Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Importance of the N Terminus of Rous Sarcoma Virus Protease
for Structure and Enzymatic Function
GISELA W. SCHATZ, JEFFREY REINKING, JONATHAN ZIPPIN, LINDA K. NICHOLSON,
ANDVOLKER M. VOGT*
Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York 14853
Received 23 October 2000/Accepted 19 February 2001
All retrovirus proteases (PRs) are homodimers, and dimerization is essential for enzymatic function. The dimer is held together largely by a short four-stranded antiparallel beta sheet composed of the four or five N-terminal amino acid residues and a similar stretch of residues from the C terminus. We have found that the enzymatic and structural properties of Rous sarcoma virus (RSV) PR are exquisitely sensitive to mutations at the N terminus. Deletion of one or three residues, addition of one residue, or substitution of alanine for the N-terminal leucine reduced enzymatic activity on peptide and protein substrates 100- to 1,000-fold. The purified mutant proteins remained monomeric up to a concentration of about 2 mg/ml, as determined by dynamic light scattering. At higher concentrations, dimerization was observed, but the dimer lacked or was deficient in enzymatic activity and thus was inferred to be structurally distinct from a wild-type dimer. The mutant protein lacking three N-terminal residues (⌬LAM), a form of PR occurring naturally in virions, was examined by nuclear magnetic resonance spectroscopy and found to be folded at concentrations where it was monomeric. This result stands in contrast to the report that a similarly engineered monomeric PR of human immunodeficiency virus type 1 is unstructured. Heteronuclear single quantum coherence spectra of the mutant at concentrations where either monomers or dimers prevail were nearly identical. However, these spectra differed from that of the dimeric wild-type RSV PR. These results imply that the chemical environment of many of the amide protons differed and thus that the three-dimensional structure of the ⌬LAM PR mutant is different from that of the wild-type PR. The structure of this mutant protein may serve as a model for the structure of the PR domain of the Gag polyprotein and may thus give clues to the initiation of proteolytic maturation in retroviruses.
All retroviruses encode a protease (PR) that functions to cleave the structural polyprotein Gag and the enzymatic poly-protein Pol into their constituent mature poly-proteins found in infectious virions. PR is translated as a domain of the Gag, Gag-Pro, or Gag-Pro-Pol polyprotein, depending on the ret-rovirus genus (reviewed in references 42 and 43). Cleavage of the viral polyproteins is initiated late in assembly, during or after budding of the nascent virion from the plasma mem-brane, and leads to the morphological maturation that gives rise to infectious virus. The first cleavage in maturation is believed to be an autocatalytic event in which the PR domain excises itself from the polyprotein (6, 10, 12, 24, 35, 48). What triggers this process remains unknown.
Retrovirus PRs are aspartate PRs, with the catalytic site positioned between the two identical subunits that make up the enzymatically active PR dimer. Thus, dimerization is a requi-site for enzyme function. It has been suggested elsewhere that dimerization of the PR domains of two neighboring molecules of the polyprotein is the initial and rate-limiting step of a proteolytic cascade that results in maturation (26; reviewed in reference 43). That dimerization cannot be triggered simply by mass action, by the PR domains becoming concentrated in the nascent virion, is best illustrated by the type B and type D retroviruses (prototypes, mouse mammary tumor virus and
Mason-Pfizer monkey virus, respectively). In these viruses, the immature viral core becomes fully assembled in the cytoplasm, and only after transport to the plasma membrane and budding is proteolysis initiated.
High-resolution structures of numerous retrovirus PRs are known, and in all cases a key feature of the dimer is a short, four-stranded antiparallel beta sheet formed by the first and last few residues at the N and C termini (reviewed in reference 45). A large fraction of the dimer interface surface is provided by this structural feature, and preventing its formation blocks enzymatic activity, as shown previously for Rous sarcoma virus (RSV) (18) and human immunodeficiency virus type 1 (HIV-1) (3, 36, 46). By contrast with the highly detailed knowledge of the structural and enzymatic features of mature dimeric PRs, little is known about PR domains as part of the polyproteins in which they are initially embedded. In some studies, PR pre-cursors of this type have been reported to be inactive or poorly active in vitro, but these findings depend on the virus and on the details of the conditions used (10, 24, 29, 37, 47, 48). Assessing the enzymatic activity of a PR domain embedded in a larger segment of polypeptide can be difficult because the PR domain may excise itself during the assay, thereby generating fully active dimeric PR. Even a small amount of mature PR will initiate a feedback loop in which more PR is generated by cleavage intrans,leading ultimately to complete processing of the precursor. Structural studies of retrovirus PRs and larger PR-containing polypeptides are complicated by their limited solubility and by the invariable need to renature these proteins from inclusion bodies after expression inEscherichia coli.
* Corresponding author. Mailing address: Department of Molecular Biology and Genetics, Biotechnology Building, Cornell University, Ithaca, NY 14853. Phone: (607) 255-2443. Fax: (607) 255-2428. E-mail: [email protected].
4761
on November 9, 2019 by guest
http://jvi.asm.org/
The avian sarcoma and leukemia viruses (ASLV; prototype, RSV) have been an important model system to study PR struc-ture and function. Indeed, the first crystal strucstruc-ture of a ret-rovirus PR was determined for avian myeloblastosis virus, a strain of ASLV (16). Next to HIV-1 PR, ASLV PR probably has been the best-studied retrovirus PR from an enzymatic point of view (1, 7, 13, 20, 21, 34, 38). Among retroviruses, the ASLV are unusual in that PR is translated as the C-terminal domain of Gag and thus is present in virions in amounts equimolar with those of the other Gag proteins. We showed previously that purified RSV NC-PR protein, consisting of the joined NC and PR domains as they are found in RSV Gag, has very little enzymatic activity and at low concentrations behaves as a monomer in solution (37). These observations suggested that amino acid sequences upstream of PR interfere with dimerization and thus that the RSV Gag protein is analogous to a zymogen. Consistent with this notion, we found in trans-fection experiments that a cleavage site mutation at the NC-PR junction prevented proper excision of PR from Gag and com-pletely blocked the appearance of mature Gag and also Pol proteins in virions (6, 35, 39). However, in these mutant virions about 25% of the Gag molecules were cleaved near the NC-PR junction, generating an inactive PR species truncated by three amino acid residues at its N terminus. The same truncated species also is found in wild-type virions (32). This result im-plies that the only activity of the PR domain as part of the polyprotein is to cleave itself out and that all other cleavages in Gag and Pol are mediated by the mature PR.
The PR domain of a single Gag molecule must exist in a monomeric state, at least transiently and perhaps until the time when proteolysis is initiated late in assembly. Thus, knowledge of the three-dimensional structure of a monomeric PR might provide insight into the process of dimerization. Based on initial results with the NC-PR protein in vitro and the⌬LAM protein in mutant virions (35, 37), we decided to analyze the effects of N-terminal mutations on RSV PR activity, dimeriza-tion, and three-dimensional structure. The results presented here show that even minor changes at the N terminus block enzymatic activity, and they do so by preventing proper dimer-ization. From nuclear magnetic resonance (NMR) spectros-copy of the mutant⌬LAM, we infer that this monomeric form of PR is folded, unlike a similar mutant HIV-1 protein (26), and that its structure differs significantly from that of wild-type PR analyzed in parallel.
MATERIALS AND METHODS
Purification of recombinant PRs.Eight PR mutations were constructed by PCR mutagenesis and subcloned into the pET11 expression plasmid (Novagen) by standard techniques, and the sequence was confirmed. Plasmids were prop-agated inE. coliDH5␣and transformed into BL21 cells for protein expression.
