JOURNAL OFVIROLOGY, July 2008, p. 6434–6446 Vol. 82, No. 13 0022-538X/08/$08.00⫹0 doi:10.1128/JVI.02455-07
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
HLA Class I-Driven Evolution of Human Immunodeficiency Virus
Type 1 Subtype C Proteome: Immune Escape and Viral Load
䌤
Christine M. Rousseau,
1* Marcus G. Daniels,
2Jonathan M. Carlson,
3,4Carl Kadie,
4Hayley Crawford,
5Andrew Prendergast,
5Philippa Matthews,
5Rebecca Payne,
5Morgane Rolland,
1Dana N. Raugi,
1Brandon S. Maust,
1Gerald H. Learn,
1David C. Nickle,
1Hoosen Coovadia,
6Thumbi Ndung’u,
6Nicole Frahm,
7Christian Brander,
7Bruce D. Walker,
7,8Philip J. R. Goulder,
5,6,7Tanmoy Bhattacharya,
2,9David E. Heckerman,
1,4Bette T. Korber,
2,9and James I. Mullins
1,10Department of Microbiology, University of Washington, Seattle, Washington1; Los Alamos National Laboratory, Los Alamos, New Mexico2; Department of Computer Science and Engineering, University of Washington, Seattle, Washington3;
Machine Learning and Applied Statistics Group, Microsoft Research, Redmond, Washington4; Department of Pediatrics, Nuffield Department of Medicine, Oxford, England5; HIV Pathogenesis Program, Nelson Mandela School of
Medicine, University of KwaZulu-Natal, Durban, South Africa6; Partners AIDS Research Center, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts7;
Howard Hughes Medical Institute, Chevy Chase, Maryland8; Santa Fe Institute, Santa Fe, New Mexico9; and Department of Medicine,
University of Washington, Seattle, Washington10
Received 14 November 2007/Accepted 11 April 2008
Human immunodeficiency virus type 1 (HIV-1) mutations that confer escape from cytotoxic T-lymphocyte (CTL) recognition can sometimes result in lower viral fitness. These mutations can then revert upon transmission to a new host in the absence of CTL-mediated immune selection pressure restricted by the HLA alleles of the prior host. To identify these potentially critical recognition points on the virus, we assessed HLA-driven viral evolution using three phylogenetic correction methods across full HIV-1 subtype C proteomes from a cohort of 261 South Africans and identified amino acids conferring either susceptibility or resistance to CTLs. A total of 558 CTL-susceptible and -resistant HLA-amino acid associations were identified and organized into 310 immunological sets (groups of individual associations related to a single HLA/epitope combination). Mutations away from seven susceptible residues, including four in Gag, were associated with lower plasma viral-RNA loads (q< 0.2 [whereqis the expected false-discovery rate]) in individuals with the corresponding HLA alleles. The ratio of susceptible to resistant residues among those without the corresponding HLA alleles varied in the order Vpr > Gag > Rev > Pol > Nef > Vif > Tat > Env > Vpu (Fisher’s exact test;P<0.0009 for each comparison), suggesting the same ranking of fitness costs by genes associated with CTL escape. Significantly more HLA-B (2;Pⴝ3.59ⴛ10ⴚ5) and HLA-C (2;Pⴝ
4.71ⴛ10ⴚ6) alleles were associated with amino acid changes than HLA-A, highlighting their importance in driving viral evolution. In conclusion, specific HIV-1 residues (enriched in Vpr, Gag, and Rev) and HLA alleles (particularly B and C) confer susceptibility to the CTL response and are likely to be important in the development of vaccines targeted to decrease the viral load.
Human immunodeficiency virus type 1 (HIV-1) infection can elicit strong human leukocyte antigen (HLA) class I-me-diated immune responses from HIV-specific cytotoxic T lym-phocytes (CTL) (55, 66), which are thought to be important mediators of disease progression. However, viral sequence changes in critical amino acid residues of HLA-presented epitopes and immediately surrounding regions can be selected for their ability to effectively reduce the potency of the CTL response during the course of infection (escape variants) (5, 6, 10, 30, 37, 54, 57). The selective pressure of this CTL escape can be balanced by viral fitness constraints (16, 20, 27, 42, 45, 46, 53), with several studies finding CTL escape variants that
were less fit than the original strains (7, 11, 16, 27, 40, 45, 46, 62). Importantly, studies of humans and of macaques have shown that some escape variants revert to the original se-quence when infecting a host with a different HLA genotype (16, 27, 40), again suggesting that CTL escape in these in-stances was associated with a loss in viral fitness. Additional evidence demonstrates that despite reduced fitness, CTL es-cape variants can be transmitted from one host to another (2, 28, 29, 40, 41, 47), suggesting that they can persist in a popu-lation with common HLA alleles. Indeed, viral loads have been found to be correlated with HLA supertype frequencies, sug-gesting a population-wide selection for CTL escape mediated by the more common HLA alleles (65).
The identification of viral variants predicted to have higher viral fitness has important implications for vaccine design. A vaccine that elicits immune responses to the fittest viral vari-ant(s) and blocks common immune escape routes may not necessarily provide sterilizing immunity but might be able to
* Corresponding author. Mailing address: Department of Microbi-ology, University of Washington, 1959 NE Pacific Street, Box 358070, Seattle, WA 98195-8070. Phone: (206) 732-6102. Fax: (206) 732-6167. E-mail: [email protected].
䌤Published ahead of print on 23 April 2008.
6434
on November 8, 2019 by guest
http://jvi.asm.org/
produce a sufficient immune response to slow disease progres-sion (50). Such a vaccine might also reduce secondary trans-mission of the virus, which has been shown to be correlated with plasma viral-RNA loads (18, 58, 61). This type of vaccine could have dramatic public health benefits.
Because CTL epitopes are HLA restricted, the sequence changes of common viral escape pathways are characteristic of the HLA genotype of the infected host. Recently, novel com-putational methods exploiting this fact have been developed to identify amino acid changes associated with HLA genotypes in large cohorts of HIV-1-infected individuals (8, 12). With these methods, bias due to viral lineage can be taken into account using phylogenetic correction, allowing a more accurate assess-ment of HLA associations than previous efforts (39, 48). These methods can predict whether specific amino acid residues are generally susceptible or resistant to the CTL response medi-ated by the corresponding HLA allele. However, a simple picture of escape and reversion could not explain the HLA-amino acid associations found by Bhattacharya et al. (8), as particular substitutions were found to change roles in terms of resistance or susceptibility depending on the specific patient’s CTL tested. Furthermore, overlapping epitopes presented by different HLA molecules can place conflicting pressures on particular amino acid residues. Detailed analysis of the HIV-1 epitope SLYNTVATL has revealed substantial complexity of cross-recognition, escape, and reversion (32).
Because HLA alleles that are common in a given population can drive the fixation of the corresponding escape variants, consensus sequence peptides used in laboratory assays are likely to encode some viral escape variants incapable of elicit-ing a cellular response in vitro (3, 4). This could prevent the identification of certain epitopes when using consensus pep-tides, whereas novel computational methods may be better able to identify them.
Previous studies used phylogenetic correction to investigate HLA-amino acid associations in single genes or subgenomic fragments, primarily from subtype B sequences (8, 12). A high density of associations was found in Nef compared to protease, reverse transcriptase (RT), and Vpr, suggesting that HIV-1 proteins experience differential selection pressures in response to CTL recognition (12). Here, we provide a comparison of all HIV-1 proteins, allowing a more comprehensive assessment of viral evolution in CTL epitopes. Furthermore, we have focused our analysis on HIV-1 subtype C, the most globally prevalent and rapidly expanding subtype. We analyzed 261 full-length HIV-1 genomes from a cross-sectional South African cohort, investigating the relationship of HLA class I alleles and amino acid evolution to identify residues predicted to be either sus-ceptible or resistant to HLA class I-mediated immune re-sponses. The amino acid changes identified were further eval-uated for their association with the plasma viral-RNA load. Three different phylogenetic correction methods were utilized because of their complementary strengths. We also focused in greater depth on proteins for which additional sequences (in Gag, Pol, Env, and Nef) and corresponding host HLA infor-mation were available. We organized the identified associa-tions into immunological sets in an attempt to identify the most important residues and genes as candidates for a CTL-based HIV-1 vaccine.
