies were recently identified (reviewed in reference 30). The physiological activities of chemokines are mediated by the selective recognition and activation of chemo-kine receptors belonging to the seven-membrane-domain G-pro-tein-coupled receptor superfamily (GPCRs). In addition, chemo-kines also bind to glycosaminoglycans (GAGs) through distinct binding sites. Chemokine binding to GAGs on cells, particularly endothelial cells, results in chemotactic chemokine gradients that allow correct presentation of chemokines to leukocytes, therefore enabling target cells to cross the endothelial barrier and migrate into tissues (reviewed in reference 10).
Stromal cell-derived factor 1 (SDF-1)/CXCL12 is a member of the CXC chemokine family and is a key regulator of B-cell lymphopoiesis, hematopoietic stem cell mobilization, and leu-kocyte migration (reviewed in reference 10). CXCL12 was originally thought to mediate these processes through the
sin-gle receptor CXCR4 (9). However, later studies demonstrated that RDC-1/CXCR7 is also a receptor for CXCL12 (6, 11). CXCL12 has also been shown to block HIV-1 infection (5). There are two known human splice variants of CXCL12, re-ferred to as CXCL12␣and CXCL12(27). The genomic struc-ture of the CXCL12 gene revealed that human CXCL12␣and CXCL12are encoded by a single gene and arise by alterna-tive splicing. The cDNAs corresponding to CXCL12␣ and CXCL12encode proteins of 89 and 93 amino acids, respec-tively. A third splice variant, classified as CXCL12␥, has been identified in rats (14). The human equivalent of CXCL12␥was recently identified among other splice variants of CXCL12 (33). The novel human splice variants CXCL12␥, CXCL12ε, CXCL12␦, and CXCL12 (also reported as CXCL12 [33]) are expressed through alternative splicing events that result in different exons being added to the same first three exons. Therefore, all six splice variants of CXCL12 are identical in the first 88 amino acid residues from the amino terminus.
It has been demonstrated that CXCL12␣ and - are ex-pressed in numerous tissues, with the highest expression levels in the liver, pancreas, and spleen (33). The mRNA encoding CXCL12␥was detectable in the adult human heart but hardly detectable in several other tissues. On the other hand, CXCL12␦, -ε, and -could be detected in several human adult and fetal tissues, with the pancreas expressing the highest lev-els (33). Recent studies have demonstrated the tissue expres-sion of CXCL12␥in the adult heart (24). We previously dem-onstrated that CXCL12␥ is the most potent anti-HIV-1 inhibitor, with the weakest chemotactic activity and no detect-able enhancing activity for hematopoietic progenitor cell sur-vival or replating capacity (2). The first three exons present in the CXCL12␥splice variant are identical to those found in
* Corresponding author. Present address: Department of Biomedi-cal Sciences and Center of Excellence for Infectious Diseases, Paul L. Foster School of Medicine, Texas Tech University Health Sciences Center, 5001 El Paso Drive, El Paso, TX 79905. Phone: (915) 783-1245. Fax: (915) 783-1271. E-mail: [email protected].
䌤Published ahead of print on 16 December 2009.
2563
on November 8, 2019 by guest
http://jvi.asm.org/
demonstrated that a mutation of CXCL12␣in the KHLK domain reduces the antiviral activity at least 50 percent without affecting the chemotactic activity (4, 29). It was proposed that chemokine binding to HSPG might concentrate the chemokine near the CXCR4 receptor or form a haptotactic chemokine gradient.
In this study, we analyzed the mechanism of the potent antiviral activity of CXCL12␥. We examined the role of the additional BBXB domains of CXCL12␥in the observed bio-logical activities of CXCL12␥. Mutations in CXCL12␥were introduced to knock out the BBXB domains either individually or in combination. We analyzed receptor internalization and binding affinities of the mutant chemokines for CXCR4 and HSPG. The results demonstrate that the potent anti-HIV ac-tivity of CXCL12␥is due to its efficient binding and internal-ization of CXCR4. The results provide important insight into the structure-function relationship of CXCL12␥ and suggest that determinants other than the BBXB domains are involved in the observed biological activities of CXCL12␥.
MATERIALS AND METHODS
Cells and other reagents.All cell lines were obtained from the American Type Culture Collection (Rockville, MD). The HeLa, TZM-bl, LM(tk⫺), and Sog9 cell lines were maintained in Dulbecco modified Eagle medium (DMEM; Qual-ity Biologicals, Gaithersburg, MD) containing 10% fetal bovine serum (FBS) and antibiotics. The TZM-bl cell line, previously designated JC53-bl (clone 13), is a HeLa cell line that expresses endogenous CXCR4. The parental cell line (JC.53) stably expresses large amounts of CD4 and CCR5. The TZM-bl cell line is CD4⫹ CXCR4⫹CCR5⫹(21, 31) and was generated from JC.53 cells by introducing separate integrated copies of the luciferase and-galactosidase (-Gal) genes under the control of the HIV-1 promoter. The TZM-bl indicator cell line enables simple and quantitative analysis of HIV by use of either-Gal or luciferase as a reporter. The CEM T lymphoblast cell line was maintained in RPMI 1640 containing 10% FBS and antibiotics. Human peripheral blood mononuclear cells (PBMCs) were prepared by Ficoll-Hypaque fractionation of cell concentrates obtained from healthy donors seronegative for hepatitis B virus and HIV. PBMCs were activated with 10g/ml phytohemagglutinin for 3 days in RPMI 1640 containing 10% FBS.
The 12G5 monoclonal anti-CXCR4 antibody conjugated to phycoerythrin (PE) was purchased from BD Biosciences Pharmingen (Franklin Lakes, NJ). AMD3100 was a gift from AnorMed Corp. (Langley, British Columbia, Canada). The CXCL12 expression vectors were previously constructed in our lab (2), using the pET32a expression plasmid from Novagen (Madison, WI). An antibody to heparin sulfate (HepSS-1) and its isotype were purchased from U.S. Biologicals. The HepSS-1 antibody recognizes an epitope in heparan sulfate glycosamino-glycan (HS-GAG). This epitope is closely related to the O-sulfated and N-acetylated glucosamine residue linked to glucuronic acid in HS-GAG. The an-tibody recognizes heparan sulfate in a variety of fresh and established human, mouse, monkey, rat, hamster, and chicken cell lines.