E. coliBL21 cells were grown in 2YT medium containing ampicillin and chlor-amphenicol. For isotope labeling with15N and13C, cells were grown in MT-9 CN
medium (Martek) and with15N alone in M9 medium containing [15N]ammonium
chloride (Isotec). After induction with 1 mM isopropyl thiogalactoside (IPTG) at
A600⫽0.4, cultures were incubated for another 4 h in rich medium or overnight in minimal medium. Inclusion bodies were collected from sonicated cell lysates, washed two times in 20 mM Tris-HCl (pH 7.5)–5 mM EDTA–2 M urea–2% Triton X-100–10 mM dithiothreitol (DTT)–1 mM phenylmethylsulfonyl fluoride, and then washed once in the same solution without urea and Triton X-100. The washed inclusion bodies were dissolved in 20 mM Tris-HCl (pH 7.5)–7 M urea– 10% glycerol–5 mM EDTA by stirring at room temperature for 30 min and diluted to 1 M urea with 20 mM Tris-HCl (pH 8)–20 mM NaCl–5 mM EDTA–
10% glycerol, and insoluble material was removed by centrifugation. The super-natant was passed over a DEAE column in the same buffer, and the flowthrough containing the PR was collected and dialyzed into 20 mM Na phosphate (pH 6)–100 mM NaCl–1% glycerol–0.4 M urea–10 mM DTT. After removal of any precipitate, the supernatant was concentrated with an Ultrafree centrifugal filter unit (molecular mass cutoff, 3.5 kDa; Millipore), and the protein concentration and purity were assessed byA280andA260, using the molar extinction coefficient for PR, and by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Coomassie blue staining. Molar concentrations of PR as used in this paper refer to the total concentration of the PR polypeptide as if it were monomeric.
The viral PR (vPR) serving as a control was purified from avian myeloblastosis virus (Life Sciences, Inc., St. Petersburg, Fla.) by chloroform-methanol extrac-tion (34). TheE. coli-derived wild-type PR, dPR, was obtained by self-cleavage of His-ePR, a protein carrying the sequence MAHHHHHHAPAVS N-terminal to the PR coding region. PAVS is the sequence of the C terminus of NC, i.e., it occurs naturally at this position in Gag. His-ePR was purified either as done for the other PR mutants or by metal chelate chromatography on Ni-nitrilotriacetic acid (Qiagen). In either case, the final step was dialysis into 20 mM NaPO4(pH
7.0)–100 mM NaCl–1% glycerol–0.4 M urea. The protein was incubated in this condition at 37°C for 5 h to allow self-cleavage. We found that self-cleavage was more efficient at this pH and low salt than at more acid pH values and higher salt, the conditions normally used for retrovirus PRs. The resulting dPR was dialyzed into the same buffer but with a pH of 6.0, concentrated, and then assayed for activity on protein and peptide substrates as described below.
Enzyme assays.Activity of wild-type and recombinant PRs was measured on protein and peptide substrates. Standard reaction mixtures contained 100 mM 3-(N-morpholino)propanesulfonic acid (MOPS; pH 6), 0.8 M NaCl, 5 mM EDTA, and 6% glycerol, to which was added 5g of the substrate protein
⌬MBD⌬PR, in a final reaction volume of 30l. The substrate is the RSV Gag protein missing the 83-amino-acid membrane binding region of MA at the N terminus and missing the entire 124 amino acid residues comprising PR at the C terminus. From 1 to 10g (approximately 2 to 20M PR) of each PR was added to the reaction mixture, and incubation was carried out at 39°C for 0.25 to 60 min for vPR or dPR and 10 min to 10 h for the mutants. The reaction products were separated by SDS-PAGE and visualized by Coomassie blue staining. Peptide assays were based on cleavage of the decapeptide PFQAY/PLREA, which cor-responds to the cleavage site between the reverse transcriptase and integrase domains in the RSV Pol protein. The same conditions as for the protein sub-strate assays were used, except that glycerol was omitted. The concentration of peptide was 0.4 mM, and the concentration of PR was 0.07 to 7M for vPR and dPR and 14M for the mutant PRs. The reactions were stopped by addition of trifluoroacetic acid to 10% and stored at⫺20°C until they were analyzed by reverse-phase high-pressure liquid chromatography (HPLC) on a C18Vidac
column with absorbance at 210 nm recorded. Reaction products were eluted at a flow rate of 1 ml/min with a 0 to 60% acetonitrile gradient in 0.1% trifluoro-acetic acid.
DLS and cross-linking analysis.Mutant and wild-type PRs were tested for dimerization status by dynamic light scattering (DLS) on a DynaPro MSTC molecular sizing instrument (Protein Solutions, Inc.). Samples at concentrations ranging from 0.5 to 5 mg/ml were passed through a 20-nm-pore-size Whatman Anotop Plus filter disk, and then 15l was loaded into the DLS cuvette and illuminated by a laser with a wavelength of 780 to 830 nm. The time scale of the scattered light intensity fluctuations at 25°C was analyzed using the autocorre-lation function of the company’s software. From the transautocorre-lational diffusion co-efficient, the hydrodynamic radius, Rh, can be derived from the Stokes-Einstein
relationship. A standard curve of globular proteins of known mass allows esti-mation of the molecular weight from the Rhof the sample protein with an
accuracy of⫾10% for globular molecules.
Cross-linking was carried out on vPR or⌬LAM at 1 mg/ml. Samples were incubated with the homobifunctional cross-linking reagent dimethylsuberimidate (DMS) at pH 8.0 and 2 mM or with 1,6-bismaleimidohexane (BMH) at pH 6.0 and 2 or 10 mM (both reagents from Pierce Chemical Company). DMS has a spacer arm length of 11 Å and is reactive toward primary amino groups, whereas BMH has a spacer arm length of 16 Å and reacts with sulfhydryl groups. DMS was allowed to react at room temperature for 2 h in 50 mM triethanolamine buffer (pH 8.1) and was quenched with 0.1 M glycine for 20 min before addition of sample buffer for SDS-PAGE. BMH was allowed to react at room tempera-ture for 30 min in 20 mM NaPO4buffer (pH 6) and was quenched by addition of
50 mM DTT.
NMR.⌬LAM and dPR protein samples for NMR analysis were prepared as described above with [15N]ammonium chloride (Isotec) as the sole nitrogen
source. Sterile D2O (Martek Biosciences) was added to the sample to a final
on November 9, 2019 by guest
http://jvi.asm.org/
concentration of 10% (vol/vol), and the samples were placed in microcell NMR tubes (Shigemi, Inc.). NMR experiments were performed at 298 K using a Varian Inova 600-MHz spectrometer equipped with a triple resonance probe with a
z-axis pulsed-field gradient. The15N-1H heteronuclear single quantum
coher-ence (HSQC) pulse sequcoher-ence from Lewis Kay’s library (University of Toronto) was employed, which includes water-flip-back pulses to avoid saturation transfer (14) as well as sensitivity enhancement in conjunction with gradient coherence selection (19, 27).