MATERIALS AND METHODS
Study subjects.A cross-sectional cohort of HIV-1-infected individuals from KwaZulu-Natal province of South Africa was ascertained with informed consent as previously described (35). Because of the cross-sectional nature of the study design, most individuals were expected to be in the chronic phase of infection. All individuals were sampled prior to antiretroviral therapy. This study was approved by the internal review boards of the University of Washington, Los Alamos National Laboratories, Massachusetts General Hospital, and the University of KwaZulu-Natal.
Molecular methods. The sequences of full-length viral genomes, some of
which (n⫽244) were reported previously (60), were obtained by PCR
amplifi-cation of plasma-derived viral RNA, followed by cloning and sequencing. This data set was enlarged using the same techniques, and the sequences have been reported in association with this paper. Hypermutated genomes, intersubtype recombinants, and sequences with no corresponding HLA information were not
included in the analysis (n⫽11). Among the remaining sequences (n⫽261), all
were subtype C except two (one subtype B and one subtype A). Additionalgag,
pol,env, andnefsequences were obtained using population sequencing as
pre-viously described (16, 35, 39, 40). HLA genotypes were obtained as prepre-viously described (35). The CTL responses of the study participants were experimentally determined using gamma interferon enzyme-linked immunospot (ELISPOT) assays with a set of overlapping peptides from the consensus of HIV-1 subtype C sequences, as previously described (35). From the ELISPOT data, HLA-peptide associations were defined as those from at least one individual who reacted at least once to the peptide.
Sequence and phylogenetic analyses.Viral sequences were codon aligned using MacClade 4.08 (43). The 261 full-length HIV-1 genomes were divided into fragments of 1,000 nucleotides overlapping by 50 nucleotides (ignoring gene boundaries) to minimize the impact of intrasubtype recombination on phyloge-netic reconstructions. Each partition was used to generate a maximum likelihood phylogenetic tree, as previously described (8). Columns within the alignment that
were⬎10% gaps were considered null for the construction of the trees. The trees
were then evaluated using three distinct methods, maximum likelihood character state analysis followed by a likelihood ratio test (MLL) (available at http://www .microsoft.com/mscorp/tc/computational-tools.mspx), maximum likelihood char-acter state analysis followed by a Fisher test (MLF) (8), and parsimony charchar-acter state analysis followed by a Fisher test (parsimony) to identify HIV-1 amino acid changes associated with the HLA genotype. Because each method evaluated HLA and amino acid associations at each amino acid site individually, the results were compiled based on the gene to which the amino acid belonged. The results for overlapping reading frames were thus partitioned to the gene corresponding to the reading frame used.
In all three methods, only unambiguous matches were tabulated. For HLA alleles, two-digit genotypes were excluded as nonmatches when they shared the same first two digits of a four-digit HLA allele being considered. They were excluded because the true genotype might have been identical to the four-digit allele if the precision of the two-digit HLA genotype test had been to four-digit resolution. For amino acid comparisons, gaps were considered unknown and were excluded. Because ambiguous codons could result in multiple amino acid translations, they were also excluded. Ambiguous codons were present only in the extended data set, which included sequences derived from population se-quencing (16, 35, 39), whereas the full-length genome sequences were derived from unambiguous cloned sequences (60). In the MLF method, however, amino acid ambiguities from gaps and IUPAC ambiguities occurred in only a small number of cases because missing or partial data were inferred from the tree. This method inferred the parental sequence on leaf nodes with gaps. Thus, change was not often observed in these parts of the tree.
Two of the three methods (MLL and MLF) have been previously described (8, 12, 14). Both of these methods involved the evaluation of single-amino-acid changes and their relationship with the HLA genotype. For this study, overlap-ping 9-mers were also evaluated for an association with the HLA genotype using MLL and MLF.
The third method (parsimony) was developed to provide a more simplified approach and was compared to the others. For the parsimony method, the nucleotide sequence was translated into a protein sequence using MacClade 4.08 (43). The maximum likelihood trees of each 1,000-base-pair partition were used to make a parsimony-based reconstruction and inference of the amino acid sequences at the ancestral nodes. Using PAUP* 4.0b11 (64), the changes from the most recent nodal ancestor were recorded. Fisher’s exact test was used to evaluate the relationship of these sequence changes and HLA genotypes using STATA 8.0 (StataCorp LP, College Station, TX). Similar to the other two
methods, the resultingPvalues were used to calculateqvalues (the expected
on November 8, 2019 by guest
http://jvi.asm.org/
false-discovery rates), and only the associations withqvalues of⬍0.20 were considered.
Evaluation of significance.For all three methods, theP values from the two-digit and four-digit HLA genotypes were analyzed separately for the
calcu-lation ofqvalues. In addition, the calculation ofqvalues was performed
sepa-rately on each protein. Contingency tables were used for MLF and parsimony, and the odds ratio determined the predicted impact of the amino acid residue evaluated. The following expressions for the odds ratios indicated the impact:
[(A3!A and HLA)/(A3!A and !HLA)]/[(A3A and HLA)/(A3A and !HLA)]
(odds ratio⬎1, escape [susceptible]; odds ratio⬍1, repulsion [resistant]) and
[(!A3A and HLA)/(!A3A and !HLA)]/[(!A3!A and HLA)/(!A3!A and
!HLA)] (odds ratio⬎1, attraction [resistant]; odds ratio⬍1, reversion
[suscep-tible]). A and !A indicate the presence and absence, respectively, of a given amino acid residue. Similarly HLA and !HLA indicate the presence and absence, respectively, of the HLA allele. The arrows indicate “changes to.” The MLL
method used four statistical models for escape, reversion, attraction, and repul-sion (14).
For all three methods, one can interpret these terms as follows: escape means that in the presence of the HLA, there was pressure to change away from A (A to !A); reversion means that in the absence of the HLA, there was pressure to change to A (!A to A); attraction means that in the presence of the HLA, there was pressure to change to A (!A to A); and repulsion means that in the absence of the HLA, there was pressure to change away from A (A to !A). For both the escape and reversion cases, the given amino acid was most likely to be contrib-uting to the susceptible form of the epitope. Attraction and repulsion indicate amino acids likely to be associated with the immunologically resistant form of the epitope (Fig. 1).
Associations were placed into the following categories: raw associations (all unique associations) and individual associations (reducing “escape” or “rever-sion” to “susceptible” and “attraction” or “repul“rever-sion” to “resistant”; also
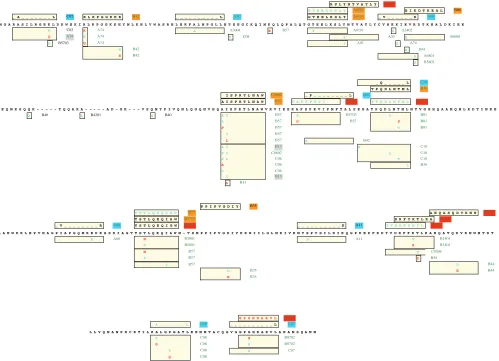
remov-FIG. 1. Epitope map of p17 and p24 of Gag showing individual associations and immunological sets. The reference sequence was the consensus of the full-length HIV-1 sequences from South Africa. All significant individual associations identified are shown below the consensus sequence. Blue amino acids reflect the susceptible form and red the resistant form. Shaded boxes indicate immunological sets. The size of the immunological set was determined by both the individual associations and validation data. These sets were chosen to minimize the number of epitopes that explained the observed data, taking linkage disequilibrium and 9-mer overlap into account. Only significant 9-mer regions with two or more associations are shown. For example, if a susceptible and a resistant residue were at a single site, both are shown, or if more than one site was involved in the variation that makes the 9-mer association distinctive, it is shown. If there were conserved amino acids between the HLA-associated variant sites within the 9-mer, they are indicated by small gray letters. When linked HLAs were associated with the same site, the HLA with the lowestPvalue is shown. An HLA in green indicates that there was a gamma interferon ELISPOT reactivity pattern associated with that HLA that overlapped the associated amino acid change. In the validation area (above the consensus sequence), fuchsia shading over the HLA label indicates motifs, orange indicates B list epitopes, red indicates A list epitopes, and blue indicates predicted epitopes. Similar to the individual associations, epitopes and motifs that were inferred to be susceptible or resistant are also labeled with blue or red letters, respectively. In the association area (below the consensus sequence), a gray background indicates that the association came from linkage disequilibrium. Associations in sites with⬎90% gaps are not shown. See http://www.hiv.lanl.gov/content/immunology/hlatem/index .html for maps of remaining HIV-1 proteins and the extended data set.
