0022-538X/10/$12.00 doi:10.1128/JVI.01394-10
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Tetrameric Ring Formation of Epstein-Barr Virus Polymerase
Processivity Factor Is Crucial for Viral Replication
䌤
Sanae Nakayama,
1Takayuki Murata,
1Yoshihiro Yasui,
2Kazutaka Murayama,
3Hiroki Isomura,
1Teru Kanda,
1and Tatsuya Tsurumi
1*
Division of Virology, Aichi Cancer Center Research Institute, 1-1, Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan1;
Division of Virology, Aichi Prefectural Institute of Public Health, Nagoya 462-8576, Japan2; and
Division of Biomedical Measurements and Diagnostics, Graduate School of
Biomedical Engineering, Tohoku University, Sendai 980-8575, Japan3
Received 5 July 2010/Accepted 24 September 2010
The Epstein-Barr virus BMRF1 DNA polymerase processivity factor, which is essential for viral genome replication, exists mainly as a C-shaped head-to-head homodimer but partly forms a ring-shaped tetramer through tail-to-tail association. Based on its molecular structure, several BMRF1 mutant viruses were con-structed to examine their influence on viral replication. The R256E virus, which has a severely impaired capacity for DNA binding and polymerase processivity, failed to form replication compartments, resulting in interference of viral replication, while the C95E mutation, which impairs head-to-head contact in vitro, unexpectedly hardly affected the viral replication. Also, surprisingly, replication of the C206E virus, which is expected to have impairment of tail-to-tail contact, was severely restricted, although the mutant protein possesses the same in vitrobiochemical activities as the wild type. Since the tail-to-tail contact surface is smaller than that of the head-to-head contact area, its contribution to ring formation might be essential for viral replication.
Epstein-Barr virus (EBV), a human gammaherpesvirus har-boring a 172-kb double-stranded DNA (dsDNA) genome, is associated with several human cancers, including Burkitt’s lym-phoma and nasopharyngeal carcinoma (NPC) (14). EBV has two alternative life styles, latent and productive (lytic). Infec-tion is primarily latent with no producInfec-tion of virus particles (14), but a switch to productive replication is triggered by expression of the BZLF1 gene product as a result of various stimuli (20). BZLF1 is a lytic replication origin binding protein which also transactivates various viral promoters (17), leading to an ordered cascade of viral gene expression. In the viral productive cycle, the EBV genome is amplified more than 100-fold by utilizing the viral replication machinery (15), which works at replication forks to synthesize leading and lagging strands of the concatemeric EBV genome (15).
The DNA polymerase processivity factor of EBV, BMRF1, associates with the polymerase catalytic subunit, BALF5, to enhance the polymerase processivity and exonuclease activities of the holoenzyme (51, 52), and it is the major early phospho-protein expressed during EBV productive replication (7–9, 24–26, 29, 50). Judging from immunostaining data, together with the finding that almost all abundantly expressed BMRF1 proteins bind to double-stranded DNA (10), the factor not only acts at replication forks for polymerase processivity but also is widely distributed on newly synthesized EBV genomic DNA. Furthermore, it can transcriptionally activate the EBV BHLF1 promoter, one of two divergent early promoters
lo-cated within the lytic origin of viral DNA replication, oriLyt (55), and enhance BZLF1-mediated transcriptional activation of the BALF2 promoter (39).
From our recent resolution of the crystal structure of C-terminally truncated BMRF1 protein (38), the molecular struc-ture shares structural similarity with other processivity factors, such as herpes simplex virus type 1 (HSV-1) UL42, human cytomegalovirus (HCMV) UL44, and human proliferating cell nuclear antigen (PCNA). Most BMRF1 proteins form a C-shaped head-to-head homodimer, but some form ring-C-shaped tetramers through tail-to-tail association (Fig. 1). In general, processivity factors are associated with their cognate DNA polymerases on the template during replication. These pro-teins, which are also known as “sliding clamps,” include PCNA from eukaryotes (19, 27) and archaebacteria (34), the sub-unit ofEscherichia coliDNA polymerase III (4), and gp45 from the T4 (35) and RB69 (47) bacteriophages. They assemble as toroidal, ring-shaped structures, forming a central channel to accommodate the template DNA. However, the herpesvirus polymerase processivity factors display different molecular as-semblies. The HCMV UL44 forms a dimer in crystal structure as well as in solution. In contrast, the HSV-1 UL42 directly binds to DNA as a monomer (44). Electron microscopy obser-vations have revealed that BMRF1 adopts a ring-shaped struc-ture (32) which is almost twice as large as the previously re-ported PCNA ring structure.
In our previous study (38), several BMRF1 mutants were prepared: the C95E, H141F, and C206E mutations are pre-dicted to affect the dimer interface, and the K19E, K29E, R87E, K99E, and R256E mutations are in the putative DNA binding region. Some were mapped on the molecular surface, as shown in Fig. 1.In vitroDNA binding assays suggested that basic amino acid residues (Lys19, Lys29, Arg87, Lys99, and
* Corresponding author: Mailing address: Division of Virology, Ai-chi Cancer Center Research Institute, 1-1, Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan. Phone and fax: 81-52-764-2979. E-mail: [email protected].
䌤Published ahead of print on 6 October 2010.
12589
on November 8, 2019 by guest
http://jvi.asm.org/
Arg256) on the concave surface of the C-shaped head-to-head
dimer play important roles in interactions with double-stranded DNA. A monomeric C95E mutant, which is impaired in head-to-head homodimerization, showed decreased DNA
binding activityin vitro, suggesting that dimer formation en-hances its DNA binding. The mutant, however, retained poly-merase processivity activity (38) and could still enhance BZLF1-mediated transactivation of the BALF2 promoter (39). The C206E mutant, whose mutation should prevent the tail-to-tail contact, still formed a head-to-head dimer retaining DNA binding activity and polymerase processivity (38). Al-though the H141F mutant exhibited reduced polymerase pro-cessivity, it was still able to bind DNA and to dimerize. In contrast, DNA binding and processivity activities were dramat-ically inhibited by the R256E mutation.
For the present study, we constructed recombinant viruses with point mutation C95E, H141F, R256E, or C206E in BMRF1 and examined their effects on productive replication. Levels of viral DNA synthesis, processing of replication inter-mediates, and progeny virus production of the C95E and H141F viruses were comparable to those of wild-type and revertant viruses. On the other hand, the R256E and C206E mutant viruses exhibited severe reductions in viral DNA rep-lication, maturation, and progeny virus production. These re-sults indicate that thein vitrodata do not necessarily reflectin vivophenotypes and suggest that integrity of the tail-to-tail contact of BMRF1 is important for efficient viral productive replication.
MATERIALS AND METHODS
Cells.HEK293 cells were grown and maintained in Dulbecco modified Eagle medium (DMEM) (Sigma) supplemented with 10% fetal calf serum (FCS) at 37°C in a humidified atmosphere containing 5% CO2. Akata(⫺) cells were cultured in RPMI 1640 medium containing 10% FCS.