RESULTS
Activity of N-terminally mutated versions of PR.In earlier work, we showed that an RSV PR truncated by three amino acid residues at its N terminus lacked detectable activity, as evidenced by the complete absence of mature Gag and Pol proteins in virions (6, 35, 39). Also, it was reported elsewhere that the specific activity of wild-type PR expressed in E. coli
was only approximately 10% of that of the wild-type PR when isolated from virions (4, 34, 37), the attenuation of activity being attributed to the lack of removal of the initiating methi-onine residue inE. coli. Consistent with this interpretation, in RSV PR-Pol fusion proteins created with an initiating Met residue in front of the PR domain, the Met residue was not removed and the proteins were enzymatically inactive when highly expressed in insect cells (39). These several observations suggested that the proper N terminus is critical for PR func-tion. In order to more carefully characterize the role of the N terminus, we constructed several additional mutant PRs (Fig. 1).⌬LAM lacks the three N-terminal amino acid residues and thus is equivalent to the truncated PR formed in virions when the cleavage site at the NC-PR junction is mutated (35). The same truncated species also is found in wild-type virions, rep-resenting about 7% of the total PR (32).⌬L deletes only Leu1, the first residue of PR. L1A, L1V, and L1I change the N-terminal leucine residue as indicated. As controls for these mutants, three different forms of wild-type PR were used. PR purified from avian myeloblastosis virus virions, designated vPR, was the standard in all assays. The PR polypeptide ex-pressed inE. coliwith an initiating methionine residue proxi-mal to Leu1 is designated rPR. This form of PR is poorly active, by inference because of failure of methionine removal
inE. coli(see below). We engineered a version of PR, called
His-ePR, that processes itself slowly in vitro to liberate wild-type PR with the correct N-terminal Leu1. In this protein, the four natural residues preceding the PR sequence, PAVS, as well as a histidine tag are positioned proximal to the coding sequence. His-ePR was purified in the same manner as the other mutant PRs and incubated under conditions found to maximize the weak autocatalytic processing activity, and the resulting derived PR (dPR) was repurified. As found for other retrovirus PRs, all of the PRs studied here were located almost exclusively in inclusion bodies and thus had to be solubilized in 7 M urea before refolding. Control experiments with vPR showed that denaturation in urea followed by renaturation upon removal of urea did not compromise the specific activity (data not shown).
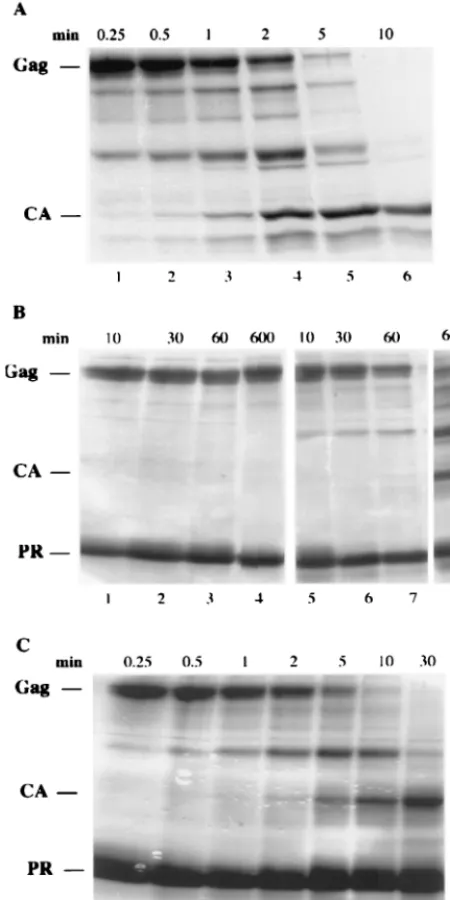
To estimate the activity of the mutant PRs, two assays were used. The first is semiquantitative and visualizes cleavage of the substrate protein⌬MBD⌬PR, a truncated RSV Gag pro-tein missing 83 residues from the N terminus and the 124 PR residues from the C terminus (8, 17). This protein, which contains several natural cleavage sites, was purified after ex-pression inE. coli,and its processing after incubation with PR was monitored by SDS-PAGE and staining. In typical experi-ments, incubation of 1g of vPR with 10g of substrate led to 50% cleavage in about 2 min and complete cleavage in 10 to 20 min (Fig. 2A). Complete cleavage was defined by a gel profile in which the mature CA polypeptide was the largest prominent species visible on the stained gel. In the same time period, 1g of the mutant protein⌬LAM, L1A, L1V, L1I, or⌬L failed to process the substrate protein (data not shown). However, with a 10-fold increase in PR and longer incubation times, limited cleavage was observed for L1V, with intermediate cleavage products barely evident at 12 min and substantial amounts of CA evident by 10 h (Fig. 2B). Somewhat higher levels of activity were observed for L1I (data not shown). No activity was detectable for the mutant proteins L1A (Fig. 2B) and⌬L and⌬LAM (data not shown) in similar assays. However, in a more sensitive assay in which Western blotting with anti-CA serum was used instead of staining,⌬LAM did manifest de-tectable activity, estimated at ⬃0.1% of the activity of vPR (data not shown). For rPR, 10g of enzyme gave complete cleavage by 30 min (Fig. 2C). We attempted to quantitate these results by visually gauging the intensity of bands from gels like those shown in Fig. 2 and estimating at what time and enzyme concentration 50% of the substrate protein had been con-verted into smaller polypeptides. Based on the assumptions that enzyme activity is directly proportional to the amount of PR in the reaction and that the enzyme remains stable during the incubation period, we derived semiquantitative estimates of the residual activities of the different PRs: dPR was as active as vPR, rPR was 5 to 10% as active, and the mutants ranged from 2 to 3% for L1I to undetectable by the staining assay for L1A and⌬LAM (Table 1).
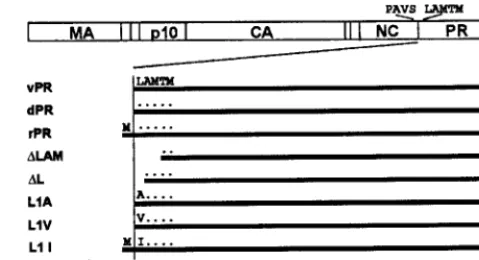
[image:3.612.56.296.74.204.2]Knowledge of the actual N-terminal sequence of the several PR species is clearly critical for interpreting their enzymatic activities. InE. coli,the second amino acid residue determines if the initiating methionine will be removed (15). Loss of Met generally occurs if the side chain is small but not if it is large. We determined the N-terminal sequence for theE. coli -de-rived PR proteins and found them to obey this rule. Thus, the FIG. 1. Schematic structure of PR mutants. The rectangle at the
top depicts the RSV Gag protein and its major domains, with the amino acid sequence at the NC-PR cleavage site shown above. The horizontal bars represent the several wild-type and mutant PRs stud-ied. The wild-type amino acid residues at the N terminus are indicated, with dots indicating identity and the absence of any symbol indicating a deletion of those residues. The N-terminal sequence of all proteins was determined experimentally.
on November 9, 2019 by guest
http://jvi.asm.org/
initiating Met was removed when the second residue was Ala (L1A and ⌬L), Val (L1V), or Thr (⌬LAM), but Met was retained when the second residue was Leu (rPR) or Ile (L1I) (Table 1). N-terminal sequence analysis is rarely quantitative, however, reflecting only the predominant polypeptide species. Thus, a minor species of PR comprising less than about 5% of the total protein would not have been evident in these analyses. Therefore, for the proteins that retained the Met, rPR and L1I,
it is unclear to what extent the low activity observed reflects intrinsic activity of the major species bearing an N-terminal Met or alternatively reflects a low level of a fully active species with the Met removed.