6436 ROUSSEAU ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.585.45.544.69.430.2]ing associations involving a two-digit HLA allele when the same association involving a four-digit allele shared the same first two digits) plus site associations (site with any association) and immunological sets (described below).
The top 274 results from each of the three methods were compared using receiver operating characteristic curves (available at http://www.hiv.lanl.gov /content/immunology/hlatem/study5/index.html) that considered each individual association (after those that were due to linkage disequilibrium were removed) a true-positive result if it was embedded in or within 3 amino acids of a known epitope. Two methods at a time were subjected to a permutation test in which each result was randomly swapped between the two methods. The area under the receiver operating characteristic curve of the real results was then compared to the area under the curve for the permuted results.
Amino acid entropy was calculated using methods previously described (38). The Wilcoxon signed-rank test was used to evaluate the relationship of amino acid entropy and the presence of an HLA association per amino acid site.
Estimation of the minimum number of known or potential epitopes required to explain the HLA-amino acid association data and the creation of epitope maps.The significant associations from both the single-amino-acid comparisons and the 9-mer comparisons were combined with validation data sets, which included all known epitopes, the reactive peptides identified with ELISPOT reactions, and known epitope motifs (MotifScan [http://www.hiv.lanl.gov/content /immunology/motif_scan/motif_scan] and EpiPhred [http://www.codeplex.com /MSCompBio]), each with the corresponding HLA genotype. This information was grouped into maps of putative immunological sets, defined as the minimum number of epitopes required to explain the observed HLA-amino acid associa-tion patterns, taking 9-mer overlap, HLA linkage disequilibrium, and HLA ambiguity into account. Immunological sets were defined as a group of HLA-amino acid associations within the same HLA allele and in the same epitope region. For example, a set of one or more individual associations with the same or a related HLA that were very near one another in terms of sites in the sequence were considered to be members of the same immunological set. HLA alleles were considered related based on two-digit and four-digit resolution (i.e., B35 and B3501) or HLA linkage disequilibrium. The same definitions were used by Brumme et al. (12).
The identification of immunological sets was automated. The associations and validation data sets were grouped into candidate sets by sliding a 13-amino-acid window over the region. The HLA that had the most significant association with the amino acid patterns in the set was considered the founder HLA. If two HLA alleles were in linkage disequilibrium and an associated amino acid was contra-dictory (the susceptible amino acid for one was the resistant amino acid for the other), they were split into separate sets. Known or potential epitopes (based on the presence of anchor motifs) with appropriate HLA-presenting molecules that overlapped onto the grouped associations were mapped onto the immunological sets. Immunological sets might include only a single amino acid association or, alternatively, if there were complex escape alternatives for an epitope, multiple associations. They were constrained to be no longer than 16 amino acids (for example, a 10-amino-acid epitope and 3 amino acids on either side) to allow for processing mutations (34). To be conservative, we chose to evaluate three sites flanking each epitope because they were the fewest sites with the most likelihood of involvement in cleavage (34).
We scanned for single HIV amino acid associations with HLA as described above using the MLL and MLF methods. We also scanned with amino acid patterns in sets of contiguous 9-mers (epitope length fragments) that might be associated with the HLA. If multiple escape routes were available to a given HLA-presented epitope, then by scanning for HLA-associated patterns in con-tiguous 9-mers, we would have greater statistical power to identify the variety of forms. The 9-mer references used in 9-mer association tests, like single amino acids, represented all observed patterns in the input alignments and tree ances-try. We took the output at the immunological-map-processing stage and, when
possible, reduced the 9-mer with HLA associations to potentially smallern-mers
representing just those amino acids that varied and the intervening conserved amino acids.
Following Brumme et al. (12), a hierarchal validation scheme was applied to the grouping of immunological sets. The HIV immunology database has two lists of experimentally defined epitopes: those that are optimally defined based on a set of stringent criteria and assembled in an annual review article (22), known as the “A list,” and the full database based on all of the epitopes that can be assembled from the experimental literature, known as the “B list” (http://www .hiv.lanl.gov/content/immunology/). The validation datasets were ranked into A list first and B list second. The third ranking, known as “motif,” included all known motifs from the HIV database (MotifScan [http://www.hiv.lanl.gov /content/immunology/motif_scan/motif_scan]) and epitopes predicted using Epitope Predictor (http://www.codeplex.com/MSCompBio). The “best” kind of
available validation data was selected for each immunological set (A⬎B⬎motif),
and all others were removed. The removal of these identical columns simply served to make the maps easier to read. Maps for all proteins and lists of associations can be viewed at http://www.hiv.lanl.gov/content/immunology/hlatem/study5/index.html. The ratio of susceptible to resistant amino acids at each site association was calculated using individuals without the corresponding HLA allele, taking link-age disequilibrium into account. The HLA alleles were reduced to two-digit designations, with the exception of the A68 and B15 alleles, which were kept at four digits because they represented more than one HLA supertype.
Determination of expected validations.To determine the number of valida-tions expected due to chance, the individual associavalida-tions were reduced to unique site associations in each immunological set. These sites were randomized 250 times. The number of sites found embedded in or within 3 amino acids of a known epitope were determined in both the original data set and the randomized datasets. The proportion of associations from the original data set was then compared to the randomized one for each gene.
Evaluation of amino acid changes associated with plasma viral-RNA loads.At each amino acid site with an HLA association, the viral loads of individuals with each corresponding HLA and a change away from the susceptible residue
(A3!A) was compared to the viral loads of individuals with no change away from
the susceptible residue using the Wilcoxon signed-rank test.qvalues were
de-termined from the resultingPvalues using QVALUE (63). The results with aq
value of⬍0.2 were considered significant. The same analysis was repeated using
the resistant instead of the susceptible residues.
Evaluation of extended data sets.Additional sequences in Gag, Pol, Env, and Nef were available for evaluation (16, 35, 39, 40). Extended data sets were generated containing these additional sequences, as well as the full-length ge-nome sequences in the corresponding regions. Because the additional sequences had variable ends due to inconsistent sequencing runs and/or different primers,
only amino acid sites with⬍10% missing data were included in the analysis,
which served to trim the alignment under consideration to the core region of the added sequences. The results of the analysis of the extended data set were tabulated and compared to those obtained from the full-length genome analysis over the respective protein regions.
Nucleotide sequence accession numbers.Accession numbers for full-length genomes, protein alignments, and host HLA information are available at http: //www.hiv.lanl.gov/content/immunology/hlatem/study5/index.html.
RESULTS
HLA-amino acid associations and immunological sets. Full-length HIV-1 genomes (n⫽261) from a cross-sectional South African cohort were screened for amino acid changes associ-ated with HLA alleles. Single amino acids, as well as 9-mers, were evaluated using phylogenetic correction methods that took the viral lineage into account. Amino acids were catego-rized into those predicted to confer susceptibility or resistance to the corresponding HLA-mediated immune response. As defined in Materials and Methods, susceptible residue catego-ries included negative correlations, i.e., the amino acid was enriched when the corresponding HLA was absent from the host or the amino acid was infrequent when the HLA allele was present within the host. Resistant categories included pos-itive correlations, i.e., the amino acid was infrequent when the HLA allele was absent from the host or the amino acid was enriched when the HLA allele was present within the host.
