J
OURNAL OFV
IROLOGY, Apr. 2011, p. 3356–3366
Vol. 85, No. 7
0022-538X/11/$12.00
doi:10.1128/JVI.02105-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
The Herpes Simplex Virus Immediate-Early Ubiquitin Ligase ICP0
Induces Degradation of the ICP0 Repressor Protein E2FBP1
䌤
Yayoi Fukuyo,
1,2Nobuo Horikoshi,
3Alexander M. Ishov,
4Saul J. Silverstein,
5and Takuma Nakajima
2*
California Pacific Medical Center Research Institute, 475 Brannan Street, Suite 220, San Francisco, California 94107
1; Section of
Bacterial Pathogenesis, Graduate School, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8549,
Japan
2; Division of Molecular Radiation Biology, Department of Radiation Oncology, University of Texas Southwestern Medical Center,
323 Harry Hines Blvd., Dallas, Texas 75390-8807
3; University of Florida College of Medicine Cancer Center, P.O. Box 103633,
Gainesville, Florida 32610
4; and Department of Microbiology and Immunology, College of Physicians and
Surgeons, Columbia University, 701 W. 168th St., New York, New York 10032
5Received 5 October 2010/Accepted 7 January 2011
E2FBP1/hDRIL1, a DNA-binding A/T-rich interaction domain (ARID) family transcription factor, is
ex-pressed ubiquitously in human tissues and plays an essential role in maintaining the proliferation potential of
passage-limited human fibroblasts by dissociating promyelocytic leukemia nuclear bodies (PML-NBs). This
effect on PML-NBs is similar to that of viral immediate-early gene products, such as infected cellular protein
0 (ICP0) from human herpes simplex virus 1 (HSV-1), which also disrupts PML-NBs to override the intrinsic
cellular defense. Here we report that E2FBP1 inhibits accumulation of ICP0 RNA and, at the same time, is
degraded via ICP0’s herpes ubiquitin ligase 2 (HUL-2) activity upon HSV-1 infection. These reciprocal
regulatory roles of ICP0 and E2FBP1 are linked in an ARID-dependent fashion. Our results suggest that
E2FBP1 functions as an intrinsic cellular defense factor in spite of its PML-NB dissociation function.
E2FBP1 was cloned independently by several laboratories as
an enhancer for E2F1/DP1 complex-mediated transcriptional
activation (65) and was shown to be a human homologue of the
Drosophila
development-related transcription factor dead
ringer (DRI) (34). E2FBP1 is evolutionally conserved from
yeast to vertebrates (24) and is a member of the DNA-binding
A/T-rich interaction domain (ARID) family. ARID proteins
are implicated in transcriptional regulation, chromatin
remod-eling, cell cycle regulation, and developmental control,
includ-ing cell fate determination (72). Among ARID family
mem-bers, orthologues of E2FBP1 (i.e., ARID3a) involved in
development are found in mice, fruit flies, zebra fish, and
nematodes (73). BRIGHT, a rodent orthologue, is a B-cell
regulator of immunoglobulin heavy chain transcription (28)
whose expression is restricted to B-cell lineages, and it binds
A/T-rich sequences within matrix-associating regions (MARs)
flanking the intronic enhancer (28). BRIGHT also contains
both a nuclear localization signal (NLS) and a nuclear export
signal (NES), with nucleocytoplasmic shuttling controlled by
chromosome region maintenance 1 (CRM1) (33), and is
known to enhance transcriptional activation in the presence of
Bruton’s tyrosine kinase (Btk) (58, 70) and to modulate
chro-matin accessibility (39). In addition to Btk, interactions with
promyelocytic leukemia nuclear body (PML-NB) components
Sp100 and LYSP100B modulate BRIGHT’s transcriptional
ac-tivity (7). A REKLES motif flanking the C terminus of the
ARID is required both for homo- and heterodimer formation
and for interaction with its specific DNA target (32). Human
E2FBP1 shares some features with murine BRIGHT,
includ-ing its target DNA sequences, interaction partners, and
sub-cellular localization; however, it is expressed ubiquitously in a
broader range of tissues (34). This difference in distribution
suggests unique roles for E2FBP1 in cellular controls. In fact,
E2FBP1 contributes to cellular regulatory mechanisms,
includ-ing cell cycle start (65), rescue from oncogenic Ras
V12-induced
premature senescence (56), dissociation of PML-NBs (23),
transforming growth factor beta (TGF-

)-induced fibroblast
growth in pulmonary fibrosis (40), and p53-mediated cell cycle
arrest following DNA damage (47). Silencing of E2FBP1
ex-pression leads to NB accumulation, resulting in
PML-mediated premature senescence (23). Recently, sumoylation of
K398 in the ARID of E2FBP1 was shown to modulate its
transcriptional activity (57).
PML-NBs, alternatively described as nuclear domain 10
(ND10), typically appear in interphase nuclei as punctate
do-mains in close proximity to MARs. PML-NBs are composed of
diverse proteins, including PML, Sp100, Daxx, Rb, p53, histone
deacetylases, polymerases, and helicases, all of which
dynam-ically change their numbers and composition during the cell
cycle (2, 14). Loss of PML-NB formation as a consequence of
genomic translocation t(15;17) results in leukemogenesis
through interference with promyelocytic differentiation (8),
and thus PML-NBs are implicated in maintenance of cellular
integrity (reviewed in reference 37), whereas increases in the
size and number of PML-NBs strongly suppress cell cycle
progression and subsequently induce premature senescence
(31, 55).
PML-NBs also play a major role against viral infection. Two
major components of PML-NBs, PML and Sp100, are induced
by type I and II interferons, and abrogation of PML-NBs
results in increased viral titers. Moreover, diverse viruses target
PML-NBs at very early stages of infection, and their
compo-nents are sorted to form similar structures in the vicinity of the
* Corresponding author. Mailing address: Section of Bacterial
Pathogenesis, Graduate School, Tokyo Medical and Dental University,
1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8549, Japan. Phone:
81-35803-5456. Fax: 81-45982-7141. E-mail: tnakajima@spn1.speednet.ne.jp.
䌤
Published ahead of print on 19 January 2011.
3356
on November 7, 2019 by guest
http://jvi.asm.org/
sites of viral replication (reviewed in references 12, 61, and 66).
Among the many viral proteins targeting PML-NBs, the best
studied is infected cellular protein 0 (ICP0) of herpes simplex
virus 1 (HSV-1). ICP0 is an immediate-early (IE) protein of
HSV-1 that exhibits multiple functions, including
transcrip-tional activation and herpes ubiquitin ligase (HUL) activity
(reviewed in references 10 and 25). These functions are
regu-lated by posttranslational modifications, interactions with
cel-lular and viral proteins, and its subcelcel-lular localizations and are
probably required for both efficient lytic infection and
reacti-vation from latency (26, 27, 60, 67, 75; reviewed in references
10 and 25). In infected cells, ICP0 is initially nuclear and
subsequently translocates to the cytoplasm after the onset of
viral DNA replication (45, 68). ICP0 in the nucleus targets
PML-NBs through its RING-dependent HUL-2 activity and
ubiquitylates both PML and a sumoylated form of Sp100 to
disintegrate PML-NBs (3, 13, 50, 54). This function of ICP0 is
important for virus replication, as ICP0-null mutant viruses,
which are defective in destruction of PML-NBs, have reduced
viral yields at a low multiplicity of infection (MOI) (reviewed
in references 11, 48, 59, 61, 64, and 66). Moreover, a reduction
of either PML or Sp100 expression in human primary foreskin
fibroblasts did not affect wild-type (WT) HSV-1 replication but
increased gene expression and plaque-forming efficiency of
ICP0-null mutants (20). Simultaneous depletion of both PML
and Sp100 resulted in a significant increase in ICP0-null
mu-tant expression (18). Curiously, while high-level expression of
transduced PML resulted in increased formation of
PML-NB-like nuclear domains in Vero cells, Hep-2 cells, and
telomer-ase-transformed human foreskin fibroblasts, it did not affect
replication of WT HSV-1 (22, 44). These apparently conflicting
results suggested that components of PML-NBs contribute to
the intrinsic viral response. Recently, some isoforms of Sp100
were revealed to protect PML from degradation and to
sup-press transcription of IE genes of HSV-1, including
ICP0
,
although details of the mechanisms remain elusive (51, 52).