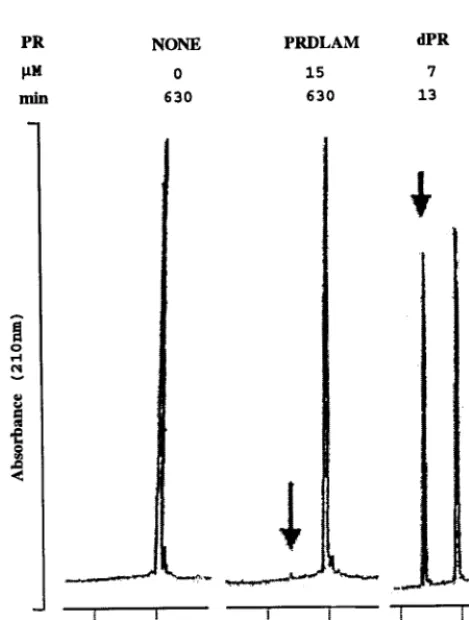
As an independent and more quantitative assay for activity, we incubated the mutant PRs with a synthetic 10-amino-acid peptide. This peptide, which includes the cleavage site between the reverse transcriptase and integrase domains of the RSV Pol protein, was reported to be the most efficiently cleaved of several Gag and Pol peptides processed by PR in vitro (41). Each PR was incubated at a final concentration of 0.1 or 0.2 mg/ml with 0.4 mM peptide, and the products and substrate were separated on a C18column by reverse-phase HPLC. The
peak height of the N-terminal product eluting at 17 min was used to estimate rate of cleavage, under conditions where less than 25% of the substrate had been used up. Representative portions of tracings showing incubations with vPR, with ⌬LAM, or without any PR are shown in Fig. 3. While 5g of vPR incubated for 13 min resulted in the appearance of a robust product peak (arrow), 10 g of⌬LAM incubated for 630 min resulted in the reproducible appearance of a barely detectable peak just above the baseline. From similar assays, we calculated approximate kcat values for three mutant
ver-sions of PR (Table 1). The results are consonant with those of the polypeptide cleavage assays, showing that ⌬L, L1A, and ⌬LAM are reduced 100- to 1,000-fold in specific activity. The
kcatvalue obtained for vPR is in the same range as reported
previously for this enzyme, 1 to 11 min⫺1(13, 21). We did not
seek to determine if the slight difference inkcatdetermined for
vPR and dPR represents an intrinsic property of these proteins or is due to experimental error.
Dimerization status of mutant PRs.In retrovirus PRs, the four-stranded beta sheet comprising the N-terminal four to five and the C-terminal four to five residues (except for the ulti-mate residue) is known to be critical for dimerization (44). For example, short peptides including the terminal residues of PR will compete at high concentrations with intermolecular beta-sheet formation, causing dissociation of the dimer and loss of activity (3, 18, 36, 46). From these results, we hypothesized that the loss or attenuation of enzyme activity in the mutant RSV PRs was due to lack of dimer formation. To address this hypothesis, we made extensive use of DLS and also carried out FIG. 2. Protein cleavage assay. Wild-type or mutant PRs were
[image:4.612.60.285.77.527.2]in-cubated with the truncated Gag protein⌬MBD⌬PR, and the time course of proteolytic processing was examined by SDS-PAGE and Coomassie blue staining. The reaction mixtures contained 100 mM MOPS (pH 6.0), 0.8 M NaCl, 1 mM EDTA, 6% glycerol, 5g of sub-strate, and either 1g of vPR or 10g of mutant PRs. The positions of the substrate and the major product, CA, as well as of PR are indicated on the left. The assay times are shown at the top of each pa-nel. (A) vPR; (B) L1A (lanes 1 to 4) and L1V (lanes 5 to 8); (C) rPR.
TABLE 1. In vitro activities of wild-type and mutant PRs
PR
constant terminusN a
% Activity on protein substrateb
% Activity on peptide substratec
kcatd (min⫺1)
rPR MLAMTM 5–10 NDf ND
L1I MIAMTM 2–3 ND ND
L1V VAMTM ⬍1 ND ND
L1A AAMTM 0e 0.3 0.009
⌬L -AMTM ⬍1 0.9 0.02
⌬LAM ---TM 0e 0.1 0.003
dPR LAMTM 100 65 2.5
vPR LAMTM 100 100 4
aN-terminal amino acid sequence was determined for each purified protein. bActivity was estimated as described in the legend to Fig. 2.
cActivity was estimated as described in the legend to Fig. 3.
dEstimated from amount of N-terminal product peak with assumptions de-scribed in the text.
eNo product was detectable by SDS-PAGE and Coomassie blue staining. fND, not determined.
on November 9, 2019 by guest
http://jvi.asm.org/
corroborative experiments with rate zonal sedimentation in glycerol gradients and with chemical cross-linking. Most exper-iments focused on the⌬LAM and L1A mutants and on vPR or dPR as controls. A limitation of DLS is that the concentration of a protein of this size must be at least⬃0.5 mg of monomer/ ml. Another limitation particular to PRs is their poor solubil-ity. We found that the mutant PRs were stably soluble to only about 3 mg/ml. vPR and dPR were less soluble than the mutant PRs, consistent with the original report that vPR could be crystallized from solutions of 1.3 mg/ml (16, 33). However, addition of 1% glycerol and 0.4 M urea, which had no effect on activity (data not shown), increased this limit to about 5 mg/ml for the mutant proteins and 2 to 3 mg/ml for wild-type PR. Most of the DLS experiments were carried out in the presence of these cosolvents, because their addition allowed the concen-tration of dPR to be increased to the level where NMR exper-iments became feasible (see below).
At pH 8.0 and 3 mg/ml,⌬LAM (Fig. 4A) appeared mono-meric by DLS, with an inferred molecular mass of⬃15 kDa. As expected, vPR appeared dimeric, with an inferred mass of⬃24 kDa (data not shown). (The actual masses of these proteins are 13.3 and 27.4 kDa, respectively, and the accuracy of mass
measurements by DLS is about⫾10% for globular proteins.) Similar results were obtained by glycerol gradient sedimen-tation. These data support the hypothesis that the mutations hindered dimer formation. Since retrovirus PRs in general and RSV PR in particular are active at acid pH but show little activity at pH 8, DLS measurements also were carried out near pH 6. We were surprised to find that under these conditions the predominant form of⌬LAM was dimeric at 4 mg/ml (Fig. 4B). However, reducing the protein concentration to 0.5 mg/ml shifted the protein to a monomeric molecular mass (Fig. 4C). These results suggested that the mutant PR is in a monomer-dimer equilibrium governed by aKdin the range of 50M for
[image:5.612.313.551.69.405.2]these solvent conditions. By definition, at a 50M monomer concentration there is an equal concentration of dimer, and thus the total concentration of PR would be 150M, corre-sponding to about 2 mg/ml. We found that the presence of 1% glycerol plus 0.4 M urea shifted the equilibrium slightly toward monomers, so that⌬LAM was largely monomeric at concen-trations as high as 3 mg/ml but became predominantly dimeric FIG. 3. Peptide cleavage assay. Samples of mutant or wild-type PR
[image:5.612.54.289.336.646.2]were incubated with 0.4 mM decapeptide corresponding to the reverse transcriptase-integrase cleavage site, under the same conditions as de-scribed for Fig. 2, except for the omission of glycerol. The reaction products were separated by reverse-phase HPLC. RepresentativeA210 traces are shown. The elution times of the decapeptide substrate and the N-terminal cleavage product were 20 and 17 min, respectively. The concentration of PR and the assay time for each reaction are shown on top.
FIG. 4. Size distribution by DLS. Samples of wild-type and mutant PR were passed through a 20-nm-pore-size filter and then submitted to DLS analysis. Representative tracings compiled by the DynaLS soft-ware of the DynaPro molecular sizing instrument are shown. The vertical axis is scattering intensity, and the (logarithmic) horizontal axis is the hydrodynamic radius (in nanometers) of the PR in the sample. Monomers and dimers of PR are not resolved, but the position of the peak is determined by a weighted average of monomers and dimers present, and the width of the main peak is indicative of the homoge-neity of the sample.
on November 9, 2019 by guest
http://jvi.asm.org/
with a further increase in concentration to 5 mg/ml near the solubility limit (Fig. 4E and F). A parallel control sample of dPR at 2 mg/ml in the same solvent was dimeric as expected (Fig. 4D). The hydrodynamic radius values determined by Dy-naLS software are summarized in Table 2.