Plasmids.The BZLF1 protein expression vector (pCAG-Z) was constructed using an In-Fusion Advantage PCR cloning kit purchased from Clontech. A PCR-amplified fragment containing the complete BZLF1-coding region was inserted into the XhoI site of pCAGGS (42). Oligonucleotide primers used for PCR were as follows: CAGpZf, 5⬘-TTGGCAAAGAATTCCTCGAGATGATG GACCCAAACTCGAC-3⬘; and CAGpZr, 5⬘-TGAGGAGTGAATTCCTCGA GTTAGAAATTTAAGAGATCCT-3⬘. The BALF4 expression vector (pcDNA-BALF4) was kindly provided by W. Hammerschmidt (41). The C-terminal FLAG-tagged intact BMRF1 expression vector (pWT-f) and BMRF1(C95E) mutant (C95E-f) were constructed as described previously (39). All point mu-tants of FLAG-tagged BMRF1 expression plasmids were generated by site-directed mutagenesis using pWT-f as a template. Complementary oligonucleo-tide primers used for site-directed mutagenesis were as follows: 5⬘-AAGGTGT CCAAGAGCCACTTCACCTGCGCC-3⬘ for H141F-f; 5⬘-CTTAGCCTCTGC GAGATTCCGGCCGTTAGC-3⬘for R256E-f; and 5⬘-CTGGGGAGGCCGAA CTCACCCTAGACTAC-3⬘for C206E-f. The inserted DNA sequence of each vector was confirmed by DNA sequencing.
Genetic manipulation of EBV BAC DNA.Bacterial artificial chromosome BAC DNA of human EBV B95-8 (B95-8/F-BAC) was provided by W. Hammer-schmidt (12). Homologous recombination was carried out inE. colias described previously (21, 23). First, EBV BAC⌬M/neo DNA was produced by inserting a marker cassette containing neomycin resistance and streptomycin sensitivity genes and kanamycin selected as described previously (36, 37, 39). Revertant EBV BAC was prepared by second recombination using wild-type BMRF1 sequence DNA as a shuttle vector, followed by streptomycin selection, as de-tailed in our recent reports (36, 37, 39). Likewise, point mutations were intro-duced using mutant BMRF1 sequences as shuttle vectors. EBV BAC DNA was then transfected into HEK293 cells, followed by hygromycin selection. Cell clones were examined if the viral DNA demonstrated latent infection and if lytic replication could be induced upon BZLF1 expression (36, 37, 39).
[image:2.585.46.284.65.583.2]Induction of lytic replication in HEK293 cells and transfection of BMRF1 plasmids. HEK293 cells in 12-well plates were transfected with 500 ng of pCAG-Z using a Microporator (Digital Bio) to induce a virus lytic cycle. To test whether impaired virus replication caused by BMRF1 knockout could be com-plemented by ectopic expression of BMRF1, FLAG-tagged, wild-type, or mutant BMRF1 plasmid was cotransfected with 200 ng of pCAG-Z into the EBV BAC⌬M/neo HEK293 cells.
FIG. 1. Mutated amino acid residues of EBV BMRF1 (amino acids [aa] 1 to 314). (A) The ring-shaped crystal structure of a tetramer of C-terminally truncated BMRF1 protein (RCSB Protein Data Bank accession no. 2Z0L) is drawn as a surface model, in which the mutated amino acid residues are displayed in colors. (B) The mutated amino acid residues are displayed in colors in a gray surface model. The partner molecule forming a homodimer is drawn as a gray ribbon model. The lower panel provides a different view of the complex.
on November 8, 2019 by guest
http://jvi.asm.org/
Antibodies.An anti-BMRF1-specific mouse monoclonal antibody (R3) was purchased from Chemicon International Inc., and an anti-FLAG-specific mouse monoclonal antibody (M2) and an anti-alpha-tubulin specific mouse monoclonal antibody were purchased from Sigma and Abcam, respectively. Affinity-purified anti-BALF5-, anti-BBLF2/3-, anti-BALF2-, anti-BZLF1-, and anti-BMRF1-spe-cific polyclonal antibodies were prepared as described previously (10, 53, 54).
Quantification of viral DNA synthesis during lytic replication.Levels of viral DNA were determined by quantitative real-time PCR (22). Lytic replication-induced HEK293 cells (3⫻105cells) were harvested and suspended in 200l of a lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.001% Triton X-100, 0.001% SDS), followed by sonication and then incubation at 50°C overnight with 50g/ml of proteinase K (Sigma). Primers and a probe within the BALF2-coding region were designed using Primer Express (Applied Biosystems). The sequences were as follows: 5⬘-GCCCGTCCGGTTGTCA-3⬘(forward primer), 5⬘-AATAT CTGGTTGTTGCCGTTGA-3⬘(reverse primer), and 5⬘-6-carboxyfluorescein (F AM)-CTGCCAGTGACCATCAACAAGTACACGG-tetramethyl rhodamine (TAMRA)-3⬘(probe). PCR was performed in 20l of aqueous solution con-taining 0.5 mM each primer, 0.2 mM labeled probe, Fast Start Universal Probe Master (Roche), and 1l of DNA mixture using the 7300 real-time PCR System (Applied Biosystems). PCR included 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 s followed by 1 min at 60°C.
Viral yield assay.Titers of infectious viral particles of recombinant EBV were measured as follows (2). Culture supernatants were harvested at 3 days after lytic induction and then filtered through a 0.45-mm-pore-size filter (Millipore). EBV-negative Akata(⫺) cells (49) were infected with serially diluted mixtures of viruses. The infected cells were harvested at 3 days postinfection and fixed with 0.5% paraformaldehyde in phosphate-buffered saline (PBS). Since the recombi-nant EBV expresses green fluorescent protein (GFP), GFP-positive cells were counted using the FACSCalibur G5 system (Becton-Dickinson) according to the manufacturer’s instructions.
PFGE.Samples for pulsed-field gel electrophoresis (PFGE) were prepared from HEK293 cells with latently infected EBV genomes as follows. Cells (1.5⫻ 105) were detached from 24-well culture plates by incubation in PBS containing 1 mM EDTA. After centrifugation at 800⫻g, the pellet was resuspended in 40
l of PBS containing 87.5 mM EDTA and 0.5% of low-melting-point melted agarose (Nippon Gene). After gelling, agarose plugs containing cells were incu-bated at 37°C for 2 overnight periods in 20 volumes of lysis buffer (10 mM Tris-HCl [pH 8.0], 100 mM EDTA, 1%N-lauroylsarcosine sodium salt, 0.1 mg/ml proteinase K [Sigma]) and washed five times in Tris-EDTA (TE) buffer. PFGE was performed using the Chromosomal DNA Electrophoresis System (Bio Craft) for 24 h at 150 V and 13°C in 0.5⫻Tris-borate-EDTA (TBE) buffer, using a pulse time of 50 s. The Lambda Ladder PFG marker (NEB) was used as a size marker. After the PFGE was run, the gel was stained with ethidium bromide and photographed. DNA separated by PFGE was transferred to a Hybond-N positively charged membrane. For the probes, DNA fragments within the W repeat region were amplified by PCR using primer pair 5⬘-TACCAGAG GGGGCCAAGAA-3⬘and 5⬘-AGGAGAGGCAGGGCCTGAA-3⬘. Labeling of probes and hybridization were performed using the DIG High Prime DNA labeling system (Roche). The images were processed with LumiVision PRO 400EX.