In this paper, we show that E2FBP1 undergoes
ICP0-in-duced ubiquitylation, that the RING/zinc finger element of
ICP0 is required for this activity, and that E2FBP1 suppresses
accumulation of ICP0 RNA. These reciprocal regulatory roles
of ICP0 and E2FBP1 are linked in an ARID-dependent
fash-ion, suggesting a role for the ARID in productive HSV-1
replication.
MATERIALS AND METHODS
Cells and viruses.hTERT-BJ1 cells (Clontech) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 18% 199 medium and 10% fetal bovine serum (FBS). Human fetal lung fibroblast TIG-3 cells were used at the indicated population doublings (PD). Hep-2 and Vero cells were maintained in DMEM containing 10% FBS. Hep-2-derived cells constitutively expressing hemagglutinin (HA)-tagged E2FBP1 or its ARID deletion mutant (⌬A) were established by introducing either pEF2HA-E2FBP1WT-IRESP or pEF2HA-E2FBP1⌬A-IRESP DNA (see the following section) cleaved at a unique ScaI site into plasmids at theblagene and were selected and maintained in the presence of 3g/ml of puromycin. HEK293FT cells (Invitrogen) were cultured in DMEM supplemented with 10% FBS, nonessential amino acid so-lution (Gibco), and 1 mM sodium pyruvate. HSV-1 wild-type strain F was propagated and titrated in Vero cells. Recombinant lentiviruses were produced using ViraPower packaging mix (Invitrogen) according to the manufacturer’s manual.
Plasmids, synthetic oligonucleotides, and antibodies.The coding sequences for two-tandem-repeat hemagglutinin (2HA)-tagged E2FBP1 (HA-E2FBP1)
and its mutants, derived from pcDNA3 HA-E2FBP1 (23), were inserted between the XhoI and XbaI sites located downstream of the human elongation factor 1␣ promoter in pEF-IRESP (29). The sequences of HA-E2FBP1 and its mutants used a bovine growth hormone polyadenylation signal inserted downstream of the mouse mammary tumor virus (MMTV) 3⬘ long terminal repeat (LTR) promoter of pMTV-dhfr (38), and then they were substituted for the U6 short hairpin RNA (shRNA) expression unit of pLenti6-GW/U6-laminshRNA
(Invitro-gen) to generate a series of pLentiMMTV-2HA-E2FBP1 plasmids. For the pQE1-E2FBP1 construct, E2FBP1 was subcloned into the pQETriSystem 1 vector (Qiagen), which encodes a (His)8 tag. pDS16 (74), pCM2/7, and
pCM11/93 (4, 5) are ICP0 expression plasmids for the WT and for deletion mutants lacking the RING/zinc finger motif and the C-terminal multimerization domain of ICP0, respectively. To construct a series of pGLIE0p-hRluc reporter plasmids for monitoring HSV-1IE-0expression, the upstream sequence of the
IE-0structural gene was amplified by PCR and subcloned between the NheI and HindIII sites of the pGL4.83 humanizedRenilla reniformisluciferase (hRluc) expression plasmid, using an In-Fusion PCR cloning system. Expression plasmids employed for HA-tagged and (His)6-tagged ubiquitin (His-Ub) were
pEF-IRESp-HA-Ub (6) and pCMV-His-Ub. Synthetic double-stranded small interfering RNA (siRNA) molecules for E2FBP1 and a nonsense control were designed using software provided by RNAi Co. Ltd. (Japan) and were synthe-sized by Japan Bio-Service Corporation and Proligo LLC (Japan). The following primary and secondary antibodies were used: ICP0 Clu 7 (42), ICP0 5H7 (Ab-cam), DRIL1 CBL665 (Bethyl Laboratories), ICP4 10F1 (Ab(Ab-cam), PML PG-M3 (SantaCruz), HA 3F10 (Roche),␣-tubulin (CHI), Alexa Fluor 488-conjugated donkey rat and mouse IgG, Alexa Fluor 555-conjugated donkey anti-mouse and anti-rabbit IgG (Invitrogen), horseradish peroxidase (HRP)-conju-gated donkey anti-mouse, anti-rabbit, and anti-rat IgG, and alkaline phosphatase (AP)-conjugated donkey anti-mouse, anti-rabbit, and anti-rat IgG (Chemicon). Introduction of foreign DNAs and siRNAs.Efficient DNA transformation of TIG-3 cells was achieved only within 45 PD, using Xfect reagent (Clontech). Otherwise, transformation of TIG-3 cells and HEK293FT cells was performed with either FuGene6 reagent (Roche) or Lipofectamine LTX (Invitrogen) ac-cording to the suppliers’ instructions. hTERT-BJ1 cells were infected with len-tiviruses expressing HA-E2FBP1 under the control of the MMTV LTR pro-moter, and stably transduced cell clones were isolated in the presence of 2g/ml blasticidin S hydrochloride. siRNA-mediated suppression of E2FBP1 was per-formed as previously described (23). For this experiment, TIG-3 cells at 47 PD were transformed twice with siRNAs and allowed to reach confluence. The cells were then plated on glass coverslips and transformed with pDS16, using FuGene6.
Infection.HSV-1 infections were carried out for 30 min at room temperature, and the diluted HSV-1 stock was replaced with prewarmed (at 37°C) medium containing 10% FBS to terminate the step. The end of the infection step was taken as 0 min postinfection (mpi). Infected cells were incubated in a CO2
incubator at 37°C for the indicated times until harvest or fixation.
Immunofluorescence microscopy.Cells grown on coverslips were fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde and 1% FBS for 20 min at room temperature. The cells were subsequently permeabilized with 0.25% Triton X-100 for 10 min, washed with PBS three times, and then stained for 4 h with anti-DRIL1 for E2FBP1 at a dilution of 1:2,000 and with 5H7 for ICP0 at a dilution of 1:20,000 at room temperature. Cells treated with primary antibodies were washed with PBS and then stained for 2 h with a secondary antibody solution containing 250 ng/ml of DAPI (4⬘,6-diamidino-2-phenylindole) and a 1:2,000-diluted mix of anti-rabbit IgG and anti-mouse IgG conjugated with Alexa 488 and Alexa 555, respectively. After being washed with PBS, specimens were mounted on glass slides with ProLong Gold antifade reagent (Molecular Probes) and subjected to fluorescence microscopy using an FV1000 laser scan-ning confocal microscope system with Fluoview software, version 1.6 (Olympus, Japan).