The ⌬LAM dimers observed at pH 6.0 and high protein concentrations must be different in specific activity and there-fore in structure from wild-type PR dimers. The logic for this conclusion is based on consideration of any monomer-dimer equilibrium and on the estimatedKdfor this reaction for
⌬LAM. In the HPLC assay, the highest concentration of⌬LAM tested for enzymatic activity was 0.2 mg/ml (equivalent to 15 M monomer), and this amount of mutant PR showed about a 1,000-fold reduction in activity compared with vPR. Assum-ing aKdof 50M, at 0.2 mg of total protein/ml about 30% of
the protein would be in dimers, vastly more than the 0.1% of activity observed. Even if theKdwere 500M, about 5% of the
protein would be dimeric at 0.2 mg/ml, far too high to explain the feeble activity observed at this concentration. Therefore, we conclude that the mutant dimers observed by DLS are severely compromised in their function and thus that they differ somehow in structure from wild-type dimers. Possibly, the mutant dimers observed by DLS, like monomers, lack enzymatic activity altogether. In that case, the tiny amount of enzymatic activity manifested by⌬LAM would be attributed to a very minor fraction of mutant protein that successfully forms a “wild-type” dimer, which would be present at much too low a level to be detectable by any physical measurements.
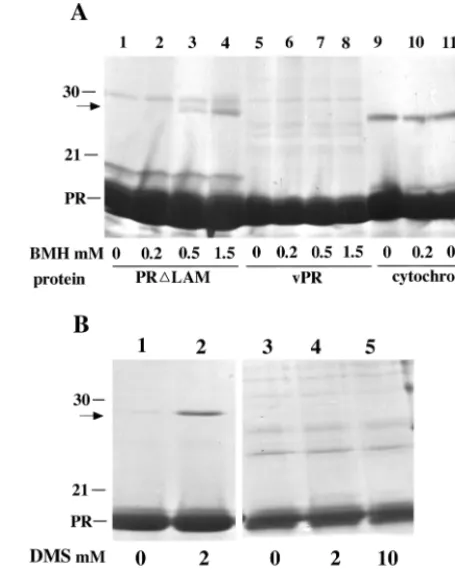
We used chemical cross-linking to provide supporting evi-dence for the existence of mutant PR monomers at alkaline pH and for structural differences between mutant and wild-type dimers at acid pH. Incubation of 1 mg of vPR/ml with 2 mM DMS, a bifunctional amino-reactive reagent, at pH 8.1 led to the appearance of a DMS-dependent dimer band by SDS-PAGE (Fig. 5B, lanes 1 and 2), as reported many years previ-ously in cross-linking experiments with intact virions (31).
[image:6.612.317.545.74.359.2]Un-der identical conditions, no specific cross-linking was observed for⌬LAM (lanes 3 to 5) or L1A, L1V, or L1I (data not shown), consistent with the conclusion from DLS and sedimentation analysis that at this pH these proteins are monomeric at the concentration tested. The chemical reaction between imidate and amino groups does not proceed well at acid pH, and hence DMS cannot be used effectively at pH 6.0. As an alternative, we chose a bifunctional reagent that reacts with sulfhydryl groups, BMH, to test for cross-linking at a pH where retrovirus PRs are active. The RSV PR polypeptide has a single sulfhy-dryl group, Cys113. In the crystal structure of the wild-type dimer, the SH groups of Cys113 and Cys113⬘are 26 Å apart, a distance that is too great to allow cross-linking by BMH, which spans only 16 Å. Consistent with this prediction, incubation of 1 mg of vPR/ml with BMH did not lead to the appearance of any BMH-dependent bands by SDS-PAGE (Fig. 5A, lanes 5 to 8). By contrast, at the same protein concentration incubation of⌬LAM yielded a distinct species migrating at the position of a dimer (lanes 1 to 4). As a specificity control, no BMH-dependent cross-linking was observed for cytochromec,which has two free SH groups (lanes 9 to 12). The fact that no dimers FIG. 5. Cross-linking analysis. (A) BMH cross-linking. Samples of 30l at 1.0 mg of protein/ml were treated for 30 min with BMH in Na-phosphate buffer (pH 6.0)–100 mM NaCl and then were quenched with 50 mM DTT. Lanes 1 to 4, PR⌬LAM; lanes 5 to 8, vPR; lanes 9 to 11, cytochromec. (B) DMS cross-linking. Samples of 20l at 1.0 mg of protein/ml were treated for 2 h with DMS in triethanolamine buffer (pH 8.1)–100 mM NaCl and then were quenched with 100 mM glycine. Lanes 1 and 2, vPR; lanes 3 to 5, PR⌬LAM. The positions of PR and molecular size markers of 30 and 21 kDa on Coomassie blue-stained SDS gels are indicated on the side, and the concentration of cross-linker in each reaction is shown at the bottom. The arrows indicate the positions of the dimers observed.
TABLE 2. Monomer or dimer status of wild-type and mutant PRs by DLS analysis
LaneconstructPR (mg/ml) pHConcn a
Peak value Mean value
Statusc Rh
(nm)b MW g
estimate (nm)Rhb MW g estimate
1 dPR 2 6.0 2.38 23 2.51 27 Dimer 2 PR-PRd 0.5 5.5 2.42 25 2.57 28 Dimer 3 ⌬LAM 3.5 8.0 1.95 15 2.11 18 Monomer 4 ⌬LAM 3.5 6.4 2.42 25 2.52 27 Dimer 5 ⌬LAM 2 6.4 2.41 25 2.40 25 Dimer 6 ⌬LAM 0.5 6.4 1.94 14 1.89 13 Monomer 7 ⌬LAMe 3 6.0 1.90 14 2.01 15 Monomer 8 ⌬LAMf 4.3 6.0 2.38 24 2.36 24 Mostly dimer 9 L1A 3.5 6.0 1.93 14 2.14 18 Monomer aBuffers were Tris-HCl (pH 8.0)–100 mM NaCl, sodium phosphate (pH 6.4)–100 mM NaCl, and sodium phosphate (pH 6.0)–100 mM NaCl–0.4 M urea–1% glycerol.
bHydrodynamic radius, R
h, as calculated by manufacturer’s software.
cMonomer or dimer status derived from the estimated molecular weight. This is calculated by DynaLS software from the mean hydrodynamic radius measured, using a standard curve of molecular weight versus Rhof globular molecules.
dCovalently linked, single-chain PR dimer (5) expressed inE. coli. eAverage of four experiments.
fAverage of two experiments. gMW, molecular mass in kilodaltons.
on November 9, 2019 by guest
http://jvi.asm.org/
were observed for vPR, even though the SH group is on the surface, further supports the conclusion that the cross-linked dimer species in⌬LAM reflects real protein-protein interac-tions. In summary, these results support the conclusion that the dimeric⌬LAM protein found at high protein concentrations differs from the wild-type dimer. We interpret the results to mean that the two Cys residues are within 16 Å of each other in the mutant dimer. However, the data do not exclude the possibility that the mutant dimer in question represents a pop-ulation of different dimeric species.
Comparison of ⌬LAM and dPR by NMR. Our long-term goal is to determine a high-resolution solution structure of a monomeric form of RSV PR, in order to gain insight into the structure of the PR domain of Gag before dimerization occurs. As a first step toward this goal, we obtained HSQC spectra for ⌬LAM and dPR proteins that had been labeled with15N. The 1H-15N HSQC experiment provides a two-dimensional
spec-trum in which the cross peak positions reflect the resonance frequencies, or chemical shifts, of both the protons and 15N
nuclei that share a covalent bond. Although resonance peaks in an HSQC can arise from NH and NH2correlations found in
some amino acid side chains, the spectrum is dominated by backbone amide correlations. Because this experiment results in a single unique cross peak for most amino acid residues, reflecting their chemical environment, the HSQC spectrum is often viewed as a “fingerprint” of the protein. Chemical shift perturbation mapping, also known as structure-activity rela-tionship by NMR, is a facile experiment wherein HSQC spec-tra are compared upon alteration of the system. Movement of peaks between HSQC spectra indicates that the residue as-signed to that peak has experienced a change in chemical environment, often indicative of a structural rearrangement or altered interaction, if there has been no change in the solvent system.