Using the three methods discussed below, we identified 948 HLA-amino acid associations categorized as reversion, escape, repulsion, and attraction (raw associations) (see Materials and Methods for definitions) with a q value of
⬍0.2. Reducing theqvalue to 0.05 resulted in a loss of 61% of these associations, 58% of which were supported by ELISPOT results. To include as many true-positive results as possible, we chose to further evaluate all associations with aqvalue of⬍0.2. The odds ratios ranged from 0 to 310 (excluding infinite odds ratios; see http://www.hiv.lanl.gov
on November 8, 2019 by guest
http://jvi.asm.org/
/content/immunology/hlatem/study5/full/index.html for a complete list). After the amino acids were recategorized as either susceptible or resistant, there were 532 individual HLA-amino acid associations (individual associations). Using a sliding window 9 amino acids in length (9-mers), 26 additional individual associations were identified, each involving a single 9-mer with two or more variable sites, for a total of 558 individual associations. These were distributed at 257 positions along the genome (site associations). After the proximities of individual associations linked to a particular HLA, overlap of 9-mers, epitope boundaries, and HLA linkage disequilibrium were taken into account, these data were organized into 310 groups of HLA-amino acid associations, each predicted to be related to single epitope /HLA combinations (immunological sets). These individual associations and immunological sets were mapped in relation to the consensus of the sequences analyzed (Fig. 1 shows p17 and p24 of Gag; see http://www.hiv.lanl.gov /content/immunology/hlatem/study5/index.html for com-plete proteome maps). The maps showed that multiple res-idues at a single site or multiple sites within or near an epitope were often involved in immune escape and rever-sion. The maps also illustrated sites with opposing selective pressures (Table 1), i.e., a single site with the same residue involved in two individual associations, one predicted to be susceptible to the CTL response mediated by one HLA allele and the other predicted to be resistant to the CTL response mediated by a different HLA allele.
The regions with the most immunological sets were at Pol amino acid position 607 with 33 sets and Nef amino acid position 83 with 28 sets, suggesting that these regions were under the most conflict due to immune pressure. The immu-nological set with the most individual associations (n⫽7) was found within the Nef cluster and included a B*44-restricted epitope (KRQEILDLW), suggesting that this epitope is under the most conflicting selective pressure. The next largest immu-nological sets were in Nef, Tat, and Pol, each of which included six individual associations involving the C*0404-restricted epitope HSQRRQDIL (also in the large Nef cluster) and the B*42-restricted epitopes QPKTPCNKCY and YPGIKVRQL, respectively.
[image:5.585.44.546.81.208.2]Association with plasma viral load.At each site association, plasma viral RNA loads in individuals with and without the susceptible residues were compared for all individuals with the corresponding HLA alleles. Under the assumption that sus-ceptible residues were identified in individuals without an ef-fective CTL response and resistant residues appeared in the presence of a CTL response (mostly ineffective due to the resistant residue), this analysis represents a comparison of the viral fitness of the susceptible to the resistant variant in the absence of an effective site-specific CTL response. Seven sites were associated with viral loads in individuals with changes away from the susceptible amino acid that were significantly lower than those in which there was no change (Table 2). Four of the seven residues were in Gag. Gag242 (Gag amino acid 242), associated with HLA-B*5801 and B*57, was previously
TABLE 1. HLA-amino acid associationsaand immunological groups in each HIV protein
Protein
No. of individual associations
No. of immunological
sets
No. of site associations/ total no. of
sites (%)
No. of sites with opposing
pressure
S/R ratiob
Env 15 12 11/960 (1.3) 0 0.398782344
Gag 93 57 48/536 (9.0) 4 4.172722191
Nef 143 58 48/230 (21.0) 13 1.067662008
Pol 197 118 85/1,024 (8.3) 12 1.844679445
Rev 32 21 21/128 (16.4) 0 3.086956522
Tat 29 17 12/104 (11.5) 1 0.540289256
Vif 36 17 13/100 (13) 3 0.687697161
Vpr 11 8 8/102 (8.0) 0 5.432038835
Vpu 2 2 2/87 (2.3) 0 0.110843373
Total 558 310 272/3,277 (8.3) 33
aAssociations found in sites with⬎10% gaps were not included in this analysis.
bRatio of susceptible to resistant residues.
TABLE 2. Amino acid changes associated with significantly lower plasma viral-RNA loads
Protein HXB2
position
Associated
HLA HLA in LD
a Susceptible
amino acid Resistant amino acids
Difference in
median VLb Pvalue qvalue Motif/epitopec
Gag p17 120 B4201 A E,G,K,M,P,T,V 70,800 6.08⫻10⫺5 0.001 Not found
Gag p24 184 B81 A01, C18 L M,S,Y 67,730 0.036 0.168 TPQDLNTML
Gag p24 242 B57, B5801 T N,S 55,550 0.011 0.168 TSTLQEQIGW
Gag p24 339 C08 P A,Q,S,T,V 41,400 0.046 0.168 r关A兴lgpgat关L/M兴
Pol 725 B4403 E A,D,G,V 46,290 0.003 0.190 QEEHEKYHSNWd
Vif 33 B1503 C0202 K G,Q,R 103,665 0.005 0.060 s关Q/K兴rasgwf关Y/F兴
Rev 17 A3001 R K,Q 43,690 0.024 0.118 关A兴vriiki关L/M兴
a
LD, linkage disequilibrium.
b
VL, viral load in copies/ml.
c
Defined epitopes are in upper case. Motifs contain anchor residues in bracketed capital letters. Lowercase letters reflect consensus residues. The associated residues are underlined and in bold.
d
A B*4403 epitope in the peptide QEEHEKYHSNW has been identified but not optimally defined. The B*4403 motif is x关EMILVD兴xxxxxx关YF兴.
6438 ROUSSEAU ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.44.541.591.683.2]found to be associated with changes in the viral load and viral fitness (11, 45). Vif33 was associated with HLA-B*1503 and
HLA-C*02. Thse two HLA alleles were in linkage disequilib-rium, and the site was embedded in a B*1503 epitope binding motif, so B*1503 was considered more likely to be driving the association. HLA-B*1503 was also previously found to be as-sociated with low viral load in a cohort infected with subtype B (24) but not in our South African population infected with subtype C (24, 35, 36). Another site we detected (Pol725, asso-ciated with B*4403) had been recognized in an earlier study evaluating HLA-restricted immunological responses to HIV-1 peptides and associations with the viral load in our cohort (36). Gag184had three HLA alleles (A*01, B*81, C*18) associated with the same single-amino-acid change. However, these three alleles were in linkage disequilibrium, and thus, the associa-tions were considered likely to be driven by a single epitope/ HLA combination. Since an HLA-B*8101 epitope had been experimentally identified in this region (36), it was considered to be the most likely candidate. Two of the remaining amino acid sites were within predicted HLA binding motifs (Gag339 and Rev17), and only one site (Gag120) had no immunological support, suggesting that it might be a novel epitope or a false-positive result. A similar analysis was carried out using the resistant residues, but no associations with aqvalue of⬍0.20 were found. Intriguingly, five HLA alleles were identified by Kiepiela et al. (35) to be associated with a low viral load in this cohort, and we found four of the five (B*8101, B*57, B*5801, and B*4201) to have specific mutations associated with a low viral load (Table 2).
Comparison of HIV-1 proteins. The highest proportion of site associations was found in Nef (Table 1), where 21% of the amino acid sites in the protein were found to be involved in one or more immunological sets. The lowest proportion of site associations was found in Env, where only 1% were found to be involved in immunological sets.
Because reversions to the more susceptible variant occur in the absence of HLA pressure, the numbers of susceptible and resistant residues at each site association were counted among the individuals without the corresponding HLA allele. To make the results independent of gene size, the ratio of suscep-tible to resistant residues was determined to reflect the amount of reversion relative to the amount of escape not balanced by reversion. The ratio of susceptible to resistant residues was on the order of Vpr⬎Gag⬎Rev⬎Pol⬎Nef⬎Vif⬎Tat⬎ Env⬎Vpu (Table 1), each with significantly more susceptible versus resistant residues than the subsequent protein (Fisher’s exact test;Pⱕ0.0009 for each comparison). Although no bias is expected in this analysis, the presence of compensatory mu-tations may inflate the significance values. These results sug-gest that there is a greater fitness cost for escape in those proteins with a greater proportion of susceptible residues.
Mechanism of escape.To investigate where the escape mu-tations occurred within epitopes, individual associations in known epitopes were evaluated. Among the HLA-assigned epitopes in the HIV database (http://www.hiv.lanl.gov/content /immunology/index.html), 73 either encompassed or were within 3 amino acids of an individual association. For 16/73 (22%) of these epitopes, an individual association was found outside the boundary of the epitope but within 3 amino acids of it, possibly reflecting aberrant epitope processing (34).
Twenty-two of the 113 (19%) inferred anchor sites and 59/478 (12%) of the internal T-cell receptor-involved sites in known epitopes were found to be associated with an HLA allele in this study. A trend toward detectable selection disproportionately affecting anchor residues was also observed (Fisher’s exact test;
P⫽0.067).