Extract preparation, Ni-NTA pulldown, and immunoprecipitation.In most instances, cells were lysed in high-salt buffer (HSB; 300 mM NaCl, 50 mM HEPES-sodium, pH 7.0, 1 mM EDTA, 0.1% NP-40, 1 mM Na3VO4) containing
protease inhibitors [PI mix; 2M MG115, 2M MG132, 1 mMN␣-p-tosyl-L -lysine chloromethyl ketone (TLCK), 400M 4-amidinophenylmethanesulfonyl fluoride hydrochloride, and 400M 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride]. Cellular debris was removed by centrifugation at 15,000⫻g, and protein concentrations of cleared lysates were measured using a bicinchoninic acid protein assay kit (Sigma-Aldrich). For Ni-nitrilotriacetic acid (Ni-NTA) pulldown, cells were disrupted with 6 M guanidine hydrochloride solution con-taining 20 mM imidazole (GHI), or cells suspended in 150l of PBS were lysed with 900l of GHI. Lysates were sonicated and mixed with 40l of 50% Ni-NTA agarose beads (Qiagen) equilibrated with GHI to collect His-tagged proteins.
on November 7, 2019 by guest
http://jvi.asm.org/
Following 1 h of gentle rocking, beads were washed three times with 1 ml GHI, followed by two washes with PBS containing 0.25% Tween 20 (PBST). After removal of the buffer, Ni-NTA agarose beads were boiled with 50l of 2⫻ Laemmli sample buffer (120 mM Tris-HCl, pH 6.8, 250 mM dithiothreitol [DTT], 3% SDS, 20% glycerol, 0.02% bromphenol blue) (36), and 25-l aliquots were analyzed by SDS-PAGE. For immunoprecipitation, cells were treated with 10M (each) proteasome inhibitors MG115 and MG132 for 15 min and then lysed with medium-salt buffer (MSB; 250 mM NaCl, 50 mM HEPES-sodium, pH 7.0, 1 mM EDTA, 0.1% NP-40, 1 mM Na3VO4) containing PI mix without
TLCK, and 2 mg of each cell lysate was incubated with 0.5g of primary antibody and 5l of 50% protein A Sepharose-FF beads (Pharmacia-GE Healthcare) for 2 h. The beads were washed four times with MSB, and antigen was released by boiling with 15l of 2⫻Laemmli sample buffer for 5 min and then subjected to immunoblotting.
Immunoblotting.Proteins separated by SDS-PAGE were transferred to Im-mobilon P polyvinylidene difluoride membranes (Millipore), and membranes were probed with a primary antibody suspended in PBS containing 0.2% I-Block blocking reagent (Tropix) for 2 h at room temperature, washed with PBST, and incubated with an appropriate secondary antibody conjugated with HRP or AP for 1 h. The membranes were subsequently washed with PBST, exposed to enhanced chemiluminescence (ECL) reagent (GE Healthcare Bioscience) or CDP-Star chemiluminescence substrate (Millipore), and detected following ex-posure to X-ray film or with a Chemidoc chemiluminescence/fluorescence im-aging instrument with Quantity One software, version 4.6.2 (Bio-Rad).
RT-qPCR analysis of transiently transformed and infected cells.TIG-3 cells (1.8⫻106
cells/dish) at 43 to 45 PD seeded in a 100-mm dish were transformed with 30g/dish of pEF-IRESP, E2FBP1WT-IRESP, and pEF2HA-E2FBP1⌬A-IRESP DNAs, using Xfect (Clontech). On the third day after seed-ing, cells were infected with HSV-1 at the stated MOI, and subsequently, infected cells were collected at various times postinfection, washed with ice-cold PBS, and suspended in 400l of PBS; aliquots of the suspension (200l) were then subjected to either genomic DNA or total RNA extraction. A mixture of genomic and viral DNAs (genomic/viral DNA) was extracted with a NucleoSpin Blood kit (Macherey-Nagel) according to the manufacturer’s instructions. Yields of genomic DNA mixtures were measured with a Nanodrop 1000 spectrophotom-eter (Thermo Fisher) and ranged from 18 to 250g. Total RNA extraction was carried out with a High Pure RNA isolation kit (Roche Applied Science). RNA yields were measured with a Nanodrop 1000 spectrophotometer and ranged from 13.5 to 23g. The resulting RNAs were converted to cDNAs with a Transcriptor High Fidelity cDNA synthesis kit. Real-time quantitative PCR (RT-qPCR) was performed on a LightCycler 480 instrument (Roche Applied Science) with either 20 ng of genomic/viral DNA or 200 ng of cDNA, using a LightCycler 480 Probes master reagent kit (Roche Applied Science) equipped with specific hydrolysis probes. The PCR program consisted of the following steps: primary denaturation at 95°C for 5 min; 45 PCR cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 1 s; and termination at 50°C for 30 s. The sequences of primers and combined hydrolysis probes shown in Table 1 were designed with ProbeFinder online software, versions 2.44 and 2.45, provided by the Roche Applied Science Assay Design Center. The genes for RNase P RNA component H1 (RPPH1; GenBank accession number NR_002312) and 18S rRNA (GenBank accession number X03205) were employed as references for normalizing copy numbers of genomic/ viral DNA (43) and expressed transcripts (53), respectively, and data were processed both by LCS480 software, version 1.5.0.39, and manually by compar-ative threshold cycle (CT) numbers as described previously (43, 63). Copy
num-bers of HSV-1 DNA were calculated as a half of the ICP0 gene number detected in the genomic/viral DNA mixture. Copy numbers of transformed plasmids in the genomic/viral DNA were calculated based on the internal ribosome entry site/ Cap-independent translation enhancer (IRES) sequence from the encephalo-myocarditis virus polyprotein gene present in pEF-x-IRESP plasmids. ICP0 RNA levels from infected HSV-1 were first normalized to 18S rRNA levels and then to the number of HSV-1 genomes (53). Every experiment was done in triplicate and repeated at least twice.
Luciferase assay.To observe the activity of theIE-0gene promoter, TIG-3, hTERT-BJ1, and Hep-2 cells were plated in 24-well culture dishes and trans-formed with a DNA mixture containing pGLIE0p-hRluc, pcycD1Pr-luc(⫺30) (carrying thePhotinus pyralisluciferase [luc] gene linked with a 31-bp fragment of rat cyclin D1 upstream sequence [GenBank accession number AF148946] without any obvious regulatory motifs), and pEF2HA-E2FBP1-IRESP (expres-sion plasmid for wild type or ARID deletion mutant of E2FBP1). Eighteen hours after transformation, cells were washed with ice-cold PBS and lysed with 100l of passive lysis buffer. Dual-luciferase assays were then performed with a dual-luciferase reporter assay system (Promega) according to the manufacturer’s manual. All assays were done in triplicate and repeated.
RESULTS
E2FBP1 interacts and colocalizes with ICP0.
Immunopre-cipitation was used to ask if E2FBP1 and ICP0 interacted.
HEK293FT cells were cotransformed with an ICP0 expression
plasmid (pDS16) and an E2FBP1 expression plasmid
(HA-E2FBP1), and cell lysates were subjected to both
immunoblot-ting and immunoprecipitation (Fig. 1A). Proteins precipitated
with anti-HA antibody included ICP0. This result suggested a
possible interaction between ICP0 and E2FBP1. The
recipro-cal immunoprecipitation with anti-ICP0 antibody was not
suc-cessful, as E2FBP1 binds with protein A-Sepharose beads
un-der nondenaturing conditions. The amount of ICP0 detected
was less than 1% of the input from the whole-cell lysate
(WCL). These data suggest that the E2FBP1 and ICP0
inter-action is either weak or unstable.
[image:3.585.42.541.82.193.2]Colocalization of endogenous E2FBP1 and ICP0 was
stud-ied by confocal microscopy. TIG-3 cells transformed with
siRNA against E2FBP1 (siE2FBP1) or with a nonsense control
(siControl) were further transformed with the ICP0 expression
plasmid pDS16. In mock-treated and siControl-treated cells
(Fig. 1B, left and right panels, respectively), E2FBP1 was
spread ubiquitously in the nucleoplasm in the absence of ICP0,
as previously reported (23). In these cells, ICP0 was found
along with endogenous E2FBP1 in subnuclear foci that were
reminiscent of enlarged PML-NBs (yellow-green signals
ob-served in both left and right panels). Colocalization of ICP0
TABLE 1. Primers and combined hydrolysis probes designed for quantitative PCR
aTarget GenBank accession no. Primer direction Sequence Probe
E2FBP1
NM_005224
Forward
GCACTCCAGCAGAACTTCCT
UPL 44
Reverse
AGAGTCCTGGCGGCTTTC
GCTGCCCA
ICP0
X04614
Forward
AGCCCCGTCTCGAACAGT
UPL 56
Reverse
ACCACCATGACGACGACTC
TGCTGTCC
IRES
M81861
Forward
TGGCTCTCCTCAAGCGTATT
UPL 41
Reverse
CCCATACAATGGGGTACCTTC
GGCTGAAG
18S rRNA
X03205
Forward
CGATTGGATGGTTTAGTGAGG
UPL 81
Reverse
AGTTCGACCGTCTTCTCAGC
GGCCCTGG
RPPH1
NR_002312
Forward
CCGGAGCTTGGAACAGACT
UPL 30
Reverse
GTAGTCTGAATTGGGTTATGAGGTC
GGCTGAGG
a
UPL, universal probe library probe provided by Roche Applied Science; IRES, cap-independent translation enhancer sequence from encephalomyocarditis virus polyprotein gene (nucleotides 335 to 834) subcloned into pEF-x-IRESP; RPPH1, RNase P RNA component H1.