Peaks within the HSQC spectrum of the ⌬LAM protein were found to be widely dispersed in both dimensions (Fig. 6A), indicative of a stably folded protein. Resonance assign-ments for the backbone at a concentration of 3 mg/ml have been reported elsewhere (33a). At concentrations necessary for solution structure determination (3 to 5 mg/ml), DLS mea-surements indicated the presence of both monomeric and dimeric PRs. Since a single set of peaks is apparent in the HSQC spectrum, the monomer-dimer equilibrium is in fast exchange with respect to the NMR time scale. Hence, the observed resonances are a population-weighted ensemble av-erage. Increasing the concentration of PR from 1 to 5 mg/ml adjusted the equilibrium from predominantly monomer to pre-dominantly dimer according to DLS data. This observation was corroborated by15N relaxation measurements at 3 and 5 mg/
[image:7.612.58.287.68.696.2]ml, which yielded a statistically significant increase of average T1/T1 ratios with an increase in concentration (data not
FIG. 6.1H-15N HSQC spectra of⌬LAM PR at 3 mg/ml with back-bone assignments labeled (A),⌬LAM PR at 1 and 5 mg/ml (black and red contours, respectively) (B), and⌬LAM PR at 5 mg/ml and dPR at
2 mg/ml (red and black contours, respectively) (C). Insets in panel B show magnified cross peaks for residues that experience the largest peak shifts upon increase in concentration. Inset boxes have dimen-sions of 0.2 ppm of1H by 0.5 ppm of15N. In panel C, solid arrows denote residues that map to alpha-helix residues 112 to 118. Hollow arrows denote residues in the mobile flap region (residues 58 to 70), and the asterisk indicates the C terminus.
on November 9, 2019 by guest
http://jvi.asm.org/
shown) indicative of increased molecular correlation, or “tumbling” time. Increased average size is manifested by subtle changes in the HSQC spectrum (Fig. 6B). No peaks move more than 0.03 ppm in the1H dimension or 0.3 ppm in the15N
dimension, but peak movement can be detected for several peaks as shown in insets of Fig. 6B. The residues that do change do not map to the wild-type PR interface but rather are widely dispersed throughout the protein.
Comparison of the HSQC spectrum of dPR with that of the predominantly dimeric⌬LAM at 5 mg/ml (Fig. 6C) revealed discernible changes for approximately a quarter of the resi-dues. Since the dPR spectrum has not been assigned, definitive assessment of whether the chemical shift of a particular residue is unchanged is not possible. Many residues appear virtually unchanged in location and intensity, comprised primarily of the sharp, solvent-exposed residues of the mobile flap region (residues 58 to 70) and the C terminus (L124). Residues 112 to 118, which comprise most of the alpha helix found in the wild-type PR dimer, do not appear to shift significantly and are all of similar intensities. Most of the loops, with the exception of the a-b loop (residues 6 to 10) and the b-c loop (residues 21 to 27) (Fig. 7), also appear to be unchanged. Although many individual residues within the beta strands do not appear to move substantially, large stretches of beta-strand secondary structure are perturbed between the two HSQCs. This does not preclude the possibility of similar beta strands in both systems, since backbone chemical shifts of extended secondary structure are exquisitely sensitive to relatively small changes of torsion angles and hydrogen bond lengths. In summary, there are real differences between the structures of dimeric⌬LAM and dPR, but the two species may share many common features.
DISCUSSION
Proteolytic processing of Gag and Pol proteins occurs late in retrovirus maturation, but the mechanism by which processing is retarded until that time is unknown. It has been assumed that a proteolytic cascade is initiated by the dimerization of some of the PR domains of the polyprotein, followed by the autocatalytic excision of PR from the polyprotein in which it is
embedded. The notion that dimerization is the rate-limiting step in initiation of proteolysis stems in part from analysis of engineered linked dimers of HIV-1 and RSV PR (2, 4, 5, 9, 11, 23, 40). Several arguments suggest that the initial cleavage in the polyprotein occurs incis, i.e., that the dimerized PR do-mains act on the same polypeptides in which the PR dodo-mains are embedded (6, 10, 25). However, there are no definitive experiments that exclude cleavage intrans, i.e., the two dimer-ized PR domains attacking a neighboring third polyprotein molecule (12). In either case, once a mature dimeric PR is formed, it would act in trans to excise other PR domains, leading to a positive feedback loop, and also to cleave the polyproteins at other sites. To gain insight into the role of dimerization in activation of proteolysis, we have studied the enzymatic and structural properties of several RSV PR mu-tants. Mutations at the N terminus were found to render PR grossly defective in enzymatic activity in vitro, in assays both of proteolytic processing and of peptide hydrolysis. These obser-vations extend those made previously in vivo in the RSV sys-tem (6, 28, 35, 39). The enzymatic defect is accounted for by the DLS observations that in the mutants the monomer-dimer equilibrium is dramatically shifted toward monomers.
More than one explanation is required to account for the observation that the several mutants do not dimerize properly. The four-stranded beta sheet comprising the N- and C-termi-nal five to six residues was known from previous studies to be of critical importance in dimer stability. For example, the beta sheet accounts for about 50% of the intersubunit ionic and hydrogen bond interactions between the two subunits (44). In the HIV-1 system, high concentrations of a peptide mimicking the N-terminal or C-terminal residues of PR inhibit enzyme activity, presumably by prying the dimer apart by competition (3, 36, 46). Thus, the extremely poor ability of⌬LAM to dimer-ize probably is due to the loss of backbone and side chain contacts between the N-terminal sequence of one subunit and the C-terminal sequence of the other. The⌬LAM dimer that apparently does form at high protein concentrations is some 1,000-fold reduced in specific activity, implying that it differs structurally from wild-type PR dimer. Comparison of the 1-and 5-mg/ml⌬LAM HSQC spectra revealed peak shifts that are not clustered about the wild-type dimer interface but rather are dispersed through other regions. If the mutant dimer is a single species, this result would imply a global folding difference compared with the wild-type dimer that would bring these dispersed regions together into a dimer interface. Alternatively, there may be multiple dimeric config-urations, which would indicate a loss of specificity in the dimer-ization process. In at least one of these putative dimers, the Cys residues must be within 16 Å to account for the cross-linking by BMH. We speculate that the mutant dimer may represent an intermediate folding state for the wild-type enzyme.
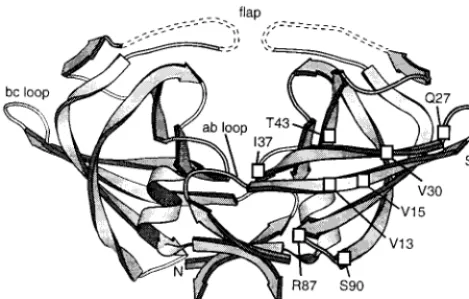
[image:8.612.57.292.73.222.2]In contrast to⌬LAM, the PR mutant L1A should be able to form the same backbone contacts of the beta sheet as wild-type PR, and therefore it must be the difference in side chains that accounts for the 1,000-fold reduction in its activity. The crystal structure of RSV PR (16) shows the Leu1 side chain to be buried in a hydrophobic pocket formed by the side chains of residues Val13, Leu36, Val84, Leu117, and Leu119 of the same subunit, plus Leu121⬘ of the other subunit. The carbon-to-carbon distances between relevant parts of these six side chains FIG. 7. Ribbon diagram of RSV PR derived from X-ray crystal
structure (16). Dashed lines denote mobile regions where no electron density was found. Amino acid residues of⌬LAM that shifted upon an increase in concentration from 1 to 5 mg/ml (Fig. 6B) are projected onto the diagram. The ribbon was created using Molscript (22).
on November 9, 2019 by guest
http://jvi.asm.org/
and the side chain of Leu1 are all less than 5.5 Å. Apparently, insertion of the Leu1 side chain into this pocket is critical for stabilization of the dimer. Either the empty pocket is unstable, causing a conformational change in the rest of the molecule that alters dimerization, or the loss of free energy associated with packing of the L1 side chain into the cavity destabilizes the dimer. It had been suggested previously that mutations of the residues involved in the conserved hydrophobic packing of retrovirus PRs would perturb enzymatic activity (44). We have not examined the L1V mutant for dimerization, but its low activity presumably reflects a very limited ability of the slightly shorter side chain of Val to fit properly into the pocket.