Immunofluorescence analysis.Immunostaining of HEK293 cells was achieved as described previously (11). Cells were harvested 24 h after lytic induction and lysed for 10 min on ice with ice-cold 0.5% Triton X-100–mCSK buffer [10 mM piperazine-N,N⬘-bis(2-ethanesulfonic acid) (PIPES) (pH 6.8), 300 mM sucrose, 1
mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5% Triton X-100] containing multiple protease inhibitors (Roche), 0.2 mM Na3VO4, 20 mM NaF, and 150 mM NaCl. After centrifugation (2,000⫻g, 3 min, 4°C), the extracted nuclear pellets were fixed with 70% methanol. The fixed cells were immunostained with anti-BMRF1 monoclonal and anti-BALF2 polyclonal antibodies, followed by secondary goat anti-mouse or rabbit IgG antibodies conjugated with Alexa Fluor 594 or 488. Slides were mounted with ProLong Gold antifade reagent with DAPI (4⬘,6⬘-diamidino-2-phenylindole) (In-vitrogen) for analysis under a fluorescence confocal microscope (Zeiss). Images were captured and processed using the Zeiss LSM Image Browser (Zeiss).
Biochemical cellular fractionation.Fractionation of HEK293 cells was per-formed as described previously (11). Cells were harvested at 3 days after lytic induction and lysed for 10 min on ice with ice-cold 0.5% Triton X-100–mCSK buffer containing multiple protease inhibitors (Roche), 0.2 mM Na3VO4, 20 mM NaF, and one of several concentrations of NaCl (50, 100, or 150 mM). The samples were then subjected to centrifugation (2,000⫻g, 3 min, 4°C) to obtain Triton X-100-extractable supernatants and extracted nuclear pellets. Each sam-ple was adjusted to the same volume by adding SDS samsam-ple buffer and boiled, and aliquots corresponding to 1.8⫻104cells per lane were applied for SDS-PAGE. The intensity of fluorescence was measured with a Lumi Vision Analyzer 2.0 (AISIN).
RESULTS
Construction of recombinant viruses.Based on our crystal structure imaging of the EBV BMRF1 (38), we previously analyzed the significance of panels of point mutations affecting biochemical properties of the protein with regard to dimer formation (38), DNA binding ability (38), transcriptional acti-vation (39), and polymerase processivityin vitro(38). We here chose four representative mutations (Table 1) out of those characterized in our previous study and generated recombi-nant viruses expressing mutated BMRF1 proteins to examine the significance of those mutations in the context of viral infection. Figure 1 illustrates a surface-and-ribbon model of tetrameric BMRF1. The mutated amino acid residues are dis-played in colors. Cys95is located at the interface of
head-to-head contact for the homodimer formation and appears to be involved in disulfide bond formation under oxidative condi-tions. C95E substitution would therefore impair dimer
forma-tionin vitro. Although the resultant monomer BMRF1 exhibits
reduced DNA binding activity in vitro, it still can act as a polymerase processivity factor (38). His141also lies at the
in-terface of head-to-head contact for homodimer formation, al-though exchange of the residue (H141F) has no effect on dimer formation but rather diminishes the processivity function (38). While Cys206is at the tail-to-tail interface, replacement of the
[image:3.585.42.544.81.178.2]residue (C206E) had no obvious effect on DNA binding and
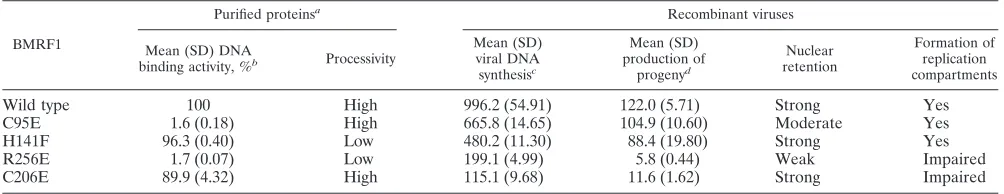
TABLE 1. Summary of BMRF1 functions
BMRF1
Purified proteinsa Recombinant viruses
Mean (SD) DNA
binding activity, %b Processivity
Mean (SD) viral DNA synthesisc
Mean (SD) production of
progenyd
Nuclear retention
Formation of replication compartments
Wild type 100 High 996.2 (54.91) 122.0 (5.71) Strong Yes
C95E 1.6 (0.18) High 665.8 (14.65) 104.9 (10.60) Moderate Yes
H141F 96.3 (0.40) Low 480.2 (11.30) 88.4 (19.80) Strong Yes
R256E 1.7 (0.07) Low 199.1 (4.99) 5.8 (0.44) Weak Impaired
C206E 89.9 (4.32) High 115.1 (9.68) 11.6 (1.62) Strong Impaired
aData are from reference 38. bPercentage of wild-type activity.
cBased on the data in Fig. 3 at 3 days after lytic induction.
dProgeny virus production levels (⫻104GFP positive cells/ml) are based on the data in Fig. 4.
on November 8, 2019 by guest
http://jvi.asm.org/
processivityin vitro(38). Arg256is situated at the DNA binding
domain, and the R256E mutation abrogates DNA binding and processivityin vitro(38).
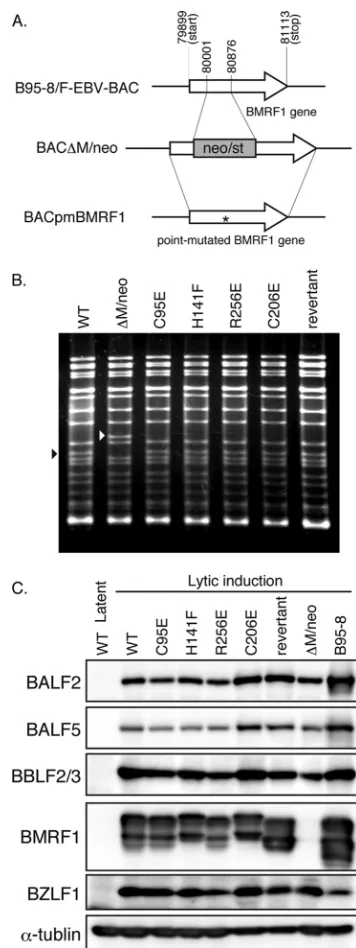
For construction of recombinant viruses harboring each point-mutated BMRF1 gene, we first constructed a BMRF1-deficient insertion mutant virus,⌬M/neo, as shown in Fig. 2A. The marker cassette containing the neomycin resistance and streptomycin sensitivity genes was inserted at nucleotides (nt) 80001 to 80876 to block the expression of BMRF1 proteins completely. The cassette was then replaced with a wild-type BMRF1 sequence or each point-mutated BMRF1 sequence to prepare revertants or viruses carrying the intended mutations, respectively (Fig. 2A).