3358
FUKUYO ET AL.
J. V
IROL.
on November 7, 2019 by guest
http://jvi.asm.org/
and endogenous E2FBP1 in nuclear foci was observed in 100%
of transformed cells (
n
⫽
40). In contrast, when cells were
pretreated with siE2FBP1, ICP0 was localized diffusely
throughout the nucleoplasm (Fig. 1B, middle panels). A diffuse
nuclear distribution of ICP0 was also observed in 100% of cells
that expressed undetectable levels of E2FBP1 (
n
⫽
16). These
data reveal that interaction of E2FBP1 and ICP0
in vivo
affects
ICP0’s nuclear distribution.
ICP0 accelerates polyubiquitylation of E2FBP1.
We next
asked if an interaction between E2FBP1 and ICP0 could be
detected in HSV-1-infected cells. Reciprocal
immunoprecipi-tations with anti-HA or anti-ICP0 were not successful (data
not shown). It is possible that other HSV immediate-early
proteins further weaken this interaction, making it difficult to
detect by immunoprecipitation. Because ICP0 is a biheaded
ubiquitin ligase (E3), we tested whether E2FBP1 was
poly-ubiquitylated during the early phase of HSV-1 infection.
HEK293FT cells were transformed with HA-E2FBP1 and
His-Ub expression plasmids and subsequently infected with
HSV-1 at an MOI of 10. Cell lysates were prepared at various
times postinfection and subjected to immunoblotting and
Ni-agarose (Ni-NTA) pulldown to check expression levels or to
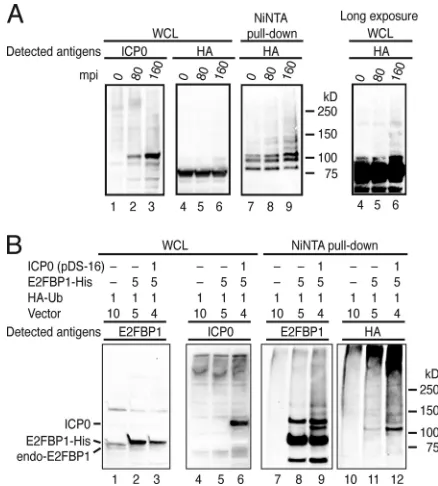
collect His-tagged ubiquitylated proteins (Fig. 2A). E2FBP1
was polyubiquitylated in uninfected cells, and the
polyubiqui-tylation level gradually increased after infection (lanes 7 to 9).
ICP0 abundance increased after 80 mpi (lanes 2 and 3),
sug-gesting to us that it was a potential E3 enzyme for E2FBP1.
[image:4.585.54.272.66.376.2]This supposition was verified by immunoblotting and
Ni-NTA pulldown of cell lysates prepared from HEK293FT cells
transformed with expression plasmids for ICP0, E2FBP1-His,
and HA-Ub. These experiments confirmed that E2FBP1 is
polyubiquitylated in the absence of ICP0 (Fig. 2B, lanes 8 and
11). However, expression of ICP0 induced greater levels of
polyubiquitylated E2FBP1, seen as slower-migrating species of
E2FBP1-His (Fig. 2B, lanes 9 and 12). Thus, E2FBP1 was
further ubiquitylated in the presence of ICP0 during the
im-mediate-early phase of HSV-1 infection. Interaction of ICP0
[image:4.585.309.528.69.312.2]FIG. 1. Interaction and colocalization of E2FBP1 and ICP0 in
HEK293FT and TIG-3 cells. (A) Interaction between E2FBP1 and
ICP0 was detected by immunoprecipitation of cell lysates. HEK293FT
cells were transformed with pDS16 (ICP0), pcDNA3-2HA-E2FBP1
(HA-E2FBP1), and pcDNA3 (empty vector) and cultivated for 18 h.
Cell were treated with 10
M (each) proteasome inhibitors MG115
and MG132 for 15 min, and lysates were prepared and subjected to
immunoblotting and immunoprecipitation. The numbers on top of the
lanes indicate the relative amounts of the plasmids. Lanes 1 to 3
received 20
g of cell lysate, whereas lanes 4 to 6 contained
coprecipi-tated materials obtained from 2 mg of cell lysates incubated with either
anti-ICP0 (5H7) or anti-HA (3F10) antibody. WCL, whole-cell lysate;
IB, immunoblotting; IP, immunoprecipitation. (B) Endogenous
E2FBP1 and ICP0 expressed from transformed plasmid DNA
colocal-ized in PML-NB-like nuclear subdomains. TIG-3 cells treated with or
without E2FBP1 siRNA (siE2FBP1) or nonsense control siRNA
(siControl) were subsequently transformed with pDS16. After 2 days,
cells were stained with anti-ICP0 (5H7) (green), anti-DRIL1 (red),
and DAPI (gray), and images were captured by confocal microscopy.
FIG. 2. ICP0 induces polyubiquitylation of E2FBP1. (A) The level
of polyubiquitylated E2FBP1 increased during the immediate-early
phase of HSV-1 infection. HEK293FT cells expressing HA-E2FBP1
and His-Ub were infected with HSV-1 at an MOI of 10. Cell lysates
were prepared at the indicated times postinfection. The left and
mid-dle panels show expression levels of ICP0 (lanes 1 to 3) and
HA-E2FBP1 (lanes 4 to 6). The right panel shows polyubiquitylated forms
of HA-E2FBP1 (lanes 7 to 9) collected from cell lysates. (B)
Poly-ubiquitylation of E2FBP1 was enhanced by expression of ICP0. The
relative ratios of plasmids transformed into HEK293FT cells are
shown at the top of the figure. Cell lysates were prepared after 20 h of
transformation and subjected to immunoblotting (lanes 1 to 6) or
Ni-NTA pulldown (lanes 7 to 12). The positions of ICP0 and
nonu-biquitylated endogenous or His-tagged E2FBP1 are indicated on the
left. mpi, minutes postinfection.
on November 7, 2019 by guest
http://jvi.asm.org/
and E2FBP1 is most probably a transient enzyme-substrate
interaction, and the polyubiquitylation process may lead to
degradation of the targeted substrate, which could explain the
inefficient recovery of E2FBP1-ICP0 complexes from infected
cell lysates.
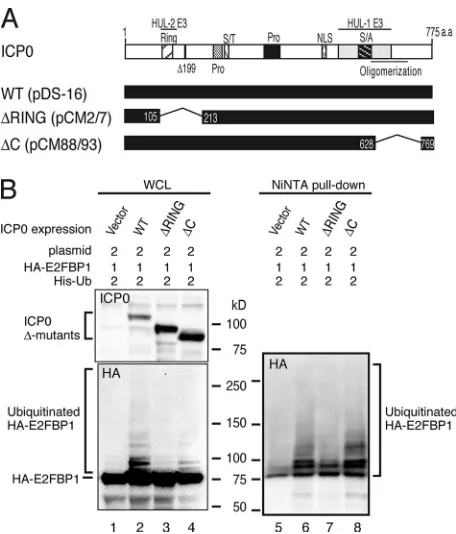
ICP0 mediates polyubiquitylation of E2FBP1 through its
RING/HUL-2 domain.