The weak activities of rPR and L1I, both of which have the initiating Met as an extra N-terminal residue, cannot be ac-counted for by either of the above arguments. We speculate that the enzymatic defect of these mutants also is due to lack of dimerization. The crystal structure of wild-type PR shows the amino group of Leu1 interacting with the oxygen atom of the amide of Asn123⬘. Any N-terminal extension would pre-vent this interaction. However, direct comparison between wild-type PR and these mutants is complicated due to the additional hydrophobic side chain of the Met residue. Because a minor species with a different N terminus would not be de-tected by N-terminal sequence analysis, it is not possible to as-sign the enzymatic activity observed for the rPR and L1I pro-teins to the major species carrying the initiating Met. Possibly, the major forms of these PRs are as diminished in activity as L1A, and the residual activity observed is due to limited re-moval of the initiating Met, thereby resulting in a small amount of wild-type PR in the case of rPR.
In summary, we suggest that the three types of mutations that cause loss of activity of RSV PR affect dimerization through different interactions. However, they all underscore the critical role played by the first few residues in stabilizing the dimeric form. Deletions at the N terminus prevent beta-sheet forma-tion. Changes in the Leu1 residue prevent proper interaction of the side chain with a hydrophobic pocket created by six other leucine and valine residues. N-terminal extensions pre-vent formation of a hydrogen bond with the penultimate Asn residue of the protein. We did not quantitate the extent of the enzymatic defect of the N-terminally extended PRs because of the difficulty in distinguishing between activity of the extended PR itself and activity of any wild-type PR that had been gen-erated therefrom by autocleavage. Based on recent results in the HIV-1 system (40), a fourth type of dimer-destabilizing mutation probably will need to be added to this list. The con-served Thr or Ser residue in the active-site sequence DTG or DSG apparently is critical for dimer stability. The hydroxyls of these two symmetrically positioned residues are a key part of the so-called “fireman’s grip,” a hydrogen bonding network that was believed to maintain the geometry of the active site (30). Strisovsky et al. (40) showed that loss of these hydroxyl groups was enough to cause dissociation of the HIV-1 PR dimer. However, activity could be maintained in the context of a linked dimer, implying that the fireman’s grip in fact is not essential for enzymatic activity. These results further demon-strate the delicate balance of forces that stabilize the dimer.
For HIV-1 PR, folding and dimer formation are hypothe-sized to be concomitant (12). Further evidence for this hypoth-esis was presented recently by Louis et al. (26). A four-residue
N-terminal deletion of HIV-1 PR was found to be largely unfolded unless a dimer-stabilizing inhibitor was added. This is in stark contrast to results reported here for the similar RSV ⌬LAM mutant, which is clearly well folded at the predomi-nantly monomeric concentration of 1 mg/ml. As the concen-tration is increased to 5 mg/ml where dimers are predominant, the HSQC remains largely unchanged. This is not consistent with a large-scale unfolded-to-folded structural reorganization. Therefore, we believe that the concomitant folding and dimer-ization model of retrovirus PRs does not extend to RSV PR. Comparison of the HSQCs of ⌬LAM and dPR reveals a number of residues that experience substantially different chemical environments. However, many structural features ap-pear to be conserved in the mutant protein, specifically includ-ing the mobile flap, alpha helix, and many of the loops and turns. Therefore, the general fold may well be similar to that of the wild-type PR. Since the N-terminally extended proteins His-ePR and the longer NC-PR (37) have a low autoprocessing activity and only barely detectable enzyme activity when mea-sured on substrates intrans, and since these proteins also are compromised in dimerization, we predict that the PR domain in these extended proteins will be found to fold like⌬LAM. This prediction remains to be tested. Furthermore, we hypoth-esize that the structure of ⌬LAM represents the prefolded status of the PR domain of the Gag polyprotein. This hypoth-esis can be addressed by comparing the⌬LAM HSQC with one obtained for an N-terminally extended construct such as His-ePR. Insight into the active-site geometry of precursor PR domains and the basis of low activity compared to mature PR may be important in understanding how proteolytic cleavage of RSV is initiated. Toward this end, we are pursuing high-reso-lution sohigh-reso-lution structures of⌬LAM PR and of extended PR forms.
ACKNOWLEDGMENTS
This work was supported by USPHS grant CA-20081 to V.M.V. and NSF grants MCB-9808727 and BIR-9512501 to L.K.N.
REFERENCES
1.Arad, G., M. Chorev, A. Shtorch, A. Goldblum, and M. Kotler.1995. Point mutation in avian sarcoma leukaemia virus protease which increases its activity but impairs infectious virus production. J. Gen. Virol.76:1917–1925. 2.Babe´, L. M., S. Pichuantes, and C. S. Craik.1991. Inhibition of HIV
pro-tease activity by heterodimer formation. Biochemistry30:106–111. 3.Babe´, L. M., J. Rose´, and C. S. Craik.1992. Synthetic “interface” peptides
alter dimeric assembly of the HIV-1 and 2 proteases. Protein Sci.1:1244– 1253.
4.Bizub, D., I. T. Weber, C. E. Cameron, J. P. Leis, and A. M. Skalka.1991. A range of catalytic efficiencies with avian retroviral protease subunits geneti-cally linked to form a single polypeptide chain. J. Biol. Chem.266:4951–4958. 5.Burstein, H., D. Bizub, and A. M. Skalka.1991. Assembly and processing of avian retroviralgagpolyproteins containing linked protease dimers. J. Virol. 65:6165–6172.
6.Burstein, H., D. Bizub, M. Kotler, G. W. Schatz, V. M. Vogt, and A. M. Skalka.1992. Processing of avian retroviral Gag polyprotein precursors is blocked by a mutation in the NC-PR cleavage site. J. Virol.66:1781–1785. 7.Cameron, C. E., B. Grinde, P. Jacques, J. Jentoft, J. Leis, A. Wlodawer, and
I. T. Weber.1993. Comparison of the substrate binding pockets of Rous sarcoma virus and human immunodeficiency virus type 1 proteinases. J. Biol. Chem.268:11711–11720.
8.Campbell, S., and V. M. Vogt.1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical par-ticles. J. Virol.71:4425–4435.
9.Cheng, Y. S. E., F. H. Yin, S. Foundling, D. Blomstrom, and C. A. Kettner. 1990. Stability and activity of human immunodeficiency virus protease: com-parison of the natural dimer with a homologous single-chain tethered dimer. Proc. Natl. Acad. Sci. USA87:9660–9664.
on November 9, 2019 by guest
http://jvi.asm.org/
10.Co, E., G. Koelsch, Y. Lin, E. Ido, J. A. Hartsuck, and J. Tang.1994. Proteolytic processing mechanisms of a miniprecursor of the aspartic pro-tease of human immunodeficiency virus type 1. Biochemistry33:1248–1254. 11.DiIanni, C. L., L. J. Davis, M. K. Holloway, W. K. Herber, P. L. Darke, N. E. Kohl, and R. A. Dixon.1990. Characterization of an active single polypeptide form of the human immunodeficiency virus type 1 protease. J. Biol. Chem. 265:17348–17354.
12.Grant, S. P., I. C. Deckman, J. S. Culp, M. D. Minnich, I. S. Brooks, P. Hensley, C. Debouck, and T. D. Meek.1992. Use of protein unfolding studies to determine the conformational and dimeric stabilities of HIV-1 and SIV proteases. Biochemistry31:9491–9498.