Comparison with subtype B.A similar study was performed on fragments of Nef (206 amino acids; n ⫽ 686), Vpr (96 amino acids;n⫽425), and protease/RT (499 amino acids;n⫽
532) from a cohort of HIV-1 infected Canadians (97.5% in-fected with subtype B) (12). We found that the South African subtype C individual associations mapped either inside or within 3 amino acids of 18/69 (26%) Nef, 2/8 (25%) Vpr, and 9/41 (22%) protease/RT immunological sets identified by the Canadian study. However, the same epitope often had differ-ent individual associations for the corresponding HLA allele, implying different common escape patterns for homologous epitopes in different HIV-1 subtypes (Fig. 2). Among the shared immunological sets, 86% (25/29) of the epitopes had different escape patterns, depending on the subtype evaluated.
Frequency of HLA associations.HLA-B and HLA-C alleles were more often identified in the individual associations than HLA-A (7%, 55%, and 38% HLA-A, -B, and -C, respectively). This was also true when the data were analyzed for each gene individually, with the exception of Vpr, which had 11 individual associations (64%, 27%, and 9% associations for HLA-A, -B, and -C, respectively) and Vpu, which had two associations, both with HLA-A. Overall, the proportion of individual asso-ciations with aq value of ⬍0.2 was significantly greater for HLA-B than for HLA-A alleles (2;P⫽3.59⫻10⫺5) and for
HLA-C than HLA-A alleles (2;P ⫽4.71⫻ 10⫺6). HLA-B
and HLA-C alleles were involved in similar proportions of individual associations with qvalues of ⬍0.2 (P, not signifi-cant). In addition, the majority (60%) of the associations in-volved an HLA allele with higher frequency than the amino acid.
Amino acid variation and HLA associations. The sites in each protein had a wide range of amino acid entropy (Env, 0 to 2.64, median⫽0.19; Gag, 0 to 1.96, median⫽0.06; Nef, 0 to 1.96, median ⫽ 0.14; Pol, 0 to 2.01, median ⫽ 0.05; Rev, 0 to 1.16, median⫽0.23; Tat, 0 to 1.79, median⫽0.16; Vif, 0 to 1.54, median ⫽ 0.096; Vpr, 0 to 1.24, median ⫽ 0.057; Vpu, 0 to 1.91, median⫽0.31). Overall, site associ-ations had significantly greater mean entropy than sites without associations (0.59 versus 0.28, respectively; Wil-coxon signed-rank test;P ⬍ 0.0001). Most individual pro-teins also demonstrated a significant relationship between amino acid entropy and site associations (Wilcoxon signed-rank test;P⬍0.0001 for Gag, Nef, Pol, Tat, and Vif andP⫽
0.0007 for Vpr), with the exception of Env and Rev (Wil-coxon signed-rank test;P, not significant for either protein).
Validation. Among the individual associations identified, 9% were supported by (embedded in or within 3 amino acids of) the optimally defined “A-list” epitopes (22), 13% by the database of reported T-cell epitopes (“B-list”), and 59% by HLA allele-specific binding motifs, and 20% did not have validation support. Because we used aqvalue cutoff of⬍0.2, we expected to see approximately 20% false positives, possibly more frequently in the group with no validation support. Sig-nificantly more associations were found to be validated by
on November 8, 2019 by guest
http://jvi.asm.org/
6440
on November 8, 2019 by guest
http://jvi.asm.org/
known epitopes compared to randomized data sets in Gag, Pol, Vif (Pⱕ0.004 for each), and Nef (P⫽0.008). No significant differences were observed for the comparisons in the other proteins; they had fewer individual associations, and we thus infer that we did not have adequate power for these compar-isons.
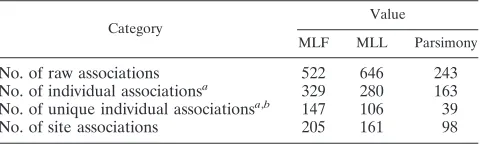
Comparison of three lineage correction methods.Maximum likelihood phylogenetic trees followed by likelihood ratio tests (MLL) or Fisher tests (MLF) or a parsimony analysis followed by a Fisher test was evaluated to identify HIV-1 amino acid changes associated with the HLA genotype. Using aqvalue cutoff of 0.2 for all methods, the MLL method found the most raw associations, although the greatest number of individual associations was found by MLF (Table 3). This was primarily due to the MLL method’s identification of more pairs of either negative (escape/reversion) or positive (attraction/repulsion) correlations, with each pair identifying the same HLA-amino acid combination. To account for correlated data, the numbers of individual associations were grouped based on sites and HLA alleles. In this case, the MLF method identified the most associations (MLF, 255; MLL, 211; parsimony, 114). On the other hand, in the Canadian data set (12), the MLL method found more individual associations grouped by site and HLA than the MLF method (MLL, 205; MLF, 177). Since the tests make different assumptions about the underlying process, the
q values do not necessarily reflect the true percentages of spurious results; therefore, finding more associations at a given
qvalue threshold does not necessarily mean that one method is superior to the others.
A similar proportion of individual associations supported by epitopes from the validation data set (A list, B list, and motifs) was found by each method (49% MLF, 49% MLL, and 54% parsimony; Fisher’s exact test;P⫽0.40). Using receiver oper-ating characteristic curves and a permutation test, no signifi-cant difference was found between the capacities of the three methods to discriminate between sites supported or not sup-ported by epitopes in the validation data set. Thus, the parsi-mony method, representing a new and more simplified ap-proach, was equivalent to the other two methods.
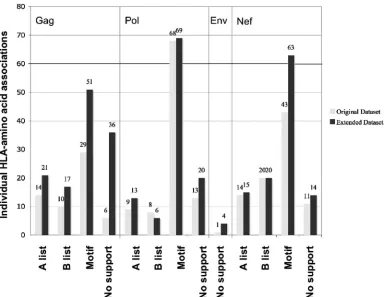
Extended sequence data set.To evaluate the impact of the number of sequences on the results of our analysis, regions where additional sequences were available from the same
co-hort (Gag [n⫽191], Pol [n⫽141], Env [n⫽84], and Nef [n⫽
91]) were reanalyzed, along with the original set of 261 se-quences, and compared to the results of the original analysis (excluding the individual associations found with the 9-mers). The number of individual associations in Env increased by 9-fold (9 versus 1), in Gag by 2.1-fold (111 versus 53), in Nef by 20% (102 versus 83), and in Pol by 11% (101 versus 90). Overall, there were 227 individual associations in the original data set in these regions, and 132 (58%) were maintained in the reanalysis with the extended data set. There were a total of 104 site associations identified in the original data set, and 77 (74%) of those were maintained in the reanalysis with the additional sequences. These changes were likely due to loss of false-positive results in the extended data set, as well as slight changes inPvalues and subsequent changes inqvalues bring-ing associations above or below the cutoffqvalue of⬍0.2.
The validation support (embedded in or within 3 amino acids of an A list epitope, B list epitope, epitope motif, or no support) for each site association was evaluated in both the original and extended data sets. To take linkage disequilibrium of the HLA alleles into account, the type of validation for each site was categorized only for the HLA with the highest level of support in each linkage disequilibrium group. The greatest number of new site associations identified in Gag and Pol were in the “no support” category, and the greatest number in Nef were in the “epitope motif” validation data (Fig. 3).
DISCUSSION
CTL responses to HIV-1 infection can lead to the evolution of escape mutations, effectively reducing the impact of the immune response and the control of disease progression. How-ever, these escape mutations can be balanced by viral fitness constraints. A vaccine that targets both the most fit viral vari-ants and their escape mutations might be effective enough to drive viral evolution toward states of lesser fitness and slow disease progression (50), potentially increasing the quality and duration of life for those infected while lowering the rate of transmission. Thus, our study and other recent work have sought to identify features of the viral proteome that might be especially important for targeting by immune responses elic-ited by vaccines.
Our approach to identifying critical immunologic features of the viral proteome was to identify associations between the HLA alleles that restrict the CTL response and amino acid variants found in the viruses infecting these individuals, along with their plasma viral loads, using the latter as a surrogate for disease status. Elements of this approach derived from earlier studies that linked HLA with specific amino acids (39, 48), refinement of these associations using phylogenetic correction (8, 14), and associations made between specific HLA alleles and specific viral proteins or the viral load (21, 24, 31, 36). Our study extends prior efforts at defining HLA-amino acid
[image:8.585.44.285.81.153.2]asso-FIG. 2. Immunological map comparing HLA-amino acid associations from subtypes B and C that were within the same epitopes. The light-yellow boxes are the subtype B sequences from a Canadian study (12), and the light-green boxes are the subtype C sequences from this study. The underlined residues represent a site with opposing selective pressures. In this case, it is mediated by the same HLA allele but is in the context of different amino acids three sites upstream.