To examine whether ICP0 E3 activity
mediates polyubiquitylation of E2FBP1, expression plasmids
for ICP0 mutants lacking either the entire RING/HUL-2
do-main (
⌬
RING) or the C-terminal half of the HUL-1 domain
(
⌬
C) (Fig. 3A) were transformed with HA-E2FBP1 and
His-Ub expression plasmids. Immunoblotting and Ni-NTA
pulldown assays revealed that highly polyubiquitylated, slowly
migrating E2FBP1 accumulated in the presence of WT and
⌬
C
ICP0 proteins (Fig. 3B, lanes 2, 4, 6, and 8). In contrast,
⌬
RING ICP0 did not enhance levels of highly
polyubiquity-lated E2FBP1 (lanes 3 and 7). These data revealed that ICP0
polyubiquitylated E2FBP1 through its RING/HUL-2 domain.
The ARID of E2FBP1 is targeted for polyubiquitylation by
ICP0.
We next asked which region of E2FBP1 was targeted for
polyubiquitylation by ICP0. Extracts from HEK293FT cells
transformed with the plasmid combinations shown in Fig. 4B
were subjected to immunoblotting and Ni-NTA pulldown
as-says. All E2FBP1 deletion mutants were polyubiquitylated,
[image:5.585.48.276.67.334.2]regardless of whether ICP0 was present (Fig. 4B, lanes 3, 5, 7,
9, 11, 13, 15, and 17). Therefore, the endogenous ubiquitin
ligase(s) targets multiple residues in E2FBP1, spanning the
entire molecule. Ectopic expression of ICP0 resulted in greater
levels of polyubiquitylated E2FBP1 and its mutants, except for
the
⌬
A mutant (Fig. 4B, lanes 4, 6, 8, 12, 14, 16, and 18).
Therefore, most target sites (i.e., lysine residues) for
ICP0-mediated ubiquitylation reside in the ARID (compare lanes 9
and 10). Among these residues, Lys398 and Lys399, present in
the Ile-Lys-Lys-Glu (IKKE) motif, are known targets for
su-moylation (57). Importantly, ICP0 significantly increased
polyubiquitylation of the
⌬
AH protein (Fig. 4B, compare lanes
15 and 16), and thus a Lys residue(s) residing outside the
ARID and the helix-loop-helix (HLH) domain might be
poly-ubiquitylated by ICP0. Because the Arg-Glu-Lys-Leu-Glu-Ser
FIG. 3. ICP0 mediates polyubiquitylation of E2FBP1 through its
RING/HUL-2 domain. (A) Schematic representation of ICP0 and its
deletion mutants. (B) ICP0 lacking the RING/HUL-2 domain fails to
induce E2FBP1 polyubiquitylation. Expression plasmids were
trans-formed into HEK293FT cells at the ratios indicated at the top of the
figure. Cell lysates were prepared after 20 h of transformation and
subjected to immunoblotting (lanes 1 to 4) (top, ICP0; bottom,
HA-E2FBP1) and Ni-NTA pulldown followed by immunoblotting (lanes 5
to 8) (HA-E2FBP1). Positions for nonubiquitylated and ubiquitylated
HA-E2FBP1 and ICP0 mutant proteins are shown to the left of the
figure.
FIG. 4. The ARID of E2FBP1 is polyubiquitylated in response to
expression of ICP0. (A) Schematic representation of HA-E2FBP1 and
its deletion mutants. (B) Polyubiquitylation of all E2FBP1 mutants but
the
⌬
A mutant was induced by ICP0. Expression plasmids for the
E2FBP1 deletion mutants shown in panel A, for His-Ub, and for ICP0
were transformed into HEK293FT cells as indicated. Cell lysates were
prepared and subjected to immunoblotting (upper panel)
(HA-E2FBP1 and ICP0) and Ni-NTA pulldown followed by
immunoblot-ting (lower panel) (HA-E2FBP1). The levels of various E2FBP1
pro-teins and ICP0 are shown in the top panel. ICP0 and nonubiquitylated
and ubiquitylated HA-E2FBP1 proteins are indicated. The bottom
panel shows the levels of ubiquitylated E2FBP1 proteins.
3360
FUKUYO ET AL.
J. V
IROL.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.300.539.70.422.2](REKLES) motif residing in the HLH domain is required for
dimerization of ARID3 members (32), the discrepancy
be-tween the ICP0-mediated polyubiquitylation statuses of
⌬
A
and
⌬
AH proteins may be explained by an additional target
site(s) besides those in the ARID. The target Lys residues
residing outside the ARID might be concealed after
REKLES-mediated homodimerization of E2FBP1. The high level of
ICP0-dependent polyubiquitylation of the
⌬
H protein (Fig. 4B,
compare lanes 11 and 12) could result from polyubiquitylation
of Lys residues residing both inside and outside the ARID.
The decrease in E2FBP1 levels after infection with HSV-1
requires the ARID.
As shown in Fig. 5A, both endogenous and
exogenous E2FBP1 levels in Hep-2 cells were decreased within
130 mpi in response to infection with HSV-1. In contrast, the
abundance of
⌬
A HA-E2FBP1, lacking the ARID, was
unaf-fected by HSV-1 infection (Fig. 5A, bottom panels). These
effects were enhanced in hTERT-BJ1 cells at 120 mpi (Fig. 5B,
upper panels). These immunofluorescence analyses support
the biochemical data shown in Fig. 4B and suggest that
E2FBP1 is degraded by ICP0 through polyubiquitylation
within the ARID.
E2FBP1’s C terminus is required for interaction with ICP0.
Together with the results shown in Fig. 1B, the colocalization
of
⌬
A E2FBP1 with ICP0 (Fig. 5A and B) suggested that these
proteins interact in the cell nucleus. Accordingly, confocal
mi-FIG. 5. Accumulation of E2FBP1 or ICP0 in HSV-1-infected cells
is affected by the level of the other. (A) Hep-2 cells were transformed
with a control (vector) expression plasmid or with expression plasmids
for wild-type (WT) or
⌬
A HA-E2FBP1. The cells were then grown on
glass coverslips and infected with HSV-1 at an MOI of 10. After
infection, cells were further cultivated for the indicated times and then
fixed and stained with anti-E2FBP1 (DRIL1; red), anti-ICP0 (5H7;
green), and DAPI (gray). (B) hTERT-BJ1 cells were infected with
recombinant lentiviruses encoding either HA-E2FBP1 or its
⌬
A
mu-tant driven by an MMTV LTR promoter. The cells were then grown on
glass coverslips, treated with 2
M dexamethasone (Dex) to induce
expression of HA-E2FBP1 for 18 h, infected with HSV-1 at an MOI of
5 in the presence of Dex, and maintained in the presence of Dex until
fixation at 120 mpi. Cells were stained with anti-HA (red), anti-ICP0
(green) (left panels), and DAPI (gray). (C) Colocalization of E2FBP1
proteins and ICP0 in HSV-1-infected hTERT-BJ1-derived cells.
hTERT-BJ1 cells were infected with recombinant lentiviruses
encod-ing the indicated mutants of HA-E2FBP1 driven by an MMTV LTR
promoter. The cells were then infected with HSV-1 and treated as
described above. Cells were stained at 120 mpi with anti-HA (red),
anti-ICP0 (green), and DAPI (gray).
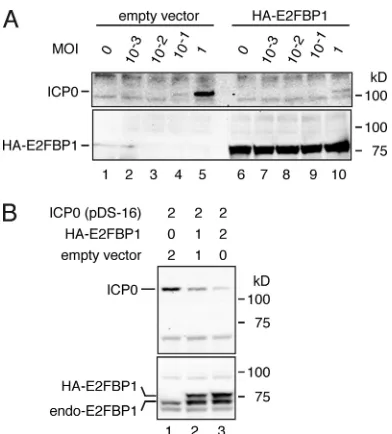
FIG. 6. Expression of endogenous E2FBP1 affects ICP0
accumu-lation, and
vice versa
, during HSV-1 infection. (A) Accumulation of
ICP0 protein during HSV-1 infection was repressed by ectopic
expres-sion of E2FBP1 in HEK293FT cells. HEK293FT cells were
trans-formed with either pcDNA3 (empty vector) or pcDNA3-2HA-E2FBP1
and infected with HSV-1 at the indicated MOIs. Cell lysates were
prepared at 120 mpi and subjected to immunoblotting with anti-ICP0
(upper panel) and anti-HA (lower panel). (B) Ectopic expression of
ICP0 is repressed by ectopic expression of E2FBP1. HEK293FT cells
were transformed with the indicated expression plasmids, and cell
lysates were prepared after 120 mpi and subjected to immunoblotting.