Mutagenesis of the BBXB domains.The pET32a expression plasmids contain-ing the CXCL12␣and CXCL12␥inserts were mutated according to protocol, using the Gene Tailor point mutagenesis system from Invitrogen. The expression plasmids, previously constructed in our lab (2), were methylated and used as templates in PCRs. The primers for the PCRs were designed in accordance with
this study are described in Fig. 1A.
Expression of recombinant CXCL12 splice variants inE. coli.The wild-type and mutated CXCL12 splice variants were purified and expressed from Esche-richia colias previously described (2). Briefly, The BL21-Gold (DE3)pLysS strain (Stratagene, La Jolla, CA) was transformed with the pET32a recombinants. Single colonies were inoculated into 5 ml of LB medium with antibiotics and shaken overnight. For large-scale expression, the overnight cultures were inoc-ulated into 1 liter of LB medium. The 1-liter culture was shaken until an optical density at 600 nm (OD600) of 0.2 was achieved. IPTG (isopropyl--D
-thiogalac-topyranoside) was added to induce expression, and the cultures were shaken for 4 h. After being shaken, the cultures were centrifuged and the pellets were stored at⫺80°C until purification.
Purification of recombinant CXCL12.Bacterial pellets for the CXCL12 splice variants were suspended in 5 ml lysis buffer per pellet and rotated at room temperature for 1 h. The lysates were then centrifuged, and the pellets contain-ing inclusion bodies were denatured in buffer containcontain-ing 6 M guanidine-HCl. The clarified supernatants were poured onto a Ni-nitrilotriacetic acid (Ni-NTA) histidine-binding column (Sigma, St. Louis, MO) equilibrated with 10 column volumes of denaturing binding buffer. Both ends of the column were immediately capped, and the columns were rotated at 4°C overnight. The denatured proteins were refolded on the column by stepwise removal of the denaturant as previously described (19). The columns were washed with decreasing levels of urea followed by increasing levels of imidazole to remove nonspecific bacterial contaminants. The proteins were eluted with 2 ml elution buffer, and the elution buffer was exchanged with phosphate-buffered saline (PBS) on a PD-10 desalting column. Protein concentrations were quantified by the Bradford assay (Bio-Rad, Hercu-les, CA) and analyzed for size and purity by SDS-PAGE. The proteins were also analyzed by Western blotting using a polyclonal antibody to the histidine tag that was present in all constructs (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The relative concentrations of the purified proteins were converted to nanomolar values by using the predicted molecular weight of each recombinant.
Chemotaxis assays.Migration of CEM T lymphoblasts was measured as pre-viously described (2). Briefly, CEM T lymphoblast cells were suspended at a density of 2⫻106cells/ml in chemotaxis medium (Iscove’s modified Dulbecco’s
medium [IMDM]) supplemented with 0.5% bovine serum albumin (BSA). The chemokines, with or without 100 nM AMD3100, were suspended in chemotaxis medium at the concentrations indicated in Fig. 2. The suspended chemokines (in 0.6 ml) were first added to the wells of a 24-well plate, followed by a 5- m-pore-size trans-well membrane for each well (Costar; Corning Incorporated, Acton, MA). The cells were added to the inside of the trans-well membrane at a volume of 0.1 ml (2⫻105
cells). The plates were then capped and incubated for 4 h at 37°C and 6% CO2. After the incubation period, the trans-well membranes were
removed from the wells and the migrated cells counted by flow cytometry. Results are reported as percentages of cells that migrated compared to total cell counts in wells with no trans-well membranes.
HIV-1 Env-mediated cell fusion assay.HIV-1 Env-mediated fusion was per-formed by a vaccinia virus-based reporter assay as previously described (1). Briefly, the target CD4⫹CXCR4⫹CCR5⫹TZM-bl cells (AIDS Reagent Pro-gram, Bethesda, MD) (12) were infected by a vaccinia virus encoding the bac-teriophage T7 RNA polymerase. The target CD4⫺CXCR4⫺LM(tk⫺) and Sog9 cells were separately coinfected with vCB-3 (encoding CD4), vYF-4 (encoding CXCR4), and vTF7-3 (encoding T7 RNA polymerase). For each assay, effector HeLa cells were coinfected by PT7-lacZ vaccinia virus (carrying thelacZreporter gene under the control of the T7 promoter) and vaccinia virus encoding either the X4 LAV Env, the R5 Ba-L Env, or the control Unc Env, an uncleaved HIV-1 Env that has its cleavage site mutated and therefore cannot engage in membrane fusion. The Unc Env was used to measure the nonspecific background in the fusion assay. The target cells were plated in a 96-well plate at 1⫻105cells per
well and treated for 1 h with the wt or mutated CXCL12 variants at increasing
on November 8, 2019 by guest
http://jvi.asm.org/
concentrations. After 1 h of incubation at 37°C and 6% CO2, the Env-expressing
HeLa cells were mixed with the target cells in a 1:1 ratio. The cell mixtures were incubated for 2.5 h at 37°C and then lysed, and the substrate chlorophenol red--D-galactopyranoside (CPRG) was added. The extent of cell fusion was assayed by measuring the amount of-galactosidase produced. When PBMCs were used as target cells, they were infected with PT7-lacZ vaccinia virus, while effector HeLa cells were coinfected with vaccinia virus encoding the T7 RNA polymerase and one of the previously listed HIV-1 Envs.
CXCR4 competitive binding assays.For CXCR4 competitive binding assays, the CD4⫹CXCR4⫹CEM T lymphocytes were used. Indirect binding experi-ments involved the use of fixed concentrations of CXCL12 mixed with escalating doses of a specific antagonist for CXCR4 (AMD3100). The cells were washed twice and suspended in cold binding buffer (50 mM HEPES, 5 mM MgCl2, 1 mM
CaCl2, 150 mM NaCl, pH 7.4, 0.5% BSA) with a 100 nM concentration of each
chemokine in the presence of increasing concentrations of AMD3100. The cells were incubated for 1 h at 4°C and washed once with cold binding buffer and once with PBS–1% BSA. To measure chemokine binding, the cells were suspended with 2g/ml fluorescein isothiocyanate (FITC)-conjugated anti-histidine-tag an-tibody or its isotype control (U.S. Biologicals) and incubated on ice for 1 h. The cells were then washed three times with PBS–1% BSA and suspended in PBS for fluorescence-activated cell sorter (FACS) analysis. The results were normalized to percent binding, with 100% being the geometric mean fluorescence intensity seen with no antagonist added. All assays were performed in triplicate and included a control with no CXCL12 or with a chemokine-like protein (MC148) to verify the background staining.