TABLE 3. Comparison of phylogenetic correction methods
Category
Value
MLF MLL Parsimony
No. of raw associations 522 646 243
No. of individual associationsa 329 280 163
No. of unique individual associationsa,b 147 106 39
No. of site associations 205 161 98
a
Reduced based on HLA linkage disequilibrium.
b
Associations found only by a single method.
on November 8, 2019 by guest
http://jvi.asm.org/
ciations using these tools (12) to the analysis of whole viral proteomes and HIV-1 subtype C, the most common subtype worldwide.
Specific amino acid changes associated with HLA alleles were identified and placed into the context of known HLA epitopes and epitope motifs, taking HLA linkage disequilib-rium into account. We identified amino acid residues that were predicted to be resistant (n⫽244) or susceptible (n⫽314) to the HLA class I-mediated immune response with the predic-tion that susceptible variants were likely to reflect reversions based on selective pressure for increased viral fitness. We also evaluated the plasma viral loads among individuals with the corresponding HLA allele and the presence of the susceptible amino acid in the viral sequence. Some of the sites with changes away from the susceptible residue were found to be significantly associated with lower viral loads, including the well-studied epitope TSTLQEQIGW (TW10 [Gag240 to Gag249]) previously shown to impact viral fitness (11, 45). In total, we identified seven susceptible residues, escape from which was associated with lower viral loads, suggesting a fitness cost of immune escape. Several factors remain unknown for this analysis and likely resulted in a bias for false-negative results. These factors include the sequence of the transmitted strain (or strains), the timing of the CTL response, the pres-ence of compensatory mutations, and the prespres-ence of coinfec-tions that impacted the viral load (such as malaria). Further-more, there may have been undetected associations due to lack of power in our study. Thus, these 7 residues probably
repre-sent those with the most robust associations. These results support the idea that a vaccine that induces the CTL response to such epitopes, alone or in combination, may be effective in reducing viral loads.
[image:9.585.100.488.67.364.2]Although the constituents of an effective vaccine immuno-gen remain elusive, the results of this study suggest greater importance of some rather than other viral proteins in eliciting suppressive, if not protective, immunological responses. In par-ticular, our results suggest a ranked hierarchy of the proteins and the fitness costs associated with immune escape. Vpr, Gag, and Rev were at the top of the list, suggesting that these proteins are best able to elicit immune responses that decrease the fitness of the virus. On the other hand, the immune re-sponses to Vpu, Env, and Tat (at the other end of the list) were largely ineffective. The high abundance of Gag compared to other HIV-1 proteins (25) makes it a logical vaccine candidate. Thus, this study, in combination with several previous reports, converges on Gag, or elements of Gag, as an important com-ponent of a vaccine immunogen. In a prior study, Gag was found to be the only protein targeted by the CTL response that was associated with lower viral loads (36), and Gag p24 has been reported to be the preferred target for HLA alleles as-sociated with protection from disease progression (9). Other studies have shown that the proportion and magnitude of the CTL response to Gag were associated with the viral load (13, 17, 68) and that CTL escape mutations in Gag were associated with reduced viral fitness (2, 20, 40, 45, 52, 53). Gag also has a large number of conserved peptide elements (59) that are less
FIG. 3. A comparison of the types of validation support for the individual associations found in the original data set (light-gray bars) and in the extended data set (black bars) that included additional sequences from Gag, Pol, Env, and Nef.
6442 ROUSSEAU ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
likely to vary in response to immune pressure. Thus, it is probable that the structural and functional conservation of Gag is such that variation in most of its CTL epitopes is not well tolerated, making it a promising gene candidate for a CTL-based vaccine. The finding that Vpr and Rev were also among the proteins with the highest fitness costs associated with immune escape emphasizes the importance of considering these auxiliary proteins as vaccine components.
This study also supports the observations that host HLA alleles provide differential impacts on the virus. We found that HLA-B alleles, followed by HLA-C, were the most commonly associated with amino acid changes and had a greater propor-tion of significant associapropor-tions than HLA-A. Our work con-firms several previous studies that have identified the impor-tance of HLA-B in driving HIV-1 evolution and impacting disease (12, 35, 56) and also underscores the importance of HLA-C in impacting HIV-1 disease. Recently, a polymorphism upstream of the HLA-C gene was found to be associated with the viral set point in a genome-wide scan (19). Because HIV-1 down regulates HLA-A and HLA-B, but not HLA-C, expres-sion (1, 15) and because HLA-C-restricted cells can have an-tiviral activity similar to that of HLA-A and HLA-B (1), it may be especially worthwhile to consider HLA-C-restricted epitopes when selecting peptides for a CTL-based vaccine.
HLA population frequencies are also critical to consider in vaccine design. In a previous study, HLA-B*1503 was found to be associated with lower viral loads in a clade B-infected pop-ulation, in which B*1503 was rare (24), but not in a clade C-infected population, in which B*1503 was common (24, 35, 36). Frahm et al. (24) concluded that fixation of escape muta-tions in subdominant epitopes was the cause of the lack of response in the subtype C cohort. We did identify an associa-tion of HLA B*1503 with low viral load in this subtype C cohort. Indeed, the resistant (escape) amino acid identified in our study was in the consensus C peptides used by Frahm et al., explaining the poor response and in accord with their hypoth-esis. This finding underscores the fact that any viral sequence used to detect CTL responses, including consensus sequences, may encode escape variants and thus preclude detection of cognate epitopes. More advanced approaches, such as toggle design (23) and others (26), are needed to reliably assess the total breadth of immune responses. We also found that iden-tical epitopes, in the context of a subtype B or subtype C data set, had different susceptible and resistant variants, implying different escape mechanisms, depending on the subtype ana-lyzed. Because the HLA frequency in each population may influence the fixation of viral variants and the viral subtype might also influence immune escape mechanisms, it is possible that different sets of epitopes will need to be considered for each subtype or each unique population.
This study employed three distinct methods for the identi-fication of HLA-amino acid associations, each using phyloge-netic correction. Each method contributed novel associations and, using known epitopes as a proxy for a gold standard (since not all epitopes have been identified), all three methods had similar predictive capacities, yet their predictions did not over-lap completely. This was, in part, due to each method capturing certain types of associations better than others. For example, MLL was able to identify more details about the associations at each site, whereas MLF was able to identify more sites.
Al-though the parsimony method identified fewer sites, there was greater validation support for those sites. Another explanation for the lack of overlap between the three studies is that each identifies a small proportion of the true associations, support-ing the idea that the union of the three methods provides the most comprehensive set of associations.
To define which associations were true positives, we used a validation set of all known epitopes. However, this validation data set, consisting of the best available information, was not an ideal standard for comparison for several reasons. We ex-pect that greater than 50% of CTL epitopes have not been experimentally identified (41). Uncertainty in epitope motifs and nonoptimized epitopes can also lead to incorrect support of an association. There also exists a bias for subtype B among known epitopes (49). Finally, compensatory mutations were not considered in the validation data set, and these can involve several amino acids or intermediate steps (16, 33, 42, 67). Thus, the associations with no validation support could have been novel epitopes, compensatory mutations, or false positives. For this reason, we did not exclude any of our significant associa-tions based on lack of validation support.
Because we did not know the exact sequence of the trans-mitted strain from each of the infected individuals in our study, we relied on phylogenetic analyses to infer the evolutionary history of the viral populations. This inference may have in-troduced error into the analysis that was likely to bias the results toward the null hypothesis, producing false-negative associations. The length of the terminal branch (the average in our study was 0.047 mutations/site/generation) reflects the amount of evolution that has taken place since the most recent ancestor and may also reflect the amount of time in the trans-mission chain from the inferred ancestor to the terminal se-quence. This time was estimated to be an average of 10 years, assuming a molecular clock, a rate of evolution of 2.5⫻10⫺5
mutations/site/generation (44), and a generation rate of 2 days (51). This estimate is likely inflated due to recombination. Undetected transmission events may have happened during this time, leading to the potential increase in detection of false-positive results using MLF and parsimony. However, the branch lengths were taken into account in the MLL method, and this method was not found to be better or worse than MLF or parsimony, suggesting that the time elapsed from the most recent ancestor of each sequence did not adversely impact our ability to identify evolving sites within known epitopes.