Expression of ICP0 is shown in the upper panel, and that of E2FBP1
is shown in the lower panel. Numbers at the top of the figure indicate
relative amounts of expression plasmids. endo-E2FBP1, endogenous
E2FBP1.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.323.519.66.283.2]3362
on November 7, 2019 by guest
http://jvi.asm.org/
croscopy was used to ask which domain of E2FBP1 was
re-quired for interaction with ICP0 (Fig. 5C). The proteins
en-coded by all deletion mutants of HA-E2FBP1, except for the
⌬
C2 mutant, showed a high degree of nuclear colocalization; in
contrast, colocalization of the
⌬
C2 protein with ICP0 was
rarely observed. These results suggested that E2FBP1’s C
ter-minus (i.e., amino acids 485 to 593) is likely required for
interaction with ICP0.
E2FBP1 represses ICP0 expression at the level of
transcrip-tion.
Because E2FBP1 was originally reported to be a
tran-scription factor (65), it was conceivable that decreased ICP0
resulted from E2FBP1-repressed transcription from the
IE-0
gene. High expression levels of
⌬
A E2FBP1 and ICP0 (Fig. 5A
and B) were probably a consequence of deletion of a
poly-ubiquitylation target region in E2FBP1 and the loss of its
function as a transcriptional repressor. HEK293FT cells
ectop-ically expressing HA-E2FBP1 were infected with HSV-1 at
various MOIs, and cell lysates were examined for ICP0 levels
by immunoblotting at 120 mpi (Fig. 6A). Accumulation of
ICP0 was detected in cells with ectopic E2FBP1 expression
only after infection at an MOI of 1 (Fig. 6A, lane 10). This level
of ICP0 was equivalent to what was seen in control cells at an
MOI of 0.1 (Fig. 6A, lane 4). Moreover, in the absence of
ectopic expression of E2FBP1, infection at an MOI of 1
re-sulted in a substantially higher level of ICP0 (Fig. 6A, lane 5).
Thus, accumulation of ICP0 was repressed in response to
ec-topic expression of E2FBP1.
To examine whether E2FBP1-repressed ICP0 expression
oc-curred in the absence of other HSV-1-derived factors, extracts
from HEK293FT cells cotransformed with an ICP0 expression
plasmid (pDS16) together with an HA-E2FBP1 expression
plasmid were subjected to immunoblotting. There was a
sig-nificant dose-dependent reduction of ICP0 levels (Fig. 6B).
The unexpectedly increased level of endogenous E2FBP1 (Fig.
6B, lanes 2 and 3) may have been a consequence of dilution of
ICP0’s E3 activity. These results led us to posit that E2FBP1
represses accumulation of ICP0 by decreasing transcription of
its RNA.
To examine the molecular basis of E2FBP1-mediated
re-pression of ICP0, TIG-3 cells at 43 PD were transformed with
HA-E2FBP1 DNA and then infected with HSV-1 at an MOI of
1. TIG-3 cells transformed with empty vector and
nontrans-formed TIG-3 cells were infected as controls. Samples were
collected at 0, 30, 75, and 130 mpi and then subjected to
RT-qPCR analyses. ICP0 RNA was detected readily at 30 mpi
and was increased in both nontransformed (mock) and empty
vector control cells (Fig. 7A). In contrast, ICP0 RNA
accumu-lation was significantly lowered in cells expressing E2FBP1.
Because ICP0 levels were unaffected after infection of cells
expressing
⌬
A E2FBP1, ICP0 RNA levels were compared in
cells expressing wild-type and
⌬
A HA-E2FBP1 after infection
with HSV-1 at an MOI of 0.5. RT-qPCR analysis revealed that
WT E2FBP1 repressed ICP0 RNA levels to a similar extent to
that in the previous experiment, whereas ICP0 RNA levels
increased in cells expressing
⌬
A E2FBP1 (Fig. 7B). Sequence
analysis of the HSV-1 F strain genome revealed the presence
of multiple ARID3 consensus and consensus-like motifs,
in-cluding 5
⬘
-GTAATTAA/G-3
⬘
and 5
⬘
-TAATTGCT-3
⬘
motifs
upstream of the
IE-0
gene (Fig. 7C). These results strongly
suggest that E2FBP1 represses expression of ICP0 as a result
of transcriptional repression. To examine this hypothesis, we
subcloned various lengths of wild-type HSV-1
IE-0
promoter
sequence 5
⬘
of a humanized
Renilla
luciferase (hRluc) coding
region in pGL4.83 (Fig. 7D) and performed dual-luciferase
assays. Ratios of hRluc to luc were calculated and aligned by
comparison with pGL4.83 activity expressed in the absence of
ectopic E2FBP1 expression (Fig. 7E). As expected, expression
from the
⫺
747 fragment retaining four ARID3 consensus
mo-tifs was suppressed by wild-type E2FBP1, while it was
unaf-fected by the
⌬
A mutant. Deletion of two upstream consensus
motifs abrogated the effect of E2FBP1, although the relative
promoter activity of this construct was decreased even in the
absence of ectopic E2FBP1. Moreover, the
⫺
1007 fusion
con-struct also diminished the suppressive effect of E2FBP1,
re-vealing how complicated the regulation of the
IE-0
promoter is
and that it is controlled not only by ARID proteins but also by
other host proteins. The precise mechanism of
E2FBP1-medi-ated repression remains to be elucidE2FBP1-medi-ated.
DISCUSSION
We report here that ICP0 depletes E2FBP1 as its HUL-2
substrate, by ubiquitin-mediated degradation, and that a major
polyubiquitylation target region is the ARID of E2FBP1.
Con-temporaneously with this event, E2FBP1 represses
accumula-tion of ICP0 transcripts in an ARID-dependent manner. As a
result of these interactions, E2FBP1 is degraded, RNA
encod-ing ICP0 is modulated, and PML-NBs are dissociated (Fig. 8).
These interactions between E2FBP1 and ICP0 suggest that
E2FBP1 contributes to the cellular defense response against
establishment of HSV-1 infection and that ICP0 works as the
first wave of attack to repel the host response by degrading this
FIG. 7. The E2FBP1 ARID is required for repression of accumulation of ICP0 transcripts. (A) E2FBP1 decreases accumulation of transcripts
encoding ICP0 from HSV-1. TIG-3 cells at 43 PD were transformed with either a plasmid expressing E2FBP1 or empty vector, incubated for 44 h,
and then infected with HSV-1 at an MOI of 1 for 30 min. Total cellular RNA was collected at the indicated times postinfection and subjected to
RT-qPCR analyses. The upper chart represents the relative accumulation of ICP0 RNA, and the lower chart shows the relative accumulation of
E2FBP1 RNA. Nontransformed TIG-3 cells (mock) served as a control for transformation. (B) E2FBP1 requires its ARID to repress accumulation
of ICP0 RNA. TIG-3 cells at 45 PD were transformed with empty vector or plasmid expressing either wild-type (WT) or
⌬
A E2FBP1, incubated
for 44 h, and then infected with HSV-1 at an MOI of 0.5 for 30 min. Total cellular RNA was then collected at the indicated times postinfection
and subjected to RT-qPCR analyses. (C) Potential ARID3-binding motifs in the
IE-0
promoter. The IRL region of HSV-1 strain F (GenBank
accession no. GU734771) was searched for ARID3-binding consensus motifs. The DNA sequence constituting the promoter for
IE-0
is shown, and
boxes identify ARID3 consensus and consensus-like motifs. (D) Schematic representation of ARID3 consensus motifs residing in the
IE-0
promoter and its truncated sequences linked upstream of the hRluc gene in reporter constructs. (E) Relative expression levels of hRluc activity
in the presence or absence of wild-type or
⌬
A mutant E2FBP1 expressed from reporter constructs.