Direct binding experiments involved the use of125
I-labeled SDF-1␣/CXCL12␣ (purchased from PerkinElmer, Boston, MA) and cold recombinant CXCL12 isoforms and mutants. CEM cells (1 million cells/tube, in triplicate), either untreated or treated with heparanase (10 mU/ml), were incubated with 0.25 nM
125
I-labeled CXCL12␣(specific activity of 2,200 Ci/mmol) and escalating con-centrations of cold CXCL12 chemokines. The cell-chemokine mixtures were incubated at 4°C for 1 h and then washed three times with PBS–0.1% BSA. The cell pellet-associated counts were measured in a gamma counter. Binding was evaluated by calculating the % chemokine binding, where 100% binding was for CEM cells incubated with the125
I-labeled SDF-1␣and no cold chemokine.
Binding to HSPG.Sog9 cells lacking HSPG were originally derived from mouse LM(tk⫺) cells (7). Sog9 cells were previously isolated as chondroitin 4-O-sulfotransferase-1-deficient cells from LM(tk⫺) cells which were partially resistant to herpes simplex virus type 1 (HSV-1) infection and defective in the expression of heparan sulfate (HSPG) because of a splice site mutation in the EXT1 gene encoding the HS-synthesizing enzyme (7).
LM or Sog9 cells were harvested with 0.2 mM EDTA in PBS and washed three times with PBS supplemented with 1 mM CaCl2and 0.1% BSA. The cells (3⫻
106
/ml) were then incubated for 1.5 h at 37°C and 5% CO2in PBS–1 mM
CaCl2–0.1% BSA in the presence of 10 mU/ml heparinase, 10 mU/ml
chondroiti-nase, or PBS. Following incubation, the cells were washed with cold PBS that contained 1% BSA and suspended in 300l PBS–1% BSA containing a 300 nM concentration of the indicated CXCL12 variants. The cells were then incubated at 4°C for 1.5 h, washed with PBS–1% BSA, and stained for 1 h with an anti-His-tag antibody conjugated to FITC. The cells were analyzed by flow cytometry.
HSPG staining.LM, Sog9, or CEM cells were harvested with 0.2 mM EDTA in PBS and washed three times with PBS supplemented with 1 mM CaCl2and
0.1% BSA. The cells (3⫻106/ml) were then incubated for 1.5 h at 37°C and 5%
CO2in PBS–1 mM CaCl2–0.1% BSA in the presence of 10 mU/ml heparinase,
10 mU/ml chondroitinase, or PBS. Following incubation, the cells were washed and stained with a monoclonal antibody to heparin sulfate (U.S. Biologicals, Swampscott, MA) and a secondary FITC-conjugated anti-mouse antibody. The cells were analyzed by flow cytometry.
CXCR4 internalization assay.To measure internalization of CXCR4, we used a previously reported method, with some modifications (28). Briefly, the CXCR4⫹CEM T cells were incubated for 30 min with 50 nM CXCL12 variants at 37°C. The cells were then centrifuged, washed once with 1 ml PBS, and then washed again with PBS containing 0.1% BSA. The cells were then incubated on ice with the anti-CXCR4 monoclonal antibody 12G5 conjugated with PE or the isotype control, suspended in PBS–1% BSA for 30 min, washed with PBS–1% BSA, PBS–0.1% BSA, and PBS consecutively, suspended in PBS only, and analyzed by flow cytometry. The same procedure was used to analyze the kinetics of CXCR4 internalization. For this analysis, the chemokine was incubated with the cells for 1 h, washed three times with IMDM–0.5% BSA, and then further FIG. 1. Predicted amino acid sequences and purification of wild-type CXCL12 and BBXB domain mutants. (A) Amino acid sequences of mature wild-type CXCL12␣and CXCL12␥proteins are indicated along with their BBXB mutations. Dashed lines indicate the nonmutated amino acids. Numbers indicate the amino acid positions relative to the first amino acid of the mature peptide. The mutants are named after the position of the BBXB domain. For example, B1 refers to the CXCL12 mutant that has its first BBXB domain mutated, and B123 refers to a mutant in which the first three BBXB domains are mutated. (B) SDS-PAGE and Coomassie brilliant blue staining of recombinant proteins purified fromE. coli. Lanes: 1, vector control; 2, CXCL12␣; 3, CXCL12␣B1; 4, CXCL12; 5, CXCL12␥; 6, CXCL12␥⌬C; 7, CXCL12␥B1; 8, CXCL12␥B12; 9, CXCL12␥B123; 10, CXCL12␥B2; 11, CXCL12␥B3; 12, CXCL12␥B23. (C) The same samples of the purified proteins were fractionated in a different SDS-PAGE gel and then blotted. The blot was probed with a polyclonal anti-histidine-tag antibody. Lane numbers correspond to the same proteins as those listed for panel B. The protein concentrations were not quantified before gel loading.
on November 8, 2019 by guest
http://jvi.asm.org/
figure legends as follows: B1, KHLK 3 AHAA for both CXCL12␣and CXCL12␥. The following mutations were con-structed from CXCL12␥: B2, 77KKEK80377AAEA80; B3, 85KRQK88385AAQA88; and␥⌬C,9CPC1139APA11.
Combi-nations of the mutations are denoted B12, B123, and B23 to indicate the locations of the mutated BBXB domains. DNA sequencing of the mutant plasmid DNAs confirmed the site mutations. Each chemokine purification procedure yielded ap-proximately 0.5 to 2.0 mg protein. The purity of the proteins was verified on a Coomassie blue-stained SDS-PAGE gel (Fig. 1B). To verify the identities of the affinity-purified chemokines, small aliquots of the purified proteins were fractionated by SDS-PAGE and blotted to a membrane, and the blot was probed with a polyclonal antibody to the histidine tag. The results of this analysis demonstrated that all purified proteins reacted with the anti-His antibodies and showed the expected molecular sizes of the mutant proteins (Fig. 1C). Mutation of the large side chain amino acids to alanine resulted in a slight decrease in molecular weight that was observed in SDS-PAGE analyses. These experiments confirmed the purity and ex-pected gel mobilities of the CXCL12 chemokine variants. In our previous study, we observed that the constructed His-tagged recombinant CXCL12␣ was indistinguishable from commercially purchased CXCL12␣in terms of chemotaxis and anti-HIV activity, suggesting that the added C-terminal His tag did not significantly alter CXCL12 biological function (2).