In summary, we have been able to identify HIV-1 amino acids that evolve in response to HLA-mediated cellular immu-nity among a South African population primarily infected with subtype C, including seven susceptible residues, escape from which was associated with lower viral loads. Moreover, we have provided a ranking of proteins based on the fitness cost of immune escape and, based on this ranking, recommend the inclusion of Gag, Vpr, and Rev as vaccine components. Our analysis also showed the importance of HLA-B and HLA-C in driving HIV-1 evolution. The information from this study can be used to design follow-up analyses to characterize CTL epitopes and viral fitness for consideration in a CTL-based vaccine. Although this study does not prove that an effective CTL-based vaccine is achievable, it provides encouragement for future research in this area.
on November 8, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
We acknowledge Zabrina Brumme and Richard Harrigan for gen-erously contributing data from the British Columbian HOMER cohort and Yi Liu and David Lockhart for helpful discussions and assistance. We also thank all of the study subjects for their participation.
This work was funded by NIH contract N01-AI-15422 (HLA typing and CTL epitope mapping to guide HIV vaccine development); U.S. Public Health Service awards AI11514, AI27005, AI058894, AI061734, and AI067077; and the University of Washington Center For AIDS Research (AI27757), including a New Investigator Award (AI047734) to C.M.R. B.T.K., T.B., and M.G.D. were funded through a Los Alamos National Laboratory directed-research grant and NIH AI061734. J.M.C., C.K., and D.E.H. were funded by Microsoft Re-search.
REFERENCES
1.Adnan, S., A. Balamurugan, A. Trocha, M. S. Bennett, H. L. Ng, A. Ali, C. Brander, and O. O. Yang.2006. Nef interference with HIV-1-specific CTL
antiviral activity is epitope specific. Blood108:3414–3419.
2.Allen, T. M., M. Altfeld, X. G. Yu, K. M. O’Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker.2004. Selection, trans-mission, and reversion of an antigen-processing cytotoxic T-lymphocyte es-cape mutation in human immunodeficiency virus type 1 infection. J. Virol.
78:7069–7078.
3.Altfeld, M., M. M. Addo, R. Shankarappa, P. K. Lee, T. M. Allen, X. G. Yu, A. Rathod, J. Harlow, K. O’Sullivan, M. N. Johnston, P. J. Goulder, J. I. Mullins, E. S. Rosenberg, C. Brander, B. Korber, and B. D. Walker.2003. Enhanced detection of human immunodeficiency virus type 1-specific T-cell responses to highly variable regions by using peptides based on autologous
virus sequences. J. Virol.77:7330–7340.
4.Altfeld, M., T. M. Allen, E. T. Kalife, N. Frahm, M. M. Addo, B. R. Mothe, A. Rathod, L. L. Reyor, J. Harlow, X. G. Yu, B. Perkins, L. K. Robinson, J. Sidney, G. Alter, M. Lichterfeld, A. Sette, E. S. Rosenberg, P. J. Goulder, C. Brander, and B. D. Walker. 2005. The majority of currently circulating human immunodeficiency virus type 1 clade B viruses fail to prime cytotoxic T-lymphocyte responses against an otherwise immunodominant
HLA-A2-restricted epitope: implications for vaccine design. J. Virol.79:5000–5005.
5.Barouch, D. H., J. Kunstman, J. Glowczwskie, K. J. Kunstman, M. A. Egan, F. W. Peyerl, S. Santra, M. J. Kuroda, J. E. Schmitz, K. Beaudry, G. R. Krivulka, M. A. Lifton, D. A. Gorgone, S. M. Wolinsky, and N. L. Letvin.
2003. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J.
Vi-rol.77:7367–7375.
6.Barouch, D. H., J. Kunstman, M. J. Kuroda, J. E. Schmitz, S. Santra, F. W. Peyerl, G. R. Krivulka, K. Beaudry, M. A. Lifton, D. A. Gorgone, D. C. Montefiori, M. G. Lewis, S. M. Wolinsky, and N. L. Letvin.2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T
lymphocytes. Nature415:335–339.
7.Barouch, D. H., J. Powers, D. M. Truitt, M. G. Kishko, J. C. Arthur, F. W. Peyerl, M. J. Kuroda, D. A. Gorgone, M. A. Lifton, C. I. Lord, V. M. Hirsch, D. C. Montefiori, A. Carville, K. G. Mansfield, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin.2005. Dynamic immune responses maintain cytotoxic T lymphocyte epitope mutations in transmitted simian
immunode-ficiency virus variants. Nat. Immunol.6:247–252.
8.Bhattacharya, T., M. Daniels, D. Heckerman, B. Foley, N. Frahm, C. Kadie, J. Carlson, K. Yusim, B. McMahon, B. Gaschen, S. Mallal, J. I. Mullins, D. C. Nickle, J. Herbeck, C. Rousseau, G. H. Learn, T. Miura, C. Brander, B. Walker, and B. Korber.2007. Founder effects in the assessment of HIV
polymorphisms and HLA allele associations. Science315:1583–1586.
9.Borghans, J. A., A. Molgaard, R. J. de Boer, and C. Kesmir.2007. HLA alleles associated with slow progression to AIDS truly prefer to present
HIV-1 p24. PLoS ONE2:e920.
10.Borrow, P., H. Lewicki, X. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A. Nelson, J. E. Gairin, B. H. Hahn, M. B. Oldstone, and G. M. Shaw.1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape
virus. Nat.Med.3:205–211.
11.Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. Desouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen.2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin
A. J. Virol.81:12608–12618.
12.Brumme, Z. L., C. J. Brumme, D. Heckerman, B. T. Korber, M. Daniels, J. Carlson, C. Kadie, T. Bhattacharya, C. Chui, J. Szinger, T. Mo, R. S. Hogg, J. S. Montaner, N. Frahm, C. Brander, B. D. Walker, and P. R. Harrigan.2007. Evidence of differential HLA class I-mediated viral
evolu-tion in funcevolu-tional and accessory/regulatory genes of HIV-1. PLoS Pathog.
3:e94.
13.Buseyne, F., D. Scott-Algara, F. Porrot, B. Corre, N. Bellal, M. Burgard, C. Rouzioux, S. Blanche, and Y. Riviere.2002. Frequencies of ex vivo-activated human immunodeficiency virus type 1-specific gamma-interferon-producing
CD8⫹T cells in infected children correlate positively with plasma viral load.
J. Virol.76:12414–12422.
14.Carlson, J., C. Kadie, S. Mallal, and D. Heckerman. 2007. Leveraging hierarchical population structure in discrete association studies. PLoS ONE
2:e591.
15.Cohen, G. B., R. T. Gandhi, D. M. Davis, O. Mandelboim, B. K. Chen, J. L. Strominger, and D. Baltimore.1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected
cells from NK cells. Immunity10:661–671.
16.Crawford, H., J. G. Prado, A. Leslie, S. Hue, I. Honeyborne, S. Reddy, M. van der Stok, Z. Mncube, C. Brander, C. Rousseau, J. I. Mullins, R. Kaslow, P. Goepfert, S. Allen, E. Hunter, J. Mulenga, P. Kiepiela, B. D. Walker, and P. J. Goulder.2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant
Hla-B*5703-restricted Gag epitope in chronic Hiv-1 infection. J. Virol.81:8346–
8351.
17.Edwards, B. H., A. Bansal, S. Sabbaj, J. Bakari, M. J. Mulligan, and P. A. Goepfert.2002. Magnitude of functional CD8⫹T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with
viral load in plasma. J. Virol.76:2298–2305.
18.Fang, G., H. Burger, R. Grimson, P. Tropper, S. Nachman, D. Mayers, O. Weislow, R. Moore, C. Reyelt, N. Hutcheon, D. Baker, and B. Weiser.1995. Maternal plasma human immunodeficiency virus type 1 RNA level: a deter-minant and projected threshold for mother to child transmission. Proc. Natl.
Acad. Sci. USA92:12100–12104.