on November 7, 2019 by guest
http://jvi.asm.org/
defense factor. Intrinsic cellular defense is initiated by proteins
interacting with PML-NBs (reviewed in references 11, 12, 41,
61, 66, and 71). However, the relationship between the
repli-cation machinery of HSV-1 and the role of PML-NBs that
impinge on viral replication has not been sorted out fully. A
highly debated issue is the significance of ICP0 disruption of
PML-NBs during viral infection. It has been reported that a
lack of PML-NB disruption as a consequence of infection with
ICP0-deficient HSV-1 interferes with replication of HSV-1 in
limited-passage human fibroblasts. However, other reports
re-vealed that high-level ectopic expression of PML in Vero cells,
Hep-2 cells, and telomerase-transformed immortalized human
foreskin fibroblasts did not affect viral replication, although the
virus accumulated in the PML-NB-like nuclear domains (22,
44). The key factor(s) contributing to intrinsic cellular defense
is therefore implied to be associated with PML-NBs (1, 9,
17–19, 21, 46). This factor(s) is likely to be targeted by ICP0
during the immediate-early phase of infection. Alternatively, it
may associate directly with HSV-1 genomes to suppress their
transcription and replication (15, 16). While attempting to
elucidate the molecular bases for this intrinsic cellular defense
mechanism, we showed that E2FBP1 interacts with PML-NBs
to dissociate them. In the absence of E2FBP1,
passage-lim-ited human fibroblasts lost their proliferation potential,
re-sulting in premature senescence accompanied by ectopic
accumulation of PML-NBs (23). Recently, human Daxx and
its partner,
␣
-thalassemia/mental retardation syndrome
X-linked (ATRX), were investigated as candidates for such
DNA-associating suppressive factors by use of an RNA
in-terference (RNAi)-mediated knockdown method, and they
were revealed to contribute to intrinsic cellular defense
(46). Both of these PML-NB-associating proteins are
in-volved in the chromatin-remodeling complex and exhibit
transcription-repressing activities (41, 49, 61, 62).
Our results identify the ARID of E2FBP1 as another target
of ICP0-mediated polyubiquitylation (Fig. 4 and 5). Since the
ARID is a highly conserved domain within the ARID protein
family (reviewed in references 35, 69, 72, and 73), other family
members are potential host targets for ICP0-mediated
degra-dation. ARID proteins are involved in multiple cellular
pro-cesses to maintain chromosomal integrity, including chromatin
remodeling, DNA repair, and transcriptional controls.
There-fore, our results may provide insight into host-virus
interac-tions, specifically into how other ARID family members
inter-act with HSV-1 and its gene products. Of possible relevance is
a report that the ARID5B transcription factor Mrf-2
sup-presses the human cytomegalovirus enhancer (30).
Finally, the ability of ICP0 to suppress the host cell cycle
during a productive infection may also be connected to the
depletion of E2FBP1. E2FBP1 activates the E2F1/DP1
com-plex to enhance transcription levels of target genes that are
important for S-phase entry (65). Therefore, depletion of
E2FBP1 should result in a delay in the G
1/S transition.
FIG. 8. Schematic representation of possible interactions between E2FBP1 and ICP0 in infected cell nuclei. ICP0 targets E2FBP1 as a HUL-2
substrate to deplete it via the ubiquitin pathway. Contemporaneously with this event, E2FBP1 targets the
IE-0
promoter to repress transcription
of ICP0 RNA. Target Lys residues in E2FBP1 reside both inside and outside the ARID. The latter sites may be occluded from ubiquitylation as
a consequence of REKLES-mediated dimerization. Interactions of PML-NBs with both E2FBP1 and ICP0 are also illustrated. K, Lys residues
available for ICP0-mediated ubiquitylation; Ub, ubiquitin moiety.
3364
FUKUYO ET AL.
J. V
IROL.
on November 7, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
We thank Naoki Inoue at the National Institute of Infectious
Dis-eases of Japan for WT HSV-1 strain F and Ikuo Morita, Miki
Yokoyama, Podyma-Inoue Katarzyna-Anna, Ichiro Nakagawa, and
Kenji Yamato at TMDU for discussions.
This study was supported by grants from the NIH Public Health
Service (AI024021 to S.J.S. and CA127378-01A1 to A.M.I.).
REFERENCES
1.Boutell, C., et al.2008. Herpes simplex virus type 1 ICP0 phosphorylation mutants impair the E3 ubiquitin ligase activity of ICP0 in a cell type-depen-dent manner. J. Virol.82:10647–10656.
2.Brand, P., T. Lenser, and P. Hemmerich.2010. Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys.3:3.
3.Chelbi-Alix, M. K., and H. de The´.1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene18:935–941.
4.Chen, J., C. Panagiotidis, and S. Silverstein.1992. Multimerization of ICP0, a herpes simplex virus immediate-early protein. J. Virol.66:5598–5602. 5.Chen, J., and S. Silverstein.1992. Herpes simplex viruses with mutations in
the gene encoding ICP0 are defective in gene expression. J. Virol.66:2916– 2927.
6.Deng, L., et al.2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell103:351–361.
7.Dent, A. L., et al.1996. LYSP100-associated nuclear domains (LANDs): description of a new class of subnuclear structures and their relationship to PML nuclear bodies. Blood88:1423–1426.
8.de The, H., et al.1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a function-ally altered RAR. Cell66:675–684.
9.Everett, R. D.2010. Depletion of CoREST does not improve the replication of ICP0 null mutant herpes simplex virus type 1. J. Virol.84:3695–3698. 10.Everett, R. D.2000. ICP0, a regulator of herpes simplex virus during lytic and
latent infection. Bioessays22:761–770.
11.Everett, R. D.2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol.8:365–374.
12.Everett, R. D., and M. K. Chelbi-Alix.2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie89:819–830.
13.Everett, R. D., et al.1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol.72:6581–6591.
14.Everett, R. D., P. Lomonte, T. Sternsdorf, R. van Driel, and A. Orr.1999. Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci.112:4581–4588.
15.Everett, R. D., and J. Murray.2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol.79:5078–5089.
16.Everett, R. D., J. Murray, A. Orr, and C. M. Preston.2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol.81:10991–11004. 17.Everett, R. D., and A. Orr.2009. Herpes simplex virus type 1 regulatory
protein ICP0 aids infection in cells with a preinduced interferon response but does not impede interferon-induced gene induction. J. Virol.83:4978–4983. 18.Everett, R. D., C. Parada, P. Gripon, H. Sirma, and A. Orr.2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol.82:2661–2672.
19.Everett, R. D., M. L. Parsy, and A. Orr.2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol.83:4963– 4977.
20.Everett, R. D., et al.2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol.80:7995–8005.
21.Everett, R. D., D. F. Young, R. E. Randall, and A. Orr.2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol.82:8871– 8881.
22.Everett, R. D., and A. Zafiropoulos.2004. Visualization by live-cell micros-copy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol.78:11411–11415.
23.Fukuyo, Y., K. Mogi, Y. Tsunematsu, and T. Nakajima.2004. E2FBP1/ hDril1 modulates cell growth through downregulation of promyelocytic leu-kemia bodies. Cell Death Differ.11:747–759.
24.Gregory, S. L., R. D. Kortschak, B. Kalionis, and R. Saint.1996. Charac-terization of the dead ringer gene identifies a novel, highly conserved family of sequence-specific DNA-binding proteins. Mol. Cell. Biol.16:792–799. 25.Hagglund, R., and B. Roizman.2004. Role of ICP0 in the strategy of
con-quest of the host cell by herpes simplex virus 1. J. Virol.78:2169–2178. 26.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient
reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240–3249.