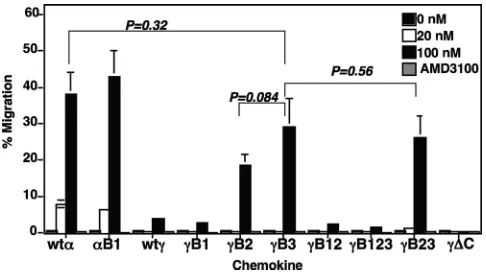
Elimination of the second and/or third BBXB domain of CXCL12␥ restores its chemotactic activity. To determine whether the purified recombinant CXCL12 proteins were bi-ologically functional, we tested them in anin vitrochemotaxis assay, using the human CEM T-lymphoblastic cell line as target cells. The purified CXCL12␣induced the expected cell migra-tion that we previously reported (2). The efficiency of the induced migration was always comparable to that observed with commercial CXCL12␣(2). Consistent with our previous data, the purified CXCL12␥induced a low level of cell migra-tion at 100 nM (Fig. 2). We used the double-cysteine mutant CXCL12␥⌬C as a negative control, since it does not induce significant chemotaxis at any concentration (Fig. 2).
Consistent with previous data in the literature (4), mutating the conserved BBXB domain (B1) in either CXCL12␣or CXCL12␥ did not result in a significant effect on their chemotactic activities (Fig. 2). The chemotactic activities of the B2, B3, and B23 mu-tants were dramatically increased, to levels comparable to those observed with wild-type CXCL12␣ (Fig. 2). The significantly higher chemotactic activities of the CXCL12␥B2 and B3 mutants were restored only when the conserved BBXB domain (B1) at positions 24 to 27 remained unaltered. The CXCL12␥mutants that lost the second and/or third BBXB domain (B2, B3, or B23)
induced significant levels of cell migration at 100 nM (Fig. 2). The chemotaxis activities of all affinity-purified CXCL12 proteins were eliminated by AMD3100 treatment, indicating that they were CXCR4 mediated (Fig. 2). The results demonstrated that the additional BBXB domains found at the novel carboxyl tail of CXCL12␥play an important role in the observed low level of chemotactic activity.
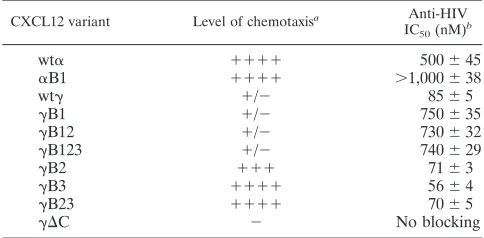
Elimination of the second and/or third BBXB domain of CXCL12␥ enhances its anti-HIV blocking effects. We previ-ously reported that CXCL12␥exhibits at least a fivefold po-tency in inhibiting HIV-1 Env-mediated fusion (2). To deter-mine the relative potencies of the C-terminal BBXB mutants of CXCL12␥, we tested their inhibitory activities in an HIV-1 Env-mediated fusion assay. A dose-response analysis was per-formed for each of the CXCL12 variants, which were preincu-bated with TZM-bl target cells (Fig. 3A and B) or human PBMCs (Fig. 3C). The results show that the CXCL12␥ mu-tants caused a dose-dependent inhibition of X4 Env fusion for both target cell types (Fig. 3A and C). The 50% inhibitory concentration (IC50) of each chemokine variant was calculated from at least three different experiments run in duplicate (Ta-ble 1). The R5 Ba-L Env was used as a negative control be-cause it has been well established that CXCL12 does not block R5 Env-mediated fusion (2). None of the CXCL12 mutants showed any inhibition of R5 Env-mediated fusion (Fig. 3B).
[image:4.585.300.543.68.204.2]We consistently observed that the B1 mutants of CXCL12␣ and CXCL12␥had significantly reduced inhibitory activities in the X4 fusion assay (Fig. 3A and C). In contrast, mutating the C-terminal BBXB domains consistently resulted in enhancement of the inhibitory effects of CXCL12␥. The calculated IC50for the B1 mutant of CXCL12␣or CXCL12␥was always higher than that of the wild-type chemokine (Table 1). The IC50of the CXCL12␥ B1 mutant was⬃9-fold higher than that of wild-type CXCL12␥.
FIG. 2. CXCL12␥mutant chemokines missing the second and/or third BBXB domain had restored chemotactic activity. The chemotac-tic activities of the indicated CXCL12 proteins were examined at increasing concentrations in a two-chamber chemotaxis assay. Chemo-taxis assays were performed as outlined in Materials and Methods, using CEM T cells. The CXCL12␥variant with the mutated cysteines at positions 9 and 11 was used as a negative control, since it does not induce cell migration. For experiments with the CXCR4-specific in-hibitor, a 100 nM concentration of the chemokine variant was com-bined with 100 nM AMD3100 in the bottom chambers. Error bars indicate standard deviations of mean values obtained from triplicate assays. This experiment is representative of three experiments done at different times with different chemokine purification sets. The indi-catedPvalues were obtained using the Studentttest.
on November 8, 2019 by guest
http://jvi.asm.org/
In contrast, the IC50s of the B2, B3, and B23 mutants, which contained the conserved BBXB domain, were consistently lower than that of wild-type CXCL12␥(Table 1). Taken together, these results suggest that the conserved BBXB domain is critical for optimal antiviral activity for both CXCL12␣and CXCL12␥and that the presence of the C-terminal BBXB domains seems to exert a positive effect on the observed potent antiviral activity of CXCL12␥.