13.Grinde, G., C. E. Cameron, J. Leis, I. T. Weber, A. Wlodawer, H. Burstein, D. Bizub, and A. M. Skalka.1992. Mutations that alter the activity of the Rous sarcoma virus protease. J. Biol. Chem.267:9481–9490.
14. Grzesiek, S., and A. Bax.1993. The importance of not saturating H2O in
protein NMR—application to sensitivity enhancement and NOE measure-ments. J. Am. Chem. Soc.115:12593–12594.
15. Hirel, P. H., J. M. Schmitter, P. Dessen, G. Fayat, and S. Blanquet.1989. Extent of amino-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. USA86:8247–8251.
16. Jasko´lski, M., M. Miller, J. K. M. Rao, J. Leis, and A. Wlodawer.1990. Structure of the aspartic protease from Rous sarcoma retrovirus refined at 2 A resolution. Biochemistry29:5889–5898.
17. Joshi, S. M., and V. M. Vogt.2000. Role of the Rous sarcoma virus p10 domain in shape determination of Gag virus-like particles assembled in vitro and withinEscherichia coli.J. Virol.74:10260–10268.
18. Katoh, I., Y. Ikawa, and Y. Yoshinaka.1989. Retrovirus protease character-ized as a dimeric aspartic proteinase. J. Virol.63:2226–2232.
19. Kay, L. E., P. Keifer, and T. Saarinen.1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with im-proved sensitivity. J. Am. Chem. Soc.114:10663–10665.
20. Konvalinka, J., I. Blaha, R. Skrabana, J. Sedlacek, I. Pichova, F. Kapralek, V. Kostka, and P. Strop.1991. Subsite specificity of the proteinase from myeloblastosis associated virus. FEBS Lett.282:73–76.
21. Kotler, M., W. Danho, R. A. Katz, J. Leis, and A. M. Skalka.1989. Avian retroviral protease and cellular aspartic proteases are distinguished by ac-tivities on peptide substrates. J. Biol. Chem.264:3428–3435.
22. Kraulis, P. J.1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr.24:946–950. 23. Kra¨usslich, H.-G.1991. Human immunodeficiency virus proteinase dimer as
a component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA88:3213–3217.
24. Louis, J. M., R. A. McDonald, N. T. Nashed, E. M. Wondrak, D. M. Jerina, S. Oroszlan, and P. R. Mora.1991. Autoprocessing of the HIV-1 protease using purified wild-type and mutated fusion proteins expressed at high levels in Escherichia coli. Eur. J. Biochem.199:361–370.
25. Louis, J. M., N. T. Nashed, K. D. Parris, A. R. Kimmel, and D. M. Jerina. 1994. Kinetics and mechanism of autoprocessing of human immunodefi-ciency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc. Natl. Acad. Sci. USA91:7970–7974.
26. Louis, J. M., G. M. Clore, and A. M. Gronenborn.1999. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol.6:868– 875.
27. Muhandiram, D. R., and L. E. Kay.1994. Gradient-enhanced triple reso-nance 3-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. B103:203–216.
28. Oertle, S., and P.-F. Spahr.1990. Role of thegagpolyprotein precursor in packaging and maturation of Rous sarcoma virus genomic RNA. J. Virol. 64:5757–5763.
29. Partin, K., G. Zybarth, L. Ehrlich, M. Decrombrugghe, E. Wimmer, and C. Carter.1991. Deletion of sequences upstream of the proteinase improves the proteolytic processing of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA88:4776–4780.
30. Pearl, L. H., and W. R. Taylor.1987. A structural model for the retroviral proteases. Nature329:351–354.
31. Pepinsky, R. B., D. Cappiello, C. Wilkowski, and V. M. Vogt.1980. Chemical cross-linking of proteins in avian sarcoma and leukemia viruses. Virology 102:205–210.
32. Pepinsky, R. B., I. A. Papayannopoulos, S. Campbell, and V. M. Vogt.1996. Analysis of Rous sarcoma virus Gag proteins by mass spectrometry indicates trimming by host exopeptidase. J. Virol.70:3313–3318.
33. Pichova, I., P. Strop, J. Sedlacek, F. Kapralek, V. Benes, M. Travnicek, L. Pavlickova, M. Soujcek, V. Kostka, and S. Foundling.1992. Isolation and biochemical characterization and crystallization of the p15Gag proteinase of myeloblastosis associated virus expressed in E. coli. Int. J. Biochem.24:235– 242.
33a.Reinking, J. L., G. W. Schatz, V. M. Vogt, and L. K. Nicholson.2001.1H,15N
and13C chemical shift assignments of a monomeric N-terminal mutant of the
Rous sarcoma viral protease. J. Biomol. NMR19:279–280.
34. Ridky, T. W., D. Bizub-Bender, C. E. Cameron, I. T. Weber, A. Wlodawer, T. Copeland, A. M. Skalka, and J. Leis.1996. Programming the Rous sarcoma virus protease to cleave new substrate sequences. J. Biol. Chem.271:10538– 10544.
35. Schatz, G. W., I. Pichova, and V. M. Vogt.1997. Analysis of cleavage site mutations between the NC and PR Gag domains of Rous sarcoma virus. J. Virol.71:444–450.
36. Schramm, H. J., H. Nakashima, W. Schramm, H. Wakayama., and N. Yamamoto.1991. HIV-1 reproduction is inhibited by peptides derived from the N- and C-termini of HIV-1 protease. Biochem. Biophys. Res. Commun. 179:847–851.
37. Se´llos-Moura, M., and V. M. Vogt.1996. Proteolytic activity of the NC-PR fragment of avian sarcoma and leukemia virus Gag protein expressed in E. coli. Virology221:335–345.
38. Stewart, L., G. W. Schatz, and V. M. Vogt.1990. Properties of avian retro-virus particles defective in viral protease. J. Virol.64:5076–5092. 39. Stewart, L., and V. M. Vogt.1993. Reverse transcriptase and protease
activ-ities of avian leukosis virus Gag-Pol fusion proteins expressed in insect cells. J. Virol.67:7582–7596.
40. Strisovsky, K., U. Tessmer, J. Langner, J. Konvalinka, and H.-G. Kra¨uss-lich.2000. Systematic mutational analysis of the active-site threonine of HIV-1 proteinase: rethinking the ‘fireman’s grip’ hypothesis. Protein Sci. 9:1631–1641.
41. Strop, P., J. Konvalinka, D. Stys, L. Pavlickova, I. Blaha, J. Velek, M. Travnicek, V. Kostka, and J. Sedlacek.199l. Specificity studies on retroviral proteinase from myeloblastosis associated virus. Biochemistry30:3437–3443. 42. Swanstrom, R., and J. W. Wills.1997. Synthesis, assembly, and processing of viral proteins, p. 263–334.InJ. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, N.Y. 43. Vogt, V. M.1996. Proteolytic processing and particle maturation. Curr. Top.
Microbiol. Immunol.214:95–131.
44. Weber, I. T.1990. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases J. Biol. Chem.265:10492–10496.
45. Wlodawer, A., and J. W. Erickson.1993. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem.62:543–585.
46. Zhang, Z. Y., R. A. Poorman, L. L. Maggiora, R. L. Heinrikson, and F. J. Kezdy.1991. Dissociative inhibition of dimeric enzymes: kinetic character-ization of the inhibition of HIV-1 protease by its carboxyl terminal tetrapep-tide. J. Biol. Chem.266:15591–15594.
47. Zybarth, G., H.-G. Kra¨usslich, K. Partin, and C. Carter.1994. Proteolytic activity of novel human immunodeficiency virus type 1 proteinase proteins from a precursor with a blocking mutation at the N terminus of the PR domain. J. Virol.68:240–250.
48. Zybarth, G., and C. Carter.1995. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J. Virol.69:3878–3884.