27.Harris, R. A., R. D. Everett, X. X. Zhu, S. Silverstein, and C. M. Preston. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reacti-vates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 63:3513–3515.
28.Herrscher, R. F., et al.1995. The immunoglobulin heavy-chain matrix-asso-ciating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev.9:3067–3082. 29.Hobbs, S., S. Jitrapakdee, and J. C. Wallace.1998. Development of a
bicistronic vector driven by the human polypeptide chain elongation factor 1alpha promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem. Biophys. Res. Commun.252: 368–372.
30.Huang, T. H., et al.1996. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res.24:1695–1701. 31.Jiang, W. Q., and N. Ringertz.1997. Altered distribution of the
promyelo-cytic leukemia-associated protein is associated with cellular senescence. Cell Growth Differ.8:513–522.
32.Kim, D., L. Probst, C. Das, and P. W. Tucker.2007. REKLES is an ARID3-restricted multifunctional domain. J. Biol. Chem.282:15768–15777. 33.Kim, D., and P. W. Tucker.2006. A regulated nucleocytoplasmic shuttle
contributes to Bright’s function as a transcriptional activator of immuno-globulin genes. Mol. Cell. Biol.26:2187–2201.
34.Kortschak, R. D., et al. 1998. The human dead ringer/bright homolog, DRIL1: cDNA cloning, gene structure, and mapping to D19S886, a marker on 19p13.3 that is strictly linked to the Peutz-Jeghers syndrome. Genomics 51:288–292.
35.Kortschak, R. D., P. W. Tucker, and R. Saint.2000. ARID proteins come in from the desert. Trends Biochem. Sci.25:294–299.
36.Laemmli, U. K.1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature227:680–685.
37.Lallemand-Breitenbach, V., and H. de The.2010. PML nuclear bodies. Cold Spring Harb. Perspect. Biol.2:a000661.
38.Lee, F., R. Mulligan, P. Berg, and G. Ringold.1981. Glucocorticoids regulate expression of dihydrofolate reductase cDNA in mouse mammary tumour virus chimaeric plasmids. Nature294:228–232.
39.Lin, D., et al.2007. Bright/ARID3A contributes to chromatin accessibility of the immunoglobulin heavy chain enhancer. Mol. Cancer6:23.
40.Lin, L., et al.2008. Cross talk between Id1 and its interactive protein Dril1 mediate fibroblast responses to transforming growth factor-beta in pulmo-nary fibrosis. Am. J. Pathol.173:337–346.
41.Lindsay, C. R., V. M. Morozov, and A. M. Ishov.2008. PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front. Biosci.13:7132– 7142.
42.Lium, E. K., C. A. Panagiotidis, X. Wen, and S. Silverstein.1996. Repression of the alpha0 gene by ICP4 during a productive herpes simplex virus infec-tion. J. Virol.70:3488–3496.
43.Livak, K. J., and T. D. Schmittgen.2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(⫺Delta Delta C(T)) method. Methods25:402–408.
44.Lopez, P., R. J. Jacob, and B. Roizman.2002. Overexpression of promyelo-cytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol.76:9355–9367.
45.Lopez, P., C. Van Sant, and B. Roizman.2001. Requirements for the nucle-ar-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol.75:3832–3840.
46.Lukashchuk, V., and R. D. Everett.2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol.84:4026–4040.
47.Ma, K., et al.2003. E2FBP1/DRIL1, an AT-rich interaction domain-family transcription factor, is regulated by p53. Mol. Cancer Res.1:438–444. 48.Maul, G. G.1998. Nuclear domain 10, the site of DNA virus transcription
and replication. Bioessays20:660–667.
49.Maul, G. G., D. Negorev, P. Bell, and A. M. Ishov.2000. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129:278–287.
50.Muller, S., and A. Dejean.1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol.73:5137–5143.
51.Negorev, D. G., O. V. Vladimirova, A. Ivanov, F. Rauscher III, and G. G. Maul.2006. Differential role of Sp100 isoforms in interferon-mediated re-pression of herpes simplex virus type 1 immediate-early protein exre-pression. J. Virol.80:8019–8029.
52.Negorev, D. G., O. V. Vladimirova, and G. G. Maul. 2009. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protec-tion from ICP0-mediated degradaprotec-tion. J. Virol.83:5168–5180.
53.Nystrom, K., et al.2004. Real time PCR for monitoring regulation of host gene expression in herpes simplex virus type 1-infected human diploid cells. J. Virol. Methods118:83–94.
on November 7, 2019 by guest
http://jvi.asm.org/
54.Parkinson, J., and R. D. Everett.2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol.74:10006–10017.
55.Pearson, M., et al. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature406:207–210.
56.Peeper, D. S., et al. 2002. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat. Cell Biol.4:148–153. 57.Prieur, A., K. Nacerddine, M. van Lohuizen, and D. S. Peeper. 2009. SUMOylation of DRIL1 directs its transcriptional activity towards leukocyte lineage-specific genes. PLoS One4:e5542.
58.Rajaiya, J., M. Hatfield, J. C. Nixon, D. J. Rawlings, and C. F. Webb.2005. Bruton’s tyrosine kinase regulates immunoglobulin promoter activation in association with the transcription factor Bright. Mol. Cell. Biol.25:2073– 2084.
59.Roizman, B.1999. HSV gene functions: what have we learned that could be generally applicable to its near and distant cousins? Acta Virol.43:75–80. 60.Russell, J., N. D. Stow, E. C. Stow, and C. M. Preston.1987. Herpes simplex
virus genes involved in latency in vitro. J. Gen. Virol.68:3009–3018. 61.Saffert, R. T., and R. F. Kalejta.2008. Promyelocytic leukemia-nuclear body
proteins: herpesvirus enemies, accomplices, or both? Future Virol.3:265– 277.
62.Salomoni, P., and A. F. Khelifi. 2006. Daxx: death or survival protein? Trends Cell Biol.16:97–104.
63.Schmittgen, T. D., and K. J. Livak.2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc.3:1101–1108.
64.Sternsdorf, T., T. Grotzinger, K. Jensen, and H. Will.1997. Nuclear dots: actors on many stages. Immunobiology198:307–331.
65.Suzuki, M., et al.1998. A novel E2F binding protein with Myc-type HLH
motif stimulates E2F-dependent transcription by forming a heterodimer. Oncogene17:853–865.
66.Tavalai, N., and T. Stamminger.2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta1783: 2207–2221.
67.Thompson, R. L., and N. M. Sawtell.2006. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J. Virol.80:10919–10930.
68.Van Sant, C., P. Lopez, S. J. Advani, and B. Roizman.2001. Role of cyclin D3 in the biology of herpes simplex virus 1 ICP0. J. Virol.75:1888–1898. 69.Webb, C. F.2001. The transcription factor, Bright, and immunoglobulin
heavy chain expression. Immunol. Res.24:149–161.
70.Webb, C. F., et al.2000. The transcription factor Bright associates with Bruton’s tyrosine kinase, the defective protein in immunodeficiency disease. J. Immunol.165:6956–6965.
71.Wileman, T.2007. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu. Rev. Microbiol.61:149–167. 72.Wilsker, D., A. Patsialou, P. B. Dallas, and E. Moran.2002. ARID proteins:
a diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ.13:95–106. 73.Wilsker, D., et al.2005. Nomenclature of the ARID family of DNA-binding
proteins. Genomics86:242–251.
74.Zhu, X. X., J. X. Chen, and S. Silverstein.1991. Isolation and characteriza-tion of a funccharacteriza-tional cDNA encoding ICP0 from herpes simplex virus type 1. J. Virol.65:957–960.
75.Zhu, X. X., J. X. Chen, C. S. Young, and S. Silverstein.1990. Reactivation of latent herpes simplex virus by adenovirus recombinants encoding mutant IE-0 gene products. J. Virol.64:4489–4498.
3366
FUKUYO ET AL.
J. V
IROL.
on November 7, 2019 by guest
http://jvi.asm.org/