19.Fellay, J., K. V. Shianna, D. Ge, S. Colombo, B. Ledergerber, M. Weale, K. Zhang, C. Gumbs, A. Castagna, A. Cossarizza, A. Cozzi-Lepri, A. De Luca, P. Easterbrook, P. Francioli, S. Mallal, J. Martinez-Picado, J. M. Miro, N. Obel, J. P. Smith, J. Wyniger, P. Descombes, S. E. Antonarakis, N. L. Letvin, A. J. McMichael, B. F. Haynes, A. Telenti, and D. B. Goldstein.2007. A whole-genome association study of major determinants for host control of
HIV-1. Science317:944–947.
20.Fernandez, C. S., I. Stratov, R. De Rose, K. Walsh, C. J. Dale, M. Z. Smith, M. B. Agy, S. L. Hu, K. Krebs, D. I. Watkins, H. O’Connor, D., M. P. Davenport, and S. J. Kent.2005. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope
ex-acts a dramatic fitness cost. J. Virol.79:5721–5731.
21.Frahm, N., S. Adams, P. Kiepiela, C. H. Linde, H. S. Hewitt, M. Lichterfeld, K. Sango, N. V. Brown, E. Pae, A. G. Wurcel, M. Altfeld, M. E. Feeney, T. M. Allen, T. Roach, M. A. St John, E. S. Daar, E. Rosenberg, B. Korber, F. Marincola, B. D. Walker, P. J. Goulder, and C. Brander.2005. HLA-B63 presents HLA-B57/B58-restricted cytotoxic T-lymphocyte epitopes and is
associated with low human immunodeficiency virus load. J. Virol.79:10218–
10225.
22.Frahm, N., P. J. R. Goulder, and C. Brander.2004. Broad HIV-1 specific CTL responses reveal extensive HLA class I binding promiscuity of
HIV-derived, optimally defined CTL epitopes, p. 1332, xiii.InB. T. M. Korber, C.
Brander, B. F. Haynes, R. Koup, J. P. Moore, B. D. Walker, and D. I. Watkins (ed.), HIV immunology and HIV/SIV vaccine databases 2003. Los Alamos National Laboratory, Los Alamos, NM.
23.Frahm, N., D. Kaufmann, K. Yusim, M. Muldoon, C. Kesmir, C. Linde, W. Fischer, T. Allen, B. Li, B. McMahon, K. Faircloth, H. Hewitt, E. Mackey, T. Miura, A. Khatri, S. Wolinsky, A. McMichael, R. Funkhouser, B. Walker, B. Korber, and C. Brander.2007. Increased sequence diversity coverage
im-proves detection of HIV-specific T Cell responses. J. Immunol.179:6638–
6650.
24.Frahm, N., P. Kiepiela, S. Adams, C. H. Linde, H. S. Hewitt, K. Sango, M. E. Feeney, M. M. Addo, M. Lichterfeld, M. P. Lahaie, E. Pae, A. G. Wurcel, T. Roach, M. A. St John, M. Altfeld, F. M. Marincola, C. Moore, S. Mallal, M. Carrington, D. Heckerman, T. M. Allen, J. I. Mullins, B. T. Korber, P. J. Goulder, B. D. Walker, and C. Brander.2006. Control of human immuno-deficiency virus replication by cytotoxic T lymphocytes targeting
subdomi-nant epitopes. Nat. Immunol.7:173–178.
25.Frahm, N., B. T. Korber, C. M. Adams, J. J. Szinger, R. Draenert, M. M. Addo, M. E. Feeney, K. Yusim, K. Sango, N. V. Brown, D. SenGupta, A. Piechocka-Trocha, T. Simonis, F. M. Marincola, A. G. Wurcel, D. R. Stone, C. J. Russell, P. Adolf, D. Cohen, T. Roach, A. St John, A. Khatri, K. Davis, J. Mullins, P. J. Goulder, B. D. Walker, and C. Brander.2004. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human
im-munodeficiency virus across multiple ethnicities. J. Virol.78:2187–2200.
26.Frahm, N., D. Nickle, C. H. Linde, R. Zun˜iga, A. Lucchetti, T. Roach, P. J. R. Goulder, B. D. Walker, T. M. Allen, B. T. Korber, J. I. Mullins, and C. Brander. 2008. Increased detection of HIV-specific T cell responses by combination of centralized sequences with comparable in vitro
immunoge-nicity. AIDS22:447–456.
27.Friedrich, T. C., E. J. Dodds, L. J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D. T. Evans, R. C. Desrosiers, B. R. Mothe, J. Sidney, A. Sette, K. Kunstman,
6444 ROUSSEAU ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
S. Wolinsky, M. Piatak, J. Lifson, A. L. Hughes, N. Wilson, D. H. O’Connor, and D. I. Watkins.2004. Reversion of CTL escape-variant immunodeficiency
viruses in vivo. Nat. Med.10:275–281.
28.Goulder, P. J., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. Korber, and B. D. Walker.2001. Evolution and transmission of stable CTL escape
mu-tations in HIV infection. Nature412:334–338.
29.Goulder, P. J., C. Pasquier, E. C. Holmes, B. Liang, Y. Tang, J. Izopet, K. Saune, E. S. Rosenberg, S. K. Burchett, K. McIntosh, M. Barnardo, M. Bunce, B. D. Walker, C. Brander, and R. E. Phillips.2001. Mother-to-child transmission of HIV infection and CTL escape through
HLA-A2-SLYNTVATL epitope sequence variation. Immunol. Lett.79:109–116.
30.Goulder, P. J., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones.1997. Late escape from an immunodominant cyto-toxic T-lymphocyte response associated with progression to AIDS. Nat. Med.
3:212–217.
31.Honeyborne, I., A. Prendergast, F. Pereyra, A. Leslie, H. Crawford, R. Payne, S. Reddy, K. Bishop, E. Moodley, K. Nair, M. van der Stok, N. McCarthy, C. M. Rousseau, M. Addo, J. I. Mullins, C. Brander, P. Kiepiela, B. D. Walker, and P. J. Goulder.2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific
CD8⫹T-cell epitopes. J. Virol.81:3667–3672.
32.Iversen, A. K. N., G. Stewart-Jones, G. H. Learn, N. Christie, C. Sylvester-Hviid, A. E. Armitage, R. Kaul, T. Beattie, J. K. Lee, Y. Li, P. Chotiyarnwong, T. Dong, X. Xu, M. A. Luscher, K. H. MacDonald, H. Ullum, B. Klarlund-Pedersen, P. Skinhøj, L. Fugger, S. Buus, J. I. Mullins, E. Y. Jones, A. van der Merwe, and A. J. McMichael.2006. Conflicting selective forces affect
CD8⫹T-cell receptor contact sites in an HLA-A2 immunodominant HIV
epitope. Nat. Immunol.7:179–189.
33.Kelleher, A. D., C. Long, E. C. Holmes, R. L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, C. Brander, G. Ogg, J. S. Sullivan, W. Dyer, I. Jones, A. J. McMichael, S. Rowland-Jones, and R. E. Phillips.2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J.
Exp. Med.193:375–386.
34.Kesmir, C., A. K. Nussbaum, H. Schild, V. Detours, and S. Brunak.2002. Prediction of proteasome cleavage motifs by neural networks. Protein Eng.
15:287–296.
35.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder.2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and
HLA. Nature432:769–775.
36.Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker, and P. Goulder.2007. CD8⫹T-cell responses to different HIV proteins have
discordant associations with viral load. Nat. Med.13:46–53.
37.Koenig, S., A. J. Conley, Y. A. Brewah, G. M. Jones, S. Leath, L. J. Boots, V. Davey, G. Pantaleo, J. F. Demarest, C. Carter, C. Wannebo, J. R. Yannelli, S. A. Rosenberg, and C. H. Lane.1995. Transfer of HIV-1-specific cytotoxic T-lymphocytes to an AIDS patient leads to selection for mutant HIV
vari-ants and subsequent disease progression. Nat. Med.1:330–336.
38.Korber, B. T., K. J. Kunstman, B. K. Patterson, M. Furtado, M. M. McEvilly, R. Levy, and S. M. Wolinsky.1994. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3
region of the envelope protein of brain-derived sequences. J. Virol.68:7467–
7481.
39.Leslie, A., D. Kavanagh, I. Honeyborne, K. Pfafferott, C. Edwards, T. Pillay, L. Hilton, C. Thobakgale, D. Ramduth, R. Draenert, S. Le