The recombinant EBV genomes were analyzed by BamHI digestion, followed by agarose gel electrophoresis (Fig. 2B). The BamHI-M fragment is present in wild-type and mutated viruses, but the corresponding band of⌬M/neo virus migrated slowly in the gel at about 1.8 kb, which is the size of the cassette.
The recombinant EBV DNAs were then introduced into HEK293 cells, and hygromycin-resistant cell colonies were screened for further analysis. The colonies formed by the wild type and each recombinant virus were comparable in size and numbers. We calculated the copy number of recombinant EBV DNA in each infected cell using quantitative real-time PCR. The average values from three independent experiments were as follows: wild type, 3.1 copies/cell; C95E, 5.3 copies/cell; H141F, 3.8 copies/cell; R256E, 5.4 copies/cell; C206E, 3.0 cop-ies/cell; revertant, 4.8 copcop-ies/cell; and⌬M/neo, 3.0 copies/cell. Exogenous expression of BZLF1 induced adequate expression of early proteins, BALF2 single-stranded DNA (ssDNA) bind-ing protein, BALF5 polymerase catalytic subunit, and BBLF2/3 helicase-primase accessory protein, as well as BMRF1 (Fig. 2C), although⌬M/neo did not produce BMRF1 at all, as expected (Fig. 2C).
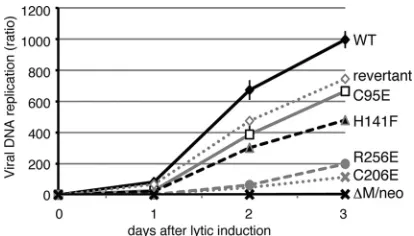
Viral DNA synthesis in recombinant viruses.When levels of viral DNA synthesis in HEK293 cells with the wild-type or recombinant viruses were examined using quantitative real-time PCR (Fig. 3), the amplification of viral DNA was found to reach nearly 1,000-fold at 3 days postinduction in cells harbor-ing the wild-type genome, whereas it was completely blocked in cells with the BMRF1 knockout⌬M/neo, as reported previ-ously (40). Viral DNA replication of the C95E mutant, which was defective in head-to-head dimerization, was at almost the same level as that of the revertant. Although the H141F mu-tant BMRF1 exhibited rather impaired polymerase
processiv-ityin vitro(38), the mutation caused only a mild reduction of
viral DNA replication in the context of infection. In contrast, although the polymerase processivity of the C206E mutantin
vitrowas not impaired at all (38), viral DNA replication was
[image:4.585.328.506.66.540.2]severely impaired in the context of infection. These results suggest thatin vitroprocessivity data do not accurately reflect the actual function in cells. In lytic replication-induced cells, other viral replication factors such as BALF2 single-stranded DNA binding protein and BBLF4/BSLF1/BBLF2-3 helicase-primase proteins might cooperate and compensate for the re-duced processivity activity of the BMRF1, especially since BALF2 can enhance the processivity of the BALF5 polymerase catalytic subunit in a manner different from that of BMRF1 (53). On the other hand, replication of the R256E mutant,
FIG. 2. Recombinant EBV BAC genome structures. (A) Schematic arrangement of the recombination of the EBV genome using neomy-cin resistance and streptomyneomy-cin sensitivity genes (neo/st). The region between nucleotides 80001 and 80876 of the EBV gene (GenBank accession no. V01555) was replaced with the neo/st genes to make BAC⌬M/neo. The BMRF1 gene, including this neo/st cassette, was replaced with each point-mutated BMRF1 and wild-type BMRF1 se-quence to construct BACpmBMRF1 and the revertant virus, respec-tively. The asterisk indicates the point-mutated site. (B) Electrophore-sis of the recombinant viruses. EBV BAC DNAs were digested with BamHI and separated in a 1.0% agarose gel. The black arrowhead indicates the BamHI-M fragment of the virus, and the white arrow-head indicates the size of BamHI-M fragments plus the marker cas-settes. (C) Expression levels of BALF2, BALF5, BBLF2/3, BMRF1, and BZLF1 proteins determined by immunoblot analysis with specific polyclonal antibodies. Lysate from HEK293 cells infected with recom-binant viruses was harvested at 48 h posttransfection with BZLF1 expression plasmids. Lysate from B95-8 cells treated with tetradeca-noyl phorbol acetate(TPA), A23187, and sodium butyrate for 48 h was included as a positive control.
on November 8, 2019 by guest
http://jvi.asm.org/
which is defective in DNA binding and polymerase processivity (38), was significantly restricted.
Production of infectious progeny viruses.To determine the yields of progeny viruses, HEK293 cells carrying each BAC DNA clone were transfected with the BZLF1 expression vec-tor to induce lytic replication and harvested at 3 days after transfection as described in Materials and Methods. The viral titer with 1.0 ml of virus solution was calculated by counting GFP-positive cells (Fig. 4). The titer of the C95E (1.05 ⫻ 106/ml) or H141F (0.84⫻106/ml) virus was almost the same as
that of wild-type (1.22⫻106/ml) or revertant (1.14⫻106/ml)
virus. Corresponding to the levels of DNA synthesis, progeny virus production was highly restricted with the R256E (0.06⫻ 106/ml) and C206E (0.12⫻106/ml) substitutions.
Processing of viral DNA intermediates. To ascertain whether each mutated BMRF1 protein influences the matura-tion of viral replicamatura-tion products, we performed pulsed-field gel electrophoresis (PFGE), followed by Southern blot analysis with EBV W-repeat sequences as probes (Fig. 5). HEK293 cells carrying the wild-type genome were transfected with the BZLF1 expression vector to induce the lytic cycle of the virus in the absence or presence of 0.4 mg/ml of phosphonoacetic acid (PAA), a specific inhibitor of viral DNA polymerase, and then harvested on days 1, 2, and 3 (Fig. 5A). In the gels, three major bands of viral DNA were detected, as was expected in other herpesviruses. It has been previously reported that sig-nificant amounts of amplified viral DNA are unable to enter the gel and are detected at wells, because huge replicated DNA forms are complicated branched and nonlinear struc-tures (18, 33, 46). We also observed similar accumulation of viral DNA (Fig. 5A). A fraction of the amplified viral DNA migrated to the same position as host chromosomal DNA, and the remainder was found at about 170 kb, which corresponds to the size of unit-length mature virion DNA (Fig. 5A). A band coinciding with circular DNA was not detectable (Fig. 5A).
We then tested cells with viruses carrying each mutation at 3 days after induction (Fig. 5B). In the C95E and H141F virus-infected cells, huge well DNA and mature unit-length virion DNA were produced, as in the cases of wild-type and revertant viruses. With the R256E and C206E mutant-infected
cells, however, markedly reduced levels of the mature unit-length viral DNA were observed in proportion to the reduced yields of the progeny viruses (Fig. 4).