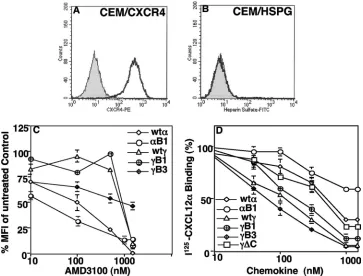
Binding of CXCL12 variants to HSPG.To investigate che-mokine binding and the role of HSPG, we used the HSPG-deficient mouse cell line Sog9 and its original HSPG⫹parent LM cell line (7) as targets in the binding assays. Additionally, binding of the CXCL12 variants was examined with L cells treated with either chondroitinase or heparanase. These en-zymes selectively degrade either chondroitin or heparan sul-fate. The levels of bound chemokines were determined by FACS analysis, using a FITC-conjugated anti-His-tag antibody that detects the His tag present in all constructs. FACS analysis confirmed the deficient expression of HSPG in Sog9 cells (Fig. 4A) and efficient HSPG expression in LM cells (Fig. 4B). Expression of HSPG on LM cells was significantly reduced after heparanase treatment (Fig. 4C) but not after
chondroiti-nase treatment (Fig. 4D). We also stained Sog9 and LM cells for CXCR4, and both cell lines were negative (data not shown). Consistent with previous data in the literature (4, 29), we observed a loss of HSPG binding of the CXCL12␣B1 mu-tant. Binding of this mutant to HSPG was comparable to that observed with the HSPG-deficient Sog9 cells (Fig. 4E). Bind-ing of wt CXCL12␥was at least 10 times more efficient than that of wt CXCL12␣(Fig. 4F). All of the BBXB mutants had reduced binding to HSPG, except for the␥B1 mutant that had lost the conserved BBXB domain (Fig. 4F). The␥B1 mutant chemokine consistently showed a significant increase in bind-ing to LM cells. The bindbind-ing of all chemokine variants was dramatically reduced after heparanase treatment but not after chondroitinase treatment (Fig. 4F). These results indicate spe-cific binding to HSPG. Among the single-site BBXB mutants, the most dramatic decrease in binding to LM cells was ob-served with the␥B3 mutant. The results suggest a critical role for this BBXB site (B3) in HSPG binding. The lowest HSPG binding was observed with the triple BBXB mutant (B123), in which all three BBXB domains were mutated (Fig. 4F). Bind-ing to Sog9 cells served as the background, since these cells are defective in HSPG expression (Fig. 4F). MC148 was used as another negative control, since it lacks BBXB domains and does not bind HSPG (Fig. 4F). We previously described the construction and purification of an MC148 chemokine-like protein that is defective in chemotaxis assays (2). The results demonstrated efficient and specific binding of wt CXCL12␥to HSPG through its BBXB domains and suggest a critical role for the B3 domain.
[image:5.585.109.472.70.188.2]Binding of CXCL12 variants to CXCR4. To determine whether CXCL12 binds to CXCR4 or HSPG on CEM cells, we first verified expression of these surface molecules by FACS analysis. The results demonstrated efficient expression of CXCR4 (Fig. 5A) and a lack of HSPG expression (Fig. 5B). Analysis of CXCL12 binding to CXCR4 revealed that a higher concentration (⬃10-fold) of AMD3100 was required to dis-place the CXCL12␥chemokine on human CEM T cells than that used to displace CXCL12␣(Fig. 5C and Table 1). The AMD3100 concentration required to result in the reduction of
TABLE 1. Biological activities of CXCL12 variants
CXCL12 variant Level of chemotaxisa Anti-HIV
IC50(nM)b
wt␣ ⫹⫹⫹⫹ 500⫾45
␣B1 ⫹⫹⫹⫹ ⬎1,000⫾38
wt␥ ⫹/⫺ 85⫾5
␥B1 ⫹/⫺ 750⫾35 ␥B12 ⫹/⫺ 730⫾32 ␥B123 ⫹/⫺ 740⫾29 ␥B2 ⫹⫹⫹ 71⫾3 ␥B3 ⫹⫹⫹⫹ 56⫾4 ␥B23 ⫹⫹⫹⫹ 70⫾5
␥⌬C ⫺ No blocking
a
The level of chemotaxis is represented by⫹signs. Chemotaxis induced by CXCL12␣is considered the highest.⫹/⫺, weak chemotaxis;⫺, no chemotaxis activity.
b
[image:5.585.42.284.564.683.2]Chemokine concentration that results in 50% inhibition of HIV-1 Env-mediated fusion. Data are means⫾standard deviations.
FIG. 3. CXCL12␥mutants missing the second and/or third BBXB domain show enhanced blocking effects in HIV-1 Env-mediated fusion assay. CXCR4⫹CCR5⫹TZM-bl cells (A and B) expressing vaccinia virus-encoded T7 RNA polymerase were used as target cells. The recombinant CXCL12 variants were added to the target cells at the indicated concentrations and incubated for 1 h at 37°C. Effector HeLa cells were coinfected with recombinant vaccinia virus carrying the PT7-lacZreporter gene and vaccinia virus encoding the X4 LAV Env (A) or the R5 Ba-L Env (B). (C) Effects of chemokine variants on X4 Env fusion with human PBMCs. Target and effector cell mixtures were incubated at 37°C, and the extent of fusion was quantified by measuring the amount of-galactosidase produced. The results are representative of at least three different experiments performed in duplicate. Error bars indicate standard deviations of mean values obtained for duplicate wells. Broken lines represent the value of the background signal obtained with the Unc Env control.
on November 8, 2019 by guest
http://jvi.asm.org/
50% of binding by the␥B1 and␥B3 mutants was not signifi-cantly different from that for wild-type CXCL12␥. The results demonstrate that CXCL12␥and its mutants had significantly increased binding affinities for CXCR4 and that the loss of any BBXB domain in CXCL12␥had little effect on the observed binding.
To confirm the previous binding results, we performed a direct binding method using125I-radiolabeled CXCL12␣. The
results of this direct binding analysis confirmed the data ob-tained with the indirect binding method. For example, higher concentrations of cold CXCL12␣ were required to result in 50% displacement of bound125I-CXCL12␣(Fig. 5D). We
con-sistently observed at least a 6 times higher binding affinity for CXCL12␥than for CXCL12␣(Fig. 5D and Table 2). Mutating the conserved BBXB domain reduced the binding efficiencies of both CXCL12␣ and CXCL12␥. However, eliminating the second and/or third BBXB domain of CXCL12␥increased the amount of chemokine binding to CEM cells (Fig. 5D and Table 2). Treating CEM cells with heparanase had no effect on the binding affinities of the CXCL12 variants (Table 2). These results confirm the higher binding affinity of CXCL12␥ for CXCR4 and demonstrate that binding of the CXCL12 variants to CEM cells occurs mainly through CXCR4.
Internalization of CXCR4 by CXCL12 variants.To deter-mine the efficiency of CXCR4 internalization induced by the CXCL12 variants, we analyzed the surface expression of CXCR4 on cells incubated with or without CXCL12 chemo-kine proteins. It has been demonstrated that CXCL12 does not block 12G5 binding to CXCR4 (28). The results of this analysis indicated that CXCL12␥induced a significantly higher level of
CXCR4 internalization. The amount of CXCR4 surface ex-pression on CEM cells incubated with CXCL12␥was significantly lower than that on cells incubated with CXCL12␣(Fig. 6A). The efficiency of CXCR4 internalization induced by the CXCL12␥ BBXB mutants depended on whether or not the conserved do-main was mutated. The second and/or third BBXB dodo-main mu-tants were not significantly different from wild-type CXCL12␥in terms of the ability to induce CXCR4 internalization. The CXCL12␥B1, CXCL12␥B12, and CXCL12␥B123 mutants in-duced less internalization of CXCR4 (Fig. 6A). Incubation of CEM cells with the CXCL12␥⌬C mutant had no significant in-ternalization effect on CXCR4 (Fig. 6A).