Nuclear retention and replication compartment formation.
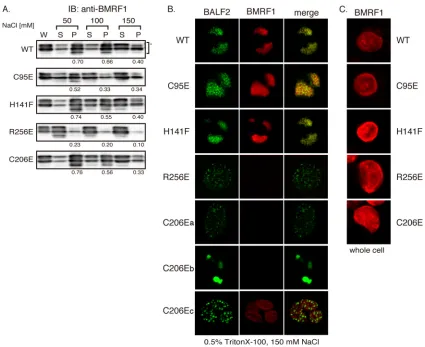
We next assessed the nuclear retention capacity of BMRF1 proteins in HEK293 cells with wild-type and recombinant vi-ruses. Cells were harvested at 2 days postinduction and lysed in 0.5% Triton X-100 solution containing 50, 100, or 150 mM NaCl. After centrifugation, the supernatant (S) and the pellet (P) fractions were separated by centrifugation (referred to as cytoplasmic and nuclear fractions, respectively). Levels of the BMRF1 proteins in each fraction were detected by immuno-blot assay (Fig. 6A). The intensity of the fluorescence was assessed with a Lumi Vision Analyzer 2.0 (AISIN), and nuclear retention was estimated by calculating the proportions of the BMRF1 proteins in the nuclear fractions [P/(S ⫹ P)]. The H141F and C206E mutant proteins behaved similarly to wild-type BMRF1, whereas the C95E protein showed slightly de-creased association with the nuclear fraction. In contrast, the R256E mutant demonstrated very weak retention in the nu-cleus (Fig. 6A). We previously demonstrated that thein vitro
DNA binding activities of C95E and R256E mutant BMRF1 proteins, as assessed by filter binding assay, were disabled moderately and completely, respectively (38). Therefore, it is likely that the nuclear retention simply reflects the DNA bind-ing activity of BMRF1 mutants.
[image:5.585.59.266.67.185.2]EBV lytic DNA replication takes place at discrete sites in nuclei, which are called replication compartments. Compo-nents of the viral DNA replication machinery, including BMRF1 and BALF2, are localized in these compartments (10). We thus examined whether BMRF1 mutants could gather to form the replication compartments in lytically in-fected cells by using immunofluorescence analysis (Fig. 6B). Cells were harvested at 24 h postinduction, fixed after treat-ment with or without 0.5% Triton X-100–mCSK buffer (150 mM NaCl), and then visualized with anti-BMRF1 mouse
[image:5.585.318.524.69.204.2]FIG. 3. Viral DNA synthesis in cells harboring BMRF1-mutated recombinant viruses after induction of lytic replication. HEK293 cells with the indicated EBV genomes were transfected with the BZLF1 expression plasmid and harvested at the indicated days. Levels of viral DNA synthesis were determined by quantitative real time-PCR assay as described in Materials and Methods and plotted as a ratio to each latency value (day 0)⫾the standard error of the mean (SEM) from three independent experiments. WT, wild type.
FIG. 4. Production of infectious progeny viruses. HEK293 cells carrying the indicated EBV genomes were transfected with both BZLF1 and BALF4 expression plasmids and harvested at 3 days post-transfection. EBV-negative Akata cells (2⫻106cells) were infected
with serially diluted (1-, 2-, or 10-fold) virus mixtures as described in Materials and Methods. The cells were analyzed by FACS analysis using FL1 and FL2 channels, and GFP-expressing cells were identified by shift of fluorescence intensity in the FL1 channel. Bars represent mean infectious viral particle units per ml (⫻104GFP-positive cells/
ml)⫾the SEM from three independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
monoclonal antibody and BALF2 rabbit polyclonal anti-body (green). Without detergent treatment, all the wild-type BMRF1 and mutants were localized throughout nuclei (Fig. 6C). When cells were treated with detergent, two BMRF1 mutants, the C95E and H141F mutants, were retained in rep-lication compartments, just like wild-type BMRF1. On the other hand, in a number of cells harboring the R256E mutation in the EBV genome, the mutant BMRF1 protein neither was retained nor colocalized with BALF2 proteins, which were distributed throughout nuclei as small dots by the treatment. The same staining pattern was observed in lytic replication-induced B95-8 cells in the presence of phosphonoacetic acid (10). Thus, formation of replication compartments was not
apparent in the R256E-infected cells. In some cells harboring the C206E mutation, the staining pattern was the same as that of the cells harboring the R256E mutation (Fig. 6B, C206Ea). However, in others, replication compartments were formed although the BMRF1 was not stained as strongly as in cells harboring the wild-type EBV genome (Fig. 6B, C206Eb). With an increase in the sensitivity of the BMRF1, staining was vis-ible as shown in Fig. 6B, C206Ec.
trans-Complementation of reduced viral replication in BMRF1-deficient virus-infected cells.So far, we have docu-mented the crucial roles of the Cys206and Arg256residues in
the context of infection, using recombinant EBV carrying des-ignated mutations. However, we still could not deny the
pos-FIG. 5. Viral replication intermediates in lytic replication-induced cells with recombinant viruses. (A) HEK293 cells carrying the wild-type EBV genome were transfected with BZLF1 expression vector and incubated with (⫹) or without (⫺) 0.4 mg/ml of phosphonoacetic acid (PAA). The cells were harvested at 1, 2, and 3 days after transfection. (B) HEK293 cells carrying the indicated EBV genomes were transfected with the BZLF1 expression plasmid and harvested 3 days thereafter. Plugs (1.5⫻105cells per plug) were prepared as described in Materials and Methods and
subjected to PFEG in a 1.0% agarose gel. Lambda ladder PFG marker (marker), restriction enzyme, IppoI-digested EBV BAC DNA (linear), and undigested EBV BAC DNA (circular) were used as size markers. Southern blots of the gels were probed with digoxigenin (DIG)-labeled EBV W-repeat fragments (see Materials and Methods).
on November 8, 2019 by guest
http://jvi.asm.org/
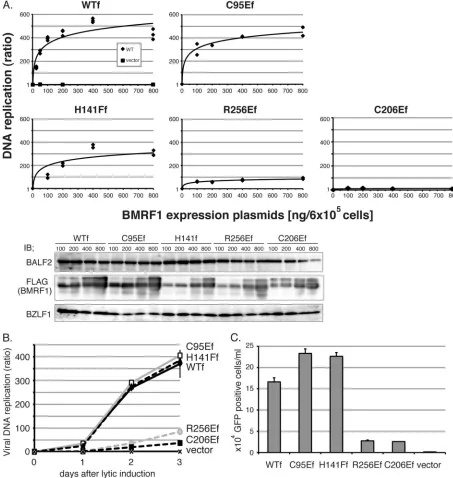
sibility that those mutant viruses might carry an additional unexpected mutation(s) in the genome. In order to preclude this possibility and further verify the significance of these res-idues, we tested whether ectopic expression of wild-type or mutant BMRF1 proteins could restore the attenuated viral replication in BMRF1 knockout virus-infected cells. HEK293 cells latently infected with BMRF1-deficient virus (BAC⌬M/ neo) were transfected with panels of the FLAG-tagged BMRF1 expression plasmid, together with the BZLF1 expres-sion plasmid. Viral DNA replication levels (Fig. 7A and B) and progeny virus production (Fig. 7C) were measured by real-time PCR and fluorescence-activated cell sorter (FACS) analysis, respectively. Overall, expression of the C95E and H141F mu-tants could complement the impaired replication of knockout BMRF1 just like wild-type BMRF1, but recovery by the R256E and C206E mutants was very limited. In addition, impaired
processing of the viral genome DNA into unit lengths with the BMRF1 knockout virus was detected upon exogenous supply of the wild-type, C95E, or H141F BMRF1 (data not shown). These results further support our conclusion that the Cys206
and Arg256residues of EBV BMRF1 are important for viral
replication, especially in the context of infection.
DISCUSSION
[image:7.585.79.504.65.412.2]The EBV BMRF1 gene product functions as the viral DNA polymerase processivity factor, playing an essential role in lytic replication (7, 16, 25, 26, 29, 50). We have recently solved the crystal structure of BMRF1, and, taking advantage of the data, several BMRF1 mutant proteins were prepared and character-ized biochemically (38, 39). Most BMRF1 exists as a C-shaped head-to-head homodimer in solution, as judged by analytical
FIG. 6. Nuclear retention of BMRF1 proteins and formation of replication compartments. HEK293 cells carrying the indicated EBV genomes were transfected with the BZLF1 expression plasmid and harvested at 48 h posttransfection. (A) Nuclear retention of BMRF1 proteins. Lytic replication-induced cells were treated with lysis buffer containing one of several concentrations of NaCl (50, 100, or 150 mM). Whole-cell lysate (W), extractable supernatants (S), and extracted nuclear pellets (P) were prepared as described in Materials and Methods. Immunoblot analysis was performed using anti-BMRF1 antibodies. The intensity of the fluorescence was counted with a Lumi Vision Analyzer 2.0 (AISIN), and nuclear retention was estimated by calculating the proportion of the BMRF1 proteins in the nuclear fraction, P/(S⫹P). (B) Formation of viral replication compartments. Infected cells were extracted with 0.5% Triton X-100–mCSK buffer and fixed with methanol. The fixed cells were immunostained with anti-BMRF1 monoclonal and anti-BALF2 polyclonal antibodies, followed by treatment with Alexa Fluor 594-conjugated anti-mouse IgG secondary antibody (BMRF1, red) or with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (BALF2, green), respectively. The right panels are the merged images. (C) The lytic replication-induced cells were harvested 24 h postinduction and fixed with methanol. The fixed cells were immunostained with anti-BMRF1 monoclonal antibody, followed by treatment with Alexa fluor 594-conjugated anti-mouse IgG secondary antibody (BMRF1, red).
on November 8, 2019 by guest
http://jvi.asm.org/
ultracentrifugation and blue native gel electrophoresis. How-ever, some C-shaped head-to-head homodimers can associate through their tail-to-tail interfaces and form tetrameric ring structures. The molecular contact surfaces within this ring
form continuous -sheets in “head-to-head” (I1-I1⬘) and “tail-to-tail” (D2-D2⬘) manners (where “prime” indicates
the neighboring molecule) (see supplemental Fig. 2 in refer-ence 38). The head-to-head contact surface area is larger
(ap-FIG. 7.trans-Complementation of impaired viral replication in BMRF1-knockout BAC⌬M/neo-harboring cells. (A)trans-complementation of viral DNA synthesis in BMRF1. Increasing amounts of FLAG-tagged BMRF1 expression vector were transfected into BAC⌬M/neo cells (6⫻105
cells) with the BZLF1 expression vector and harvested 3 days thereafter. Levels of viral DNA synthesis were determined by quantitative real time-PCR assay as described in Materials and Methods. The values from three independent experiments are plotted as ratios to values for latent cells. The solid line is the best-fit curve for the data. Cell extracts from each transfected cell preparation were electrophoresed on 10% polyacrylamide gels. Immunoblotting was carried out with anti-BALF2, -Flag, and -BZLF1 antibodies (bottom panel). (B) Viral DNA synthesis levels. A 400-ng aliquot of the indicated FLAG-tagged BMRF1 expression vector was cotransfected with the BZLF1 expression vector into BAC⌬M/neo cells (6⫻105cells), which were harvested at various time points thereafter, followed by processing as described for panel A.
(C) Production of infectious progeny viruses. A 400-ng aliquot of the indicated FLAG-tagged BMRF1 expression vector was cotransfected with 200 ng of BZLF1 and BALF4 expression vectors into HEK293 cells carrying BAC⌬M/neo (6⫻105cells), and viruses were harvested 3 days
thereafter. The viruses were then inoculated with EBV-negative Akata cells, followed by determination of infectivity by FACS analysis.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.61.514.70.548.2]proximately 930 Å2) than the tail-to-tail contact area (340 Å2).
In addition to these main-chain/main-chain interactions, disul-fide bonds (Cys95-Cys95⬘and Cys206-Cys206⬘) are present on both contact surfaces of the crystal ring structure (38, 39). Although a Cys95 mutation to a serine residue had no effect at all on the dimer formation (data not shown), replacement with a negatively charged glutamic acid, C95E, impaired dimer for-mationin vitro, with only very weak DNA binding underin vitro
specific conditions (38), strongly suggesting that head-to-head homodimer formation is mediated mainly through -sheets (I1-I1⬘) rather than through C95 residues. Since the C95E mutation had only a marginal effect on viral replicationin vivo, as shown in the present study, it would not be expected to affect dimer formation in the context of infection, unlike with thein
vitroresults. On the other hand, viral replication of the C206E
mutant, whose mutation does not affect in vitro biochemical activity of the protein, was found to be severely restricted, suggesting that tail-to-tail interactions are significant for virus replication. Since the head-to-head contact area is larger than the tail-to-tail interface, the tail-tail interaction via the Cys206
residue might be crucial for tetramer formationin vivo. There-fore, we speculate that tetrameric ring formation might be required for efficient lytic EBV replication, and this would explain why replacement of the Cys206residue at the tail-to-tail
interface had a very distinct influence on viral replication. Another possibility is that a particular viral factor might help the head-to-head association even in the absence of the Cys95
residue, while the tail-to-tail contact could not be restored by any factor. Because BALF5, the catalytic subunit of viral DNA polymerase, is estimated to associate with the contact area of the head-to-head dimer of BMRF1 (38), BALF5 might some-how enforce the dimer formation of BMRF1.