To determine the kinetics of internalization, we performed experiments involving incubation of the CXCL12 chemokine variants with CEM cells, washing, and further incubation with-out chemokine for different times. Following 3 h of incubation after washing away the chemokine, the cells preincubated with CXCL12␣restored normal expression of CXCR4. In contrast, cells preincubated with CXCL12␥restored CXCR4 expression to 50% (Fig. 6B). These results indicate that CXCL12␥ in-duces more efficient and sustained internalization of CXCR4 than does CXCL12␣.
DISCUSSION
[image:6.585.112.473.69.306.2]This study aimed at investigating the mechanism of the po-tent anti-HIV activity of CXCL12␥. We hypothesized that the extra BBXB domains in CXCL12␥play a critical role in the observed potent anti-HIV activity. Our hypothesis was based on our recent findings demonstrating that CXCL12␥is at least
FIG. 4. Binding of CXCL12 variants to HSPG⫹ and HSPG⫺ cells. Surface expression of HSPG in Sog9 cells (A), LM cells (B), heparanase-treated LM cells (C), and chondroitinase-treated LM cells (D) was verified by flow cytometry. To assess binding of the chemokines to the cell surface, the cells were incubated with a 300 nM concentration of the indicated chemokine variant and stained with antibodies against the His tag present in all chemokine constructs. Data are representative of three different experiments performed in duplicate. Error bars represent the standard errors of the duplicate results of one experiment. The graph in panel E is for the same experiment as that shown in panel F, but on a smaller scale to show the positive binding by CXCL12␣and the loss of binding by the CXCL12␣B1 mutant that lost its single BBXB domain.
on November 8, 2019 by guest
http://jvi.asm.org/
5 to 6 times more potent than CXCL12␣ in HIV blocking assays (2). The results demonstrated that mutating the C-ter-minal BBXB domains of CXCL12␥restored chemotaxis activ-ity to levels comparable to those observed with CXCL12␣. In
contrast, mutating the conserved BBXB (24KHLK27) domain
[image:7.585.111.474.69.345.2]of CXCL12␣resulted in significant decreases in CXCR4 bind-ing and anti-HIV-1 effects but had no significant effect on its chemotactic activity. These observations are consistent with
FIG. 5. Binding analysis of CXCL12␥mutants with CXCR4. CEM cells were stained with either anti-CXCR4 antibodies (A) or anti-HSPG antibodies (B). (C) To analyze CXCR4 binding, CEM cells were suspended in mixtures containing a constant 100 nM concentration of each chemokine variant (containing the His tag) and escalating doses of the CXCR4 antagonist AMD3100. The cell suspensions were incubated for 1 h at 4°C, washed, and incubated with a FITC-conjugated anti-His-tag antibody. Chemokine binding was calculated as the percent mean fluorescence intensity (MFI) of staining for the bound chemokine (wt CXCL12␣or CXCL12␥) with no AMD3100. Data are representative of two triplicate experiments. Error bars represent the standard errors of the triplicate results of one experiment. (D) Results of direct binding of125I-CXCL12␣.
The cold chemokine variants were mixed at increasing concentrations with 0.25 nM125I-CXCL12␣and added to CEM cells. The cell-chemokine
mixtures were incubated at 4°C for 1 h and then washed three times with PBS–0.1% BSA. The cell pellet-associated counts were measured in a gamma counter. Binding was evaluated by calculating the % chemokine binding, where 100% binding was that for CEM cells incubated with 0.25 nM125I-labeled CXCL12␣without any cold competitor. The results are representative of three different experiments.
TABLE 2. Summary of CXCL12 binding and CXCR4 internalization
CXCL12 variant AMD3100 concn (nM)a,b
Cold chemokine concn (nM)a,c
% Internaliz-ationd
Direct binding of
125I-CXCL12␣
125I-CXCL12␣binding to
heparinase-treated CEM cells
wt␣ 100⫾5 678⫾17 631⫾12 50–53
␣B1 Low binding ⬎1,000⫾0 ⬎1,000⫾0 35–40
wt␥ 1,000⫾50 96⫾9 89⫾7 85–90
␥B1 1,100⫾65 400⫾25 300⫾14 55
␥B12 1,100⫾70 356⫾23 321⫾13 65
␥B123 1,000⫾75 365⫾21 315⫾9 58
␥B2 1,000⫾56 71⫾8 51⫾5 89–92
␥B3 1,000⫾55 59⫾7 46⫾8 88
␥B23 1,000⫾50 64⫾9 49⫾7 87
␥⌬C ND 564⫾16 644⫾23 0
a
This experiment is representative of three experiments run at different times with different chemokine purification sets. Data are means⫾standard deviations. ND, not done.
b
Amount of AMD3100 required to cause a 50% reduction in chemokine binding.
c
Amount of cold chemokine required to displace 50% of bound125
I-labeled CXCL12.
d
Internalization is represented by the % CXCR4 downmodulation. The calculations were based on at least four different experiments performed with different lots of purified chemokine variants.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.43.542.541.674.2]the data in the literature, except for the binding data (3, 29). The CXCL12␣B1 mutant was previously reported to have the same binding affinity for CXCR4 as wt CXCL12␣(29). This discrepancy might be explained by the different structures of our BBXB mutants. For example, Amara et al. (4), Fernandez et al. (29), Sadir et al. (25), and recently, Reuda et al. (24) utilized a BBXB mutant that had the conserved KHLK domain mutated to SSLS or SHLS, while ours was mutated to AHAA. Additionally, the studies by Laguri et al. and Reuda et al. (17, 24) utilized CXCL12␥constructs that included the sequence coding for the bovine rhodopsin C9 tag (TETSQVAPA) in frame at the C terminus of CXCL12␥(17, 24). Our CXCL12␥ constructs contain a C-terminal His tag.
Decreased CXCR4 binding and internalization of CXCL12␣B1 correlated with its lower antiviral activity. In most cases, higher-affinity binding to CXCR4 correlated with a higher efficiency of receptor internalization. An exception to this was the case for CXCL12␥B1, whose binding to CXCR4 was not
signifi-results of this study suggest that in addition to the BBXB domain, other structural requirements might play a critical role in CXCL12 binding. We did not observe a direct correlation between binding to HSPG and the observed potent anti-HIV activity. However, such a correlation was true for CXCR4 binding and internalization. For example, binding to HSPG was significantly higher with CXCL12␥B1 than with wt CXCL12␥, but CXCL12␥B1 was significantly less potent in HIV blocking assays. Additionally, the␥B3 mutant showed a significant reduction in HSPG binding but was the most potent HIV-1 inhibitor. HSPG binding does not seem to modify the antiviral activity of CXCL12 or promote internalization of CXCR4.
The restored chemotaxis of the C-terminal BBXB domain mutants suggests that the C-terminal tail of CXCL12␥acts as a suppressive domain that significantly impairs its chemotaxis activity. The results suggest that the extra BBXB domains in the C terminus of CXCL12␥contribute to an altered structure of the chemokine that impairs its chemotactic activity but en-hances its anti-HIV activity. The finding that CXCL12␥ exhib-its significantly greater affinity for CXCR4 and significantly reduced chemotactic activity through CXCR4 introduces the intriguing idea that CXCL12␥ may function as a natural CXCR4 antagonist, in a similar manner to that of the bicyclam AMD3100. AMD3100 has been shown to only bind CXCR4, without engaging receptor signaling (13, 15, 26). Since abun-dant CXCL12␥protein expression has been detected in car-diac muscle, valves, and large vessels (24), we hypothesize that CXCL12␥might function as a natural antagonist. This might represent a unique mechanism of self-regulation aimed at con-trolling potentially unfavorable signaling events in the heart myocytes by CXCL12␣present in the bloodstream. Previous studies suggested that MCP-3 could act as an antagonist, since it binds CCR5 with a high affinity without inducing receptor internalization and has the ability to inhibit the functional response to MIP-1(8).
[image:8.585.43.283.71.339.2]Previous studies demonstrated that MCP-3 could compete efficiently for gp120 binding, but it was found to be a weak inhibitor of HIV infection, probably as a consequence of its inability to internalize CCR5 (8). Our study presents several lines of experimental evidence to suggest that the different antiviral activities of the BBXB mutants are associated with significant changes in the efficiency of CXCR4 internalization. First, internalization of CXCR4 was slightly impaired by CXCL12␣B1 and significantly impaired by CXCL12␥B1, the two mutants that lost the conserved BBXB domain. Second, wt CXCL12␥and CXCL12␥B1 had similar affinities for CXCR4, but CXCL12␥B1 had a significant loss in the ability to inter-nalize CXCR4 and block HIV-1. Third, the CXCL12␥B2 and
FIG. 6. CXCL12␥is more efficient at inducing CXCR4 internal-ization. (A) CEM T cells were incubated with the indicated chemokine variant (50 nM) for 30 min at 37°C. Following incubation, the cells were washed and stained with the PE-conjugated 12G5 monoclonal antibody specific for CXCR4. The control level of CXCR4 expression with no chemokine added represented 100% staining. The results are represented as percentages of the control. These results were also reproduced using a different monoclonal antibody to CXCR4 that detects an epitope in the second extracellular domain (R&D Systems). (B) CEM T cells were incubated with the indicated chemokine variant (50 nM) for 1 h at 37°C. Following incubation, the cells were washed three times and returned to the 37°C incubator. Cell samples were removed at the indicated time points and fixed in paraformaldehyde. After the final time point, the fixed cells were stained with the PE-conjugated 12G5 monoclonal antibody specific for CXCR4. The re-sults are representative of three separate experiments with different protein purifications.
on November 8, 2019 by guest
http://jvi.asm.org/
B3 mutants showed slight but significant increases in CXCR4 internalization that correlated with increased antiviral activity. Fourth, the kinetics of CXCR4 internalization indicated that CXCL12␥induced sustained CXCR4 internalization, in con-trast to CXCL12␣; CXCR4 expression was maintained at sig-nificantly lower levels with CXCL12␥3 h after removing the chemokine. Finally, despite its efficient binding to CXCR4, the double-cysteine mutant CXCL12␥⌬C showed no significant antiviral effect due to its inability to induce CXCR4 internal-ization.
The mechanisms responsible for mammalian GPCR endocy-tosis remain largely undefined (reviewed in reference 18). Long-lasting CCR5 internalization in a subset of long-term nonprogres-sors has been suggested as a mechanism for the protective effect against disease progression (20). It is critical to understand CXCR4 internalization, since it controls the temporal and spatial aspects of G protein signaling. Identifying the specific mecha-nisms that regulate internalization of CXCR4 will provide impor-tant insight into the development of new strategies to manipulate receptor signaling and will provide novel targets for designing drugs that can be used in the prevention and treatment of a wide range of human diseases, including cardiovascular disease and cancer progression (10). It is possible that CXCL12␥stabilizes a distinct conformation of the CXCR4 receptor that may signal selectively to different G proteins or promote a distinct receptor conformation that facilitates better binding to-arrestins. Alter-natively, CXCL12␥ may induce more efficient CXCR4 phos-phorylation that enhances internalization. Recent studies demon-strated that CXCR4 dimerization and -arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12␣in WHIM syndrome (16). The mechanisms that control CXCR4 endocytosis induced by CXCL12␥have yet to be elucidated fully.
ACKNOWLEDGMENTS
We thank JoAnn Trejo for helpful discussions. The following re-agents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl cells, from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc.; and AMD3100, from Anormed.
This study was supported by NIH grant RO1 A152019-01 to G.A. Q.J. was supported by a scholarship from the Chinese Scholar-ship Council, Beijing, China.
REFERENCES
1.Agrawal, L., X. Lu, J. Qingwen, Z. VanHorn-Ali, V. Nicolescue, D. McDer-mott, P. M. Murphy, and G. Alkhatib.2004. Role for CCR5⌬32 protein in resistance to R5, R5X4, and X4 human immunodeficiency virus type 1 in primary CD4⫹cells. J. Virol.78:2277–2287.
2.Altenburg, J. D., H. E. Broxmeyer, Q. Jin, S. Cooper, S. Basu, and G. Alkhatib.2007. A naturally occurring splice variant of CXCL12/stromal cell-derived factor 1 is a potent HIV-1 inhibitor with weak chemotaxis and cell survival activities. J. Virol.81:8140–8148.
3.Amara, A., S. L. Gall, O. Schwartz, J. Salamero, M. Montes, P. Loetscher, M. Baggiolini, J. L. Virelizier, and F. Arenzana-Seisdedos.1997. HIV core-ceptor downregulation as antiviral principle: SDF-1alpha-dependent inter-nalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J. Exp. Med.186:139–146.
4.Amara, A., O. Lorthioir, A. Valenzuela, A. Magerus, M. Thelen, M. Montes, J. L. Virelizier, M. Delepierre, F. Baleux, H. Lortat-Jacob, and F. Arenzana-Seisdedos.1999. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J. Biol. Chem.274: 23916–23925.
5.Arenzana-Seisdedos, F., J. L. Virelizier, D. Rousset, I. Clark-Lewis, P. Loetscher, B. Moser, and M. Baggiolini.1996. HIV blocked by chemokine antagonist. Nature383:400.
6.Balabanian, K., B. Lagane, S. Infantino, K. Y. Chow, J. Harriague, B.
Moepps, F. Arenzana-Seisdedos, M. Thelen, and F. Bachelerie.2005. The chemokine SDF-1/CXCL12 binds to and signals through the orphan recep-tor RDC1 in T lymphocytes. J. Biol. Chem.280:35760–35766.
7.Banfield, B. W., Y. Leduc, L. Esford, K. Schubert, and F. Tufaro.1995. Sequential isolation of proteoglycan synthesis mutants by using herpes sim-plex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J. Virol.69:3290–3298.
8.Blanpain, C., I. Migeotte, B. Lee, J. Vakili, B. J. Doranz, C. Govaerts, G. Vassart, R. W. Doms, and M. Parmentier.1999. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood94:1899–1905. 9.Bleul, C. C., M. Farzan, H. Choe, C. Parolin, I. Clark-Lewis, J. Sodroski, and
T. A. Springer.1996. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature382:829–833.
10.Burger, J. A., and T. J. Kipps.2006. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood107:1761–1767. 11.Burns, J. M., B. C. Summers, Y. Wang, A. Melikian, R. Berahovich, Z. Miao,
M. E. Penfold, M. J. Sunshine, D. R. Littman, C. J. Kuo, K. Wei, B. E. McMaster, K. Wright, M. C. Howard, and T. J. Schall. 2006. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med.203:2201–2213. 12.Chackerian, B., E. M. Long, P. A. Luciw, and J. Overbaugh.1997. Human
immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J. Virol.71:3932–3939.
13.Donzella, G. A., D. Schols, S. W. Lin, J. A. Este, K. A. Nagashima, P. J. Maddon, G. P. Allaway, T. P. Sakmar, G. Henson, E. De Clercq, and J. P. Moore.1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co receptor. Nat. Med.4:72–77.
14.Gleichmann, M., C. Gillen, M. Czardybon, F. Bosse, R. Greiner-Petter, J. Auer, and H. W. Muller. 2000. Cloning and characterization of SDF-1gamma, a novel SDF-1 chemokine transcript with developmentally regu-lated expression in the nervous system. Eur. J. Neurosci.12:1857–1866. 15.Hatse, S., K. Princen, G. Bridger, E. De Clercq, and D. Schols.2002.
Che-mokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett.527:255–262.
16.Lagane, B., K. Y. Chow, K. Balabanian, A. Levoye, J. Harriague, T. Planchenault, F. Baleux, N. Gunera-Saad, F. Arenzana-Seisdedos, and F. Bachelerie.2008. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood112:34–44.
17.Laguri, C., R. Sadir, P. Rueda, F. Baleux, P. Gans, F. Arenzana-Seisdedos, and H. Lortat-Jacob.2007. The novel CXCL12gamma isoform encodes an unstructured cationic domain which regulates bioactivity and interaction with both glycosaminoglycans and CXCR4. PLoS One2:e1110.
18.Marchese, A., M. M. Paing, B. R. Temple, and J. Trejo.2008. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharma-col. ToxiPharma-col.48:601–629.
19.Matsumoto, M., S. Misawa, K. Tsumoto, I. Kumagai, H. Hayashi, and Y. Kobayashi.2003. On-column refolding and characterization of soluble hu-man interleukin-15 receptor alpha-chain produced in Escherichia coli. Pro-tein Expr. Purif.31:64–71.
20.Pastori, C., B. Weiser, C. Barassi, C. Uberti-Foppa, S. Ghezzi, R. Longhi, G. Calori, H. Burger, K. Kemal, G. Poli, A. Lazzarin, and L. Lopalco.2006. Long-lasting CCR5 internalization by antibodies in a subset of long-term nonprogressors: a possible protective effect against disease progression. Blood107:4825–4833.
21.Platt, E. J., K. Wehrly, S. E. Kuhmann, B. Chesebro, and D. Kabat.1998. Effects of CCR5 and CD4 cell surface concentrations on infections by mac-rophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72:2855–2864.
22.Proudfoot, A. E., S. Fritchley, F. Borlat, J. P. Shaw, F. Vilbois, C. Zwahlen, A. Trkola, D. Marchant, P. R. Clapham, and T. N. Wells.2001. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem.276:10620–10626.
23.Rajarathnam, K., B. D. Sykes, B. Dewald, M. Baggiolini, and I. Clark-Lewis. 1999. Disulfide bridges in interleukin-8 probed using non-natural disulfide analogues: dissociation of roles in structure from function. Biochemistry 38:7653–7658.
24.Rueda, P., K. Balabanian, B. Lagane, I. Staropoli, K. Chow, A. Levoye, C. Laguri, R. Sadir, T. Delaunay, E. Izquierdo, J. L. Pablos, E. Lendinez, A. Caruz, D. Franco, F. Baleux, H. Lortat-Jacob, and F. Arenzana-Seisdedos. 2008. The CXCL12gamma chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant pro-teins. PLoS One3:e2543.
25.Sadir, R., F. Baleux, A. Grosdidier, A. Imberty, and H. Lortat-Jacob.2001. Characterization of the stromal cell-derived factor-1alpha-heparin complex. J. Biol. Chem.276:8288–8296.
26.Schols, D., J. A. Este, G. Henson, and E. De Clercq.1997. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor fusin/ CXCR-4. Antiviral Res.35:147–156.
27.Shirozu, M., T. Nakano, J. Inazawa, K. Tashiro, H. Tada, T. Shinohara, and