The Arg256 residue is located inside the ring-shaped
tet-ramer BMRF1 (Fig. 1). As the positive charge of the residue is crucial for binding to DNA, substitution with an acidic amino acid (R256E) causes complete loss of DNA binding activityin
vitro(38). In fact, viral replication was also severely restricted
by the mutation, suggesting an importance of DNA binding by the processivity factor for efficient function. The C95E mutant BMRF1, however, exhibited normal viral replication, although the mutation caused significant loss of DNA bindingin vitroat lower concentrations (38). Even so, we speculate that DNA binding is a crucial quality of BMRF1 in order for it to act as a processivity factor, because the C95E mutant BMRF1 did bind with DNA as efficiently as wild-type BMRF1 at high concentration (38), corresponding with the fact that the BMRF1 protein is abundantly present at replication compart-ments. Supporting data for this speculation are provided by BMRF1 homologues of human cytomegalovirus (HCMV) and herpes simplex virus (HSV): the DNA binding ability of HCMV UL44 (3, 31) and HSV UL42 (45) is essential for their function as processivity factors.
We then asked why DNA binding is so important for the processivity function and examined the intracellular localiza-tion of BMRF1 (Fig. 6). Interestingly, retenlocaliza-tion of the R256E mutant BMRF1 with the nuclear fraction proved to be severely impaired, and the mutant virus failed to form replication com-partments, suggesting that DNA binding ability is required for nuclear retention and proper localization to the compartments. Similarly, DNA binding ability of HCMV UL44 is required for
its nuclear retention (3). The C206E BMRF1, on the other hand, showed normal association with the nuclear fraction, but it also failed to accumulate at replication compartments. Our previous report indicated that efficient DNA replication is needed for appropriate accumulation of viral replication pro-teins to the compartments, since inhibition of viral DNA rep-lication by the addition of PAA caused similar mislocalization of viral replication factors (10). Therefore, failure of BMRF1 to accumulate at replication compartments could be the reason for impaired replication of the mutants, but we could not explain with our current data why the mutant demonstrates such impaired accumulation. Another way to account for the importance of DNA binding is that BMRF1 might not only act at viral replication forks as a polymerase processive factor but also widely attach on newly synthesized EBV genomic DNA to maintain the DNA’s integrity or to protect it from degradation (10).
The BMRF1 phenotypes of the mutantsin vitroandin vivo
are summarized in Table 1. Since the previous results for BMRF1 processivity measuredin vitrowere not always found to agree with the data presented in this paper, researchin vivo
must be accorded great importance. To give a concrete exam-ple, the C206E mutant, which is defective in the disulfide bond of the tail-to-tail contact, exhibited as efficient processivity as wild-type BMRF1in vitro, but the mutation severely affected virus replication. This is the first report that the correct con-formation of tetramer BMRF1 protein is necessary for lytic replication. With crystal structural imaging, such information might be of great value in creating novel antiviral medicines.
ACKNOWLEDGMENTS
We are grateful to W. Hammerschmidt and H. J. Delecluse for providing the EBV-Bac system, HEK293 cells, and pcDNA-BALF4 and to K. Takada for providing the Akata(⫺) cells.
This work was supported by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, Culture and Technology of Japan (20390137 and 21022055 to T.T.) and partly by the Uehara Memorial Research Fund (to T.T.).
REFERENCES
1. Reference deleted.
2.Ali, A. K., S. Saito, S. Shibata, K. Takada, and T. Kanda.2009. Distinctive effects of the Epstein-Barr virus family of repeats on viral latent gene pro-moter activity and B-lymphocyte transformation. J. Virol.83:9163–9174. 3.Alvisi, G., D. M. Roth, D. Camozzi, G. S. Pari, A. Loregian, A. Ripalti, and
D. A. Jans.2009. The flexible loop of the human cytomegalovirus DNA polymerase processivity factor ppUL44 is required for efficient DNA binding and replication in cells. J. Virol.83:9567–9576.
4.Bruck, I., and M. O’Donnell.2001. The ring-type polymerase sliding clamp family. Genome Biol.2:REVIEWS3001.
5. Reference deleted. 6. Reference deleted.
7.Chen, L. W., L. S. Lin, Y. S. Chang, and S. T. Liu.1995. Functional analysis of EA-D of Epstein-Barr virus. Virology211:593–597.
8.Chiou, J. F., and Y. C. Cheng.1985. Interaction of Epstein-Barr virus DNA polymerase and 5⬘-triphosphates of several antiviral nucleoside analogs. An-timicrob. Agents Chemother.27:416–418.
9.Cho, M. S., G. Milman, and S. D. Hayward.1985. A second Epstein-Barr virus early antigen gene in BamHI fragment M encodes a 48- to 50-kilodal-ton nuclear protein. J. Virol.56:860–866.
10.Daikoku, T., A. Kudoh, M. Fujita, Y. Sugaya, H. Isomura, N. Shirata, and T. Tsurumi.2005. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J. Virol.79:3409–3418.
11.Daikoku, T., A. Kudoh, Y. Sugaya, S. Iwahori, N. Shirata, H. Isomura, and T. Tsurumi.2006. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J. Biol. Chem. 281:11422– 11430.
12.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammer-schmidt.1998. Propagation and recovery of intact, infectious Epstein-Barr
on November 8, 2019 by guest
http://jvi.asm.org/
virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250.
13. Reference deleted.
14.Fields, B. N., D. M. Knipe, P. M. Howley, D. E. Griffin, and Lippincott Williams & Wilkins.2002. Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
15.Fixman, E. D., G. S. Hayward, and S. D. Hayward.1995. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol.69:2998– 3006.
16.Fixman, E. D., G. S. Hayward, and S. D. Hayward.1992. trans-acting re-quirements for replication of Epstein-Barr virus ori-Lyt. J. Virol.66:5030– 5039.
17.Flemington, E. K., A. E. Goldfeld, and S. H. Speck.1991. Efficient transcrip-tion of the Epstein-Barr virus immediate-early BZLF1 and BRLF1 genes requires protein synthesis. J. Virol.65:7073–7077.
18.Goldstein, J. N., and S. K. Weller.1998. In vitro processing of herpes simplex virus type 1 DNA replication intermediates by the viral alkaline nuclease, UL12. J. Virol.72:8772–8781.
19.Gulbis, J. M., Z. Kelman, J. Hurwitz, M. O’Donnell, and J. Kuriyan.1996. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell87:297–306.
20.Hammerschmidt, W., and B. Sugden.1988. Identification and characteriza-tion of oriLyt, a lytic origin of DNA replicacharacteriza-tion of Epstein-Barr virus. Cell 55:427–433.
21.Isomura, H., M. F. Stinski, A. Kudoh, T. Murata, S. Nakayama, Y. Sato, S. Iwahori, and T. Tsurumi.2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol.82:1638–1646.
22.Isomura, H., M. F. Stinski, A. Kudoh, S. Nakayama, T. Murata, Y. Sato, S. Iwahori, and T. Tsurumi.2008. A cis element between the TATA box and the transcription start site of the major immediate-early promoter of human cytomegalovirus determines efficiency of viral replication. J. Virol.82:849– 858.
23.Isomura, H., T. Tsurumi, and M. F. Stinski.2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol.78:12788–12799.
24.Kallin, B., L. Sternas, A. K. Saemundssen, J. Luka, H. Jornvall, B. Eriksson, P. Z. Tao, M. T. Nilsson, and G. Klein.1985. Purification of Epstein-Barr virus DNA polymerase from P3HR-1 cells. J. Virol.54:561–568. 25.Kiehl, A., and D. I. Dorsky.1995. Bipartite DNA-binding region of the
Epstein-Barr virus BMRF1 product essential for DNA polymerase accessory function. J. Virol.69:1669–1677.
26.Kiehl, A., and D. I. Dorsky.1991. Cooperation of EBV DNA polymerase and EA-D(BMRF1) in vitro and colocalization in nuclei of infected cells. Virol-ogy184:330–340.
27.Krishna, T. S., X. P. Kong, S. Gary, P. M. Burgers, and J. Kuriyan.1994. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell79:1233–1243.
28. Reference deleted.
29.Li, J. S., B. S. Zhou, G. E. Dutschman, S. P. Grill, R. S. Tan, and Y. C. Cheng.1987. Association of Epstein-Barr virus early antigen diffuse compo-nent and virus-specified DNA polymerase activity. J. Virol.61:2947–2949. 30. Reference deleted.
31.Loregian, A., E. Sinigalia, B. Mercorelli, G. Palu, and D. M. Coen.2007. Binding parameters and thermodynamics of the interaction of the human cytomegalovirus DNA polymerase accessory protein, UL44, with DNA: im-plications for the processivity mechanism. Nucleic Acids Res.35:4779–4791. 32.Makhov, A. M., D. Subramanian, E. Holley-Guthrie, S. C. Kenney, and J. D. Griffith.2004. The Epstein-Barr virus polymerase accessory factor BMRF1 adopts a ring-shaped structure as visualized by electron microscopy. J. Biol. Chem.279:40358–40361.
33.Martinez, R., L. Shao, J. C. Bronstein, P. C. Weber, and S. K. Weller.1996. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 215:152–164.
34.Matsumiya, S., S. Ishino, Y. Ishino, and K. Morikawa.2003. Intermolecular ion pairs maintain the toroidal structure of Pyrococcus furiosus PCNA. Protein Sci.12:823–831.
35.Moarefi, I., D. Jeruzalmi, J. Turner, M. O’Donnell, and J. Kuriyan.2000.
Crystal structure of the DNA polymerase processivity factor of T4 bacterio-phage. J. Mol. Biol.296:1215–1223.
36.Murata, T., H. Isomura, Y. Tamashita, S. Toyama, Y. Sato, S. Nakayama, A. Kudoh, S. Iwahori, T. Kanda, and T. Tsurumi.2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology389:75–81.
37.Murata, T., Y. Sato, S. Nakayama, A. Kudoh, S. Iwahori, H. Isomura, M. Tajima, T. Hishiki, T. Ohshima, M. Hijikata, K. Shimotohno, and T. Tsu-rumi.2009. TORC2, a coactivator of cAMP-response element-binding pro-tein, promotes Epstein-Barr virus reactivation from latency through interac-tion with viral BZLF1 protein. J. Biol. Chem.284:8033–8041.
38.Murayama, K., S. Nakayama, M. Kato-Murayama, R. Akasaka, N. Ohba-yashi, Y. Kamewari-Hayami, T. Terada, M. Shirouzu, T. Tsurumi, and S. Yokoyama.2009. Crystal structure of Epstein-Barr virus DNA polymerase processivity factor BMRF1. J. Biol. Chem.284:35896–35905.
39.Nakayama, S., T. Murata, K. Murayama, Y. Yasui, Y. Sato, A. Kudoh, S. Iwahori, H. Isomura, T. Kanda, and T. Tsurumi.2009. Epstein-Barr virus polymerase processivity factor enhances BALF2 promoter transcription as a coactivator for the BZLF1 immediate-early protein. J. Biol. Chem.284: 21557–21568.
40.Neuhierl, B., and H. J. Delecluse.2006. The Epstein-Barr virus BMRF1 gene is essential for lytic virus replication. J. Virol.80:5078–5081.
41.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse.2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and ef-ficiency of infection. Proc. Natl. Acad. Sci. U. S. A.99:15036–15041. 42.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for
high-expression transfectants with a novel eukaryotic vector. Gene108:193– 199.
43. Reference deleted.
44.Randell, J. C., and D. M. Coen.2004. The herpes simplex virus processivity factor, UL42, binds DNA as a monomer. J. Mol. Biol.335:409–413. 45.Randell, J. C., G. Komazin, C. Jiang, C. B. Hwang, and D. M. Coen.2005.
Effects of substitutions of arginine residues on the basic surface of herpes simplex virus UL42 support a role for DNA binding in processive DNA synthesis. J. Virol.79:12025–12034.
46.Severini, A., A. R. Morgan, D. R. Tovell, and D. L. Tyrrell.1994. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology200:428–435.
47.Shamoo, Y., and T. A. Steitz.1999. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell99:155–166.
48. Reference deleted.
49.Shimizu, N., A. Tanabe-Tochikura, Y. Kuroiwa, and K. Takada.1994. Iso-lation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J. Virol.68:6069–6073.
50.Tsurumi, T.1993. Purification and characterization of the DNA-binding activity of the Epstein-Barr virus DNA polymerase accessory protein BMRF1 gene products, as expressed in insect cells by using the baculovirus system. J. Virol.67:1681–1687.
51.Tsurumi, T., T. Daikoku, R. Kurachi, and Y. Nishiyama.1993. Functional interaction between Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit in vitro. J. Virol.67:7648–7653.
52.Tsurumi, T., T. Daikoku, and Y. Nishiyama.1994. Further characterization of the interaction between the Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit with regard to the 3⬘-to-5⬘exonucleolytic activity and stability of initiation complex at primer terminus. J. Virol.68: 3354–3363.
53.Tsurumi, T., A. Kobayashi, K. Tamai, H. Yamada, T. Daikoku, Y. Ya-mashita, and Y. Nishiyama.1996. Epstein-Barr virus single-stranded DNA-binding protein: purification, characterization, and action on DNA synthesis by the viral DNA polymerase. Virology222:352–364.
54.Yokoyama, N., K. Fujii, M. Hirata, K. Tamai, T. Kiyono, K. Kuzushima, Y. Nishiyama, M. Fujita, and T. Tsurumi.1999. Assembly of the Epstein-Barr virus BBLF4, BSLF1 and BBLF2/3 proteins and their interactive properties. J. Gen. Virol.80:2879–2887.
55.Zhang, Q., Y. Hong, D. Dorsky, E. Holley-Guthrie, S. Zalani, N. A. Elshiekh, A. Kiehl, T. Le, and S. Kenney.1996. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: effects on EBV transcription and lytic replication. J. Virol.70:5131–5142.
![FIG. 1. Mutated amino acid residues of EBV BMRF1 (amino acids[aa] 1 to 314). (A) The ring-shaped crystal structure of a tetramer of](https://thumb-us.123doks.com/thumbv2/123dok_us/156996.34140/2.585.46.284.65.583/mutated-amino-residues-bmrf-shaped-crystal-structure-tetramer